

Abstract
Biological nitrogen oxide signalling and stress is an area of extreme clinical, pharmacological, toxicological, biochemical and chemical research interest. The utility of nitric oxide and derived species as signalling agents is due to their novel and vast chemical interactions with a variety of biological targets. Herein, the chemistry associated with the interaction of the biologically relevant nitrogen oxide species with fundamental biochemical targets is discussed. Specifically, the chemical interactions of nitrogen oxides with nucleophiles (e.g. thiols), metals (e.g. hemeproteins) and paramagnetic species (e.g. dioxygen and superoxide) are addressed. Importantly, the terms associated with the mechanisms by which NO (and derived species) react with their respective biological targets have been defined by numerous past chemical studies. Thus, in order to assist researchers in referring to chemical processes associated with nitrogen oxide biology, the vernacular associated with these chemical interactions is addressed.
Keywords: nitric oxide, nitrogen oxides, nitrogen dioxide, dinitrogen trioxide, peroxynitrite, metal nitrosyls, nitrosation, nitrosylation, nitrosative stress, nitrosative signalling
Introduction
Due to the discovery of endogenous nitric oxide (NO) generation in mammalian cells, the chemical biology of NO has been an important research topic for several decades (for an initial description see, Wink and Mitchell, 1998; for more recent reviews, see Thomas et al., 2008; Toledo and Augusto, 2012) (note: ‘nitric oxide’ is the common name for the molecule NO. The systematic name for NO is nitrogen monoxide. However, in the current literature, the use of ‘nitric oxide’ predominates and will be used exclusively herein). The biological effects of nitric oxide and derived nitrogen oxide species (this is a term that collectively refers to all nitrogen oxides including NO2, N2O3, NO2−, NO3−, etc.) are a result of their fundamental chemical reactions with specific biological targets. The remarkably versatile and unique chemistry of NO and related nitrogen oxides (at times collectively referred to as ‘reactive nitrogen species’) is ideal for mediating numerous signalling networks. Indeed, cellular and physiological processes have exploited the novel and specific chemistry of NO with a variety of biological targets to evolve and develop signalling systems such as the regulation of vascular tone. Aberrant production of NO, often via misregulation of NO biosynthesis or problems associated with its availability, can lead to pathophysiological conditions. In such cases, problems with spatial and temporal NO generation can lead to disease states. The accurate description of NO chemistry and derived species with biological targets not only provides important insight into mechanisms of action but also provides guidance in the development of potential pharmacological agents/strategies to treat different diseases.
The fundamental chemistry of NO has been examined extensively. Importantly, these chemical studies involving the reactions of NO with inorganic and organic molecules have provided a framework for explaining the physiology and pathophysiology of NO. However, important aspects to consider when extrapolating purely chemical studies to an understanding of biological systems are potential differences in conditions, especially reactant concentrations and environment. Previous chemical studies have also provided a lexicon for describing distinct chemical processes. Therefore, it is important to be consistent and specific when referring to chemical phenomena in biological systems since a seemingly minor change in word usage can imply a significant difference in the chemistry. That is, use of proper chemical terms is vital to providing a rigorous description of chemical processes associated with NO biology. Improper use of terms/descriptors can be misleading and may misrepresent the intimate mechanisms of NO biology. Unfortunately, much of the current literature associated with nitrogen oxide biology/physiology uses a mixture of chemical terms to describe biological phenomena that is at times confusing, misleading and/or chemically incorrect. In this review, the biologically relevant chemistry of NO is described with an emphasis on the terminology and nomenclature of these processes.
Nitric oxide
A simple Lewis dot depiction of NO shows that it has one unpaired electron (Figure 1). Formal convention often requires that free radicals such as NO be depicted as ‘·R’ to clearly indicate the presence of the single unpaired electron. Herein, this convention has not been adopted for several reasons. First, a more rigorous understanding of the nature of the unpaired electron (by molecular orbital theory) indicates that it is not localized on either the nitrogen or oxygen but is delocalized over both atoms (although shared unevenly towards the nitrogen nucleus). Second, in the absence of any charges, it is clearly implied that NO has an unpaired electron thus it need not be shown explicitly. And finally, a similar molecule, O2, actually has two unpaired electrons and few attempts to depict this in text are seen (i.e. O2 is rarely depicted as ·O2·).
Figure 1.
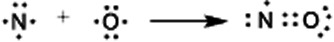
Lewis dot depiction of NO.
Although there is an internationally agreed upon nomenclature (International Union of Pure and Applied Chemistry) for both organic and inorganic compounds, common names become widely used when the formal system is cumbersome. Common names are widespread in the nitrogen oxide literature as is use of NO rather than ·NO. Here, we are attempting to clarify the nomenclature rather than remake or formalize it. Regardless, that NO has an unpaired electron is one of its most important properties. Most organic molecules have all their electrons paired in either bonds or in non-bonding orbitals (e.g. lone pairs). Such molecules repel a magnetic field and thus are called diamagnetic. In contrast, unpaired electrons are attracted to magnets, leading to paramagnetic species. The rates of reactions between diamagnetic and paramagnetic molecules are often limited by high kinetic barriers due to spin forbiddenness. Thus, NO tends to diffuse readily through the medium of a cell until encountering other paramagnetic species.
At first glance, it appears that NO would either readily gain or lose an electron in order to have both atoms obey the octet rule, which states that for molecules containing main group elements, each nucleus should be surrounded by eight bonding and/or lone pair electrons so as to adopt an inert gas electron configuration. Reduction to NO− or HNO would pair the lone electron while oxidation to NO+ would result in loss of this electron. In both cases, a Lewis structure can be drawn whereby both atoms possess an octet. However, NO is a stable molecule without a high propensity to be reduced or oxidized or even to dimerize in a biological system. The reasons for the stability of NO as a paramagnetic species are numerous and are covered elsewhere (e.g. see Fukuto et al., 2012). The important consideration is that although NO is a free radical, it is remarkably unreactive. This characteristic is critical for NO to function as a signalling molecule. In most cases, NO is produced in one cell for the purpose of eliciting a response in a neighbouring cell. This is possible due to the neutral charge and low reactivity of NO with diamagnetic species (as are most organic molecules). In biological systems, NO primarily reacts with other paramagnetic molecules as well as with transition metals. The impact of this chemistry is described below.
Nitrosation versus nitrosylation
Before proceeding further with a discussion of nitrogen oxide chemistry, it is worthwhile to first address some basic nomenclature associated with this chemistry. Especially important and a significant problem with the vernacular of NO chemical biology lies in the use of the terms ‘nitrosation’ and ‘nitrosylation’. These terms have very specific chemical definitions. Therefore, defining these terms (and their proper usage) provides important descriptors of detailed mechanisms that allude to important pharmacological and physiological insights.
The term ‘nitrosation’ refers specifically to chemical reactions involving the addition of a nitrosonium ion (NO+) to a nucleophilic group, such as an amine or thiolate. Heteronuclear diatomic molecules such as NO+ or NO have a strong tendency to react with electron donor species at the less electronegative atom, in this case nitrogen. The product can be described as a hybrid of both ionic and covalent resonance structures, with the latter formed from electron pair donation from the nucleophile to the NO+ (Reaction 1). The importance here is that the product has partial characteristics of both resonance structures.
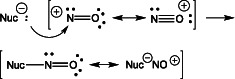 |
(1) |
In aqueous conditions, NO+ has an appreciable lifetime only under highly acidic conditions, and thus is generally not considered to be biologically relevant as an independent entity. Instead, nitrosation involves NO+ donors (species with significant ‘NO+-like’ character), formed by processes described in more detail below. Thus, the term nitrosation should be used when referring to chemical or biochemical processes that involve reactions of electrophilic NO+ donors with nucleophilic centres.
The related term ‘nitrosative stress’ refers to the indiscriminate nitrosation of biological nucleophiles that can lead to cell death and/or pathophysiological conditions. Nitrogen oxides such as NO2 and ONOO− are potent oxidants (vide infra) rather than nitrosating agents and toxicity associated with these species should not be referred to as nitrosative stress. It should be noted, however, that although NO2 itself is not a nitrosating agent, since it cannot directly participate in nitrosation chemistry, it can serve as a precursor to nitrosating species (vide infra). Therefore, ‘nitrosative stress’ should not be used as a general descriptor of all NO-mediated cellular stresses but rather used when referring specifically to stresses associated with nitrosation chemistry. Recently, nitrosation events in cellular signalling have been recognized as a new mechanism in cellular regulation (e.g. Marozkina and Gaston, 2012). ‘Nitrosative signalling’ is a non-toxic event that is part of a signal cascade to elicit a specific biological response. In cancer, for example, nitrosative stress resulting from an immune response leads to tumour eradication (e.g. Stuehr and Nathan, 1989). In contrast, nitrosative signalling in cancer cells leads to increased metastasis, proliferation and chemoresistance resulting in poor outcome in patients (e.g. Glynn et al., 2010; Ridnour et al., 2012; Switzer et al., 2012). Therefore, biological nitrosation chemistry can result in either a stress or a signalling pathway that is both context and concentration dependent.
The term ‘nitrosylation’ traditionally refers to direct addition of NO to a reactant. The term originates from chemistry that describes the coordination of NO to a metal centre to form a metal nitrosyl complex (Reaction 2) (e.g. Ford et al., 2005), with nitrosyl being the common name for NO when bound as a ligand (akin to ‘carbonyl’ being used to describe metal coordination complexes involving carbon monoxide, CO).
| (2) |
It is worth noting that metal nitrosyls can also be formed via other chemistries. For example, they can be generated by reaction of a metal complex with acidified nitrite, although this reaction may involve a change in the oxidation state of the metal in a multistep process (e.g. Ford, 2010). The first characterized and probably the most important physiological receptor for NO in mammalian systems is the metalloenzyme soluble guanylate cyclase (sGC) (e.g. Ignarro, 1999). Coordination of NO to the ferrous heme of sGC is the quintessential example of nitrosylation in biology.
In biological systems, many post-translational modifications of macromolecules are described using the suffix ‘-ylation’ such as ‘phosphorylation’, ‘methylation’ and ‘sumoylation’. With respect to NO, descriptions of its biological actions have often used the term ‘nitrosylation’ to generally describe appending an NO group to macromolecules (akin to the terms above). From a purely chemical perspective, ‘nitrosylation’ is not nearly as general a term (vide supra). Indeed, the ubiquitous use of ‘nitrosylation’ can obscure the important chemical and biological mechanisms involved in nitrogen oxide signalling. Possibly the most prevalent, established and important macromolecular signalling phenomenon is protein phosphorylation, a process that is carefully regulated and controlled via a multitude of specific kinases and phosphatases that, in turn, are controlled in numerous ways. Therefore, the use of nitro‘syl'ation to draw analogy to, for example, phosphorylation (and other established signalling systems) should be done with caution at this time since an analogous level of regulation established for these other signalling systems has not yet been demonstrated for ‘nitrosylation’.
The terms ‘nitrosation’ and ‘nitrosylation’ are often used when describing the formation of S-nitrosothiols (RS-NO, vide infra). S-nitrosothiols can be formed by translocation of an NO+ group from one sulfur to another (Reaction 3).
| (3) |
This process has been referred to as ‘transnitrosylation’ (e.g. Anand and Stamler, 2012). Since the chemistry of this transfer likely involves nucleophilic attack of a thiol at the electrophilic nitrogen atom of the S-nitrosothiol (Singh et al., 1996; Wong et al., 1998; Houk et al., 2003), this process is more appropriately termed transnitrosation (assuming the mechanism described above is being implied).
Transfer of a nitrosonium ion is presumably an important mechanism for modification of protein thiols. For example, Mitchell and Marletta (2005) have shown that the thiol redox protein thioredoxin can, with some specificity, transfer a nitrosonium species from one of its cysteines to the active site cysteine of capase-3. Thus, transnitrosation may in fact be a significant component of cellular signalling (e.g. Martinez-Ruiz and Lamas, 2007). Given the pre-existence of the terminology, their individual importance to nitrogen oxide signalling and the lack of similarity to other enzymatically controlled post-translational modifications, following the suggestion by Ford et al. (2005), we recommend that nitrosation be reserved exclusively for reactions involving transfer of NO+ and nitrosylation for direct addition of NO.
Formation of S-nitrosothiols
It has become increasingly clear that modification of both low molecular weight thiols and thiol-containing proteins occurs as a result of the biosynthesis of NO, and there are several possible chemical mechanisms for their generation. As alluded to in the above section, NO itself does not react with thiols or thiolates under biological conditions (or at the very least, this is a very slow process that generally precludes biological relevance) (Pryor et al., 1982). Modification of thiols by NO requires either prior oxidation of NO (to give an NO+-donor) or of the thiol (to give a thiyl radical, RS·).
Oxidation of NO
One-electron oxidation of NO to give free NO+ is highly unfavourable. In fact in aqueous systems, NO+ only has an appreciable lifetime in very strong acid. In purely chemical systems, NO+ can be formed by addition of nitrite (NO2−) to ∼4 M sulfuric acid, but such conditions are not physiological. However, species with NO+-like reactivity can be formed under milder conditions. For example, NO and nitrogen dioxide (NO2, another paramagnetic species) can combine to give dinitrogen trioxide (N2O3) (Reaction 4).
| (4) |
This reaction is reversible, suggesting that N2O3 can simplistically be described as an adduct of NO and NO2. However, NO2 is a reasonable one-electron oxidant, and thus N2O3 may possess some NO+NO2− character. In the presence of a nucleophile, a Nuc···NO+···NO2− intermediate would lead to transnitrosation from NO2− to the nucleophile, to produce for instance an S-nitrosothiol (Reaction 5).
| (5) |
In this way, N2O3 functions as an NO+ donor through transfer of NO+ rather than spontaneous release of free NO+.
Generation of S-nitrosothiols via reaction of thiols with N2O3 is a chemically established process. The formation of N2O3, as mentioned above, occurs when NO2 is generated in the presence of NO (Reaction 4). One mechanism to produce NO2 is through reaction of NO with O2 (autoxidation of NO; Reaction 6).
| (6) |
The paramagnetism of the reactants leads to a low kinetic barrier [high reaction rate constant, k, of 8 × 106·M−2·s−1 (Wink et al., 1993a; Keshive et al., 1996)]. However, the second order dependence on NO (rate = k[NO]2[O2]) also indicates that significant levels of N2O3 are formed only at high concentrations of NO. The high order kinetics of NO autoxidation indicates that the time required for an aerobic solution of NO to decay to half its original concentration varies hugely with concentration (on the order of hours for nanomolar and seconds for micromolar – note that this is not a true half life since this is not a first order decay process).
Under normal biological conditions/concentrations autoxidation of NO is generally considered to be too slow to be significant. However, higher levels of NO (and O2) may concentrate in lipophilic compartments (e.g. membranes) due to the favourable partitioning of these non-polar species from aqueous solution (Liu et al., 1998). Furthermore, inflammation leads to increased levels of NO. Alternatively, NO2 may be formed by a different route, such as through metal-mediated oxidation of nitrite (Thomas et al., 2008). At present, the biologically relevant mechanisms by which N2O3 is formed are not fully established.
However, that N2O3-mediated nitrosation chemistry does occur in vivo is clear and numerous studies in in vitro and in vivo systems allude to the relevance of N2O3 in mediating NO biology. Shinyashiki et al. (2001) provide just one example in which disruption of the actions of the thiol-containing, metal responsive yeast transcription factor ACE1 was observed under conditions consistent with N2O3 generation. ACE1 inhibition occurred upon addition of NO under aerobic conditions (Reactions 6 and 4), upon addition of nitrite to acidic solution (Reaction 7), or when NO and NO2 were present simultaneously (Reaction 4).
| (7) |
Since all of these conditions are amenable to N2O3 formation, it was concluded that N2O3 was the species responsible for modifying and inhibiting ACE1 activity.
Oxidation of thiols
S-nitrosothiols may also be formed by one-electron oxidation of the thiol to a thiyl radical followed by direct reaction with NO (Reactions 8 and9).
| (8) |
| (9) |
Thus oxidation of thiol to thiyl radical followed by addition of NO is termed oxidative nitrosylation. It needs to be stressed that NO does not directly react with thiols under any biological condition, therefore there is no direct nitrosylation mechanism for RSNO formation via reaction of NO with thiols (vide infra). The relevance of this nitrosylation pathway to S-nitrosothiol generation is, of course, dependent on the prevalence of thiyl radicals as well as competing reactions with thiol or O2. Significantly, many proteins/enzymes are known to possess cysteine thiyls (among other radical species) that are crucial for enzymatic activity (e.g. Stubbe and van der Donk, 1998). Thus, enzymes/proteins with existing thiyl radicals may be expected to be reactive with NO via Reaction 9. Oxidation of thiols to thiyl radicals typically is driven by the presence of strong one-electron oxidants such as HOO· and HO·. Two-electron oxidants such as H2O2 can instead produce the sulfenic acid, RSOH, which is not susceptible to nitrosylation. Both thiyl radicals and sulfenic acids can undergo further reactions to give other more stable species such as disulfides. Thus, for thiol oxidation to result in S-nitrosothiol formation requires coordination with NO biosynthesis.
NO2 is also a strong oxidant that is capable of oxidizing thiols. However, since NO and NO2 rapidly associate (Reaction 4), their simultaneous presence is commonly assumed to produce S-nitrosothiols via the nitrosation pathway. However, there have been several reports suggesting that thiols can effectively compete for the NO2 generated during autoxidation at low fluxes of NO such that S-nitrosothiol formation under these conditions may not be entirely due to a nitrosation event (Jourd'heuil et al., 2003; Koshiishi et al., 2007). This suggests that there may be more complex reaction mechanisms at low (physiological/pathophysiological) levels of NO. Regardless, it should be noted that the mechanism(s) by which NO-mediates S-nitrosothiol formation requires further investigation.
S-nitrosothiol chemistry
Both nitrosation (e.g. Reaction 5) and nitrosylation (Reaction 9) can result in production of an RSNO product. Thus, the nomenclature provides a distinction based on the chemical mechanism rather than the nature of the product. In fact, that the same product can arise from both processes is indicative of the nature of S-nitrosothiols. The occurrence of transnitrosation (Reaction 3) suggests RS−NO+ character. But, S-nitrosothiols are also suggested to function as NO or NO− donors. Moran et al. (2011) have suggested that S-nitrosothiols should be considered as a combination of covalent, zwitterionic and ion pair resonance structures, R-S-N=O, R-S+=N-O−, RS−NO+, with the R-S-N=O form generally being dominant in the resonance hybrid. Whether an S-nitrosothiol undergoes homolytic cleavage to release NO (such as by photolysis) or heterolytic cleavage, transferring either NO+ or NO− to a nucleophile or electrophile, respectively, will be highly condition-dependent. This is analogous to the chemistry already described for N2O3, which spontaneously dissociates into NO and NO2 but can also transfer NO+ to a nucleophile.
Nitrosylation of metal complexes and metalloproteins
Analysis of the interaction of NO with metal centres both in proteins and in model complexes constitutes a very large literature. The reaction of NO with ferrous (FeII) systems is rapid, and the resulting complexes are highly stable, which are ideal properties for signalling. The ferrous heme protein sGC is commonly referred to as the primary biological target for NO. This enzyme catalyzes the cyclization of GTP to cGMP (Reaction 10).
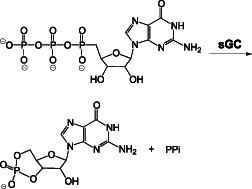 |
(10) |
Although sGC possesses basal activity, it is activated many fold by NO. Elucidation of the full mechanism of this activation process is hampered by the lack of detailed structural data for the large protein. However, it is generally accepted that nitrosylation of the regulatory ferrous heme, producing a ferrous-nitrosyl (FeII-NO) complex, leads to dissociation of the proximal histidine ligand (Traylor and Sharma, 1992). Although other small molecules such as O2 and CO also can bind to ferrous hemes, cleavage of the proximal ligand (due to a strong trans directing nature) is unique to NO. In turn, movement of the helix that includes the proximal histidine leads to structural changes that facilitate conversion of GTP to cGMP at a distant catalytic site.
NO reacts with a variety of other FeII systems including the heme centres of haemoglobin, myoglobin, NO synthase and cytochrome P450 (Khatsenko et al., 1993; Wink et al., 1993b; Stadler et al., 1994) as well as the iron-sulfur cluster of aconitase (Drapier and Bouton, 1996). In contrast to sGC, nitrosylation of most enzymes leads to regulation of function by reducing activity. For example, nitrosylation of the Co(II)(H2O) form of cobalamin (vitamin B12 derivative) results in a diminished ability for this complex to serve as a cofactor for methionine synthase (Brouwer et al., 1996).
Metal complexes can also function as NO scavengers. Haemoglobin, and to a lesser extent myoglobin, in particular is considered to regulate NO levels. In these proteins, NO reacts with the Fe(II)O2 complex to rapidly produce methaemoglobin (FeIII) and nitrate (Reaction 11, shown for haemoglobin, Hb) (Eich et al., 1996).
| (11) |
Nitrate is then excreted [or possibly recycled back to NO (Kapil et al., 2010)], and metHb can be enzymatically reduced back to the O2-binding ferrous haemoglobin.
NO also reacts with more oxidized metals. Nitrosylation of ferric (FeIII) complexes is generally considered to be reversible, due to lower binding affinities compared to the corresponding ferrous analogues (see Lim et al., 2005). However, nitrosylation of ferric heme centres can also lead to enzyme inhibition, as observed for catalase (Hoshino et al., 1993; Farias-Eisner et al., 1996). Furthermore, the resulting ferric nitrosyl complex often is written as FeII-NO+ rather than FeIII-NO, indicating that significant redistribution of electron density occurs upon binding [Reaction 12; also applicable to Mn(III)]. As with N2O3 and S-nitrosothiols, ferric nitrosyls can therefore be susceptible to nucleophilic attack (Reaction 13).
| (12) |
| (13) |
In this case, nitrosylation of the metal in the presence of a nucleophile leads to transnitrosation to the nucleophile and one-electron reduction of the metal centre. A second nitrosylation of the reduced metal centre then produces a stable ferrous nitrosyl complex (Reaction 14).
| (14) |
Together, this multistep process is called reductive nitrosylation (e.g. Gwost and Caulton, 1973; Wayland and Olson, 1973). Wade and Castro demonstrated that ferric nitrosyl complexes could nitrosate biomolecules including N-acetyl cysteine (Wade and Castro, 1990).
Nitrosation of other biological targets
Nitrosation is not limited to thiols, and the study of nitrosation chemistry is more than a century old (reviewed in Williams, 1983). Prior to the discovery that NO is biosynthesized, nitrosation of amines in the gastrointestinal tract became a concern as a potential source of carcinogens (Bartsch et al., 1990) (Reaction 15).
| (15) |
In fact, the detection of N-nitrosamines following activation of the immune system was critical to discovery of endogenous formation of NO (Stuehr and Marletta, 1985; Miwa et al., 1987; Hibbs, 1991). Formation of N-nitrosamines has also been proposed to play a role in carcinogenesis associated with inflammation (Marletta, 1988).
Primary amines (RNH2) such as found in nucleobases can also be nitrosated by either N2O3 or ferric nitrosyl complexes (Castro and Bartnicki, 1994). However, primary N-nitrosamines are unstable to further reaction as they can tautomerize. As shown in Figure 2 for cytosine, the N-nitrosamine can tautomerize and then dehydrate to produce a diazonium ion. This intermediate hydrolyzes to release N2 to produce an enol. Tautomerization to the keto form produces uracil. Importantly, Caulfield et al. (1998) have demonstrated that DNA structure plays an important role in this potentially mutagenic reaction.
Figure 2.
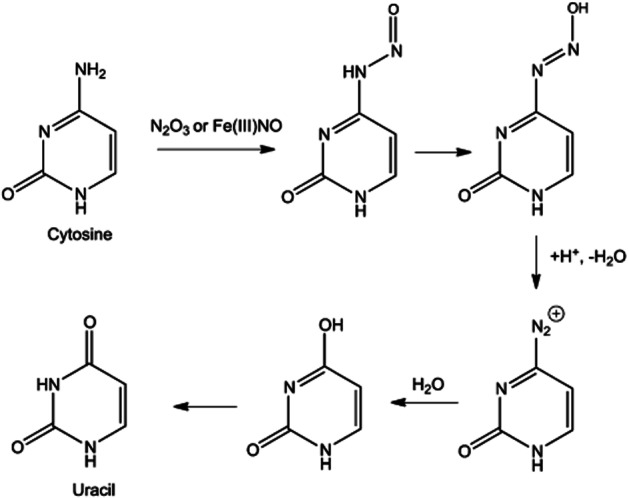
Nitrosation of cytosine leading to the conversion to uracil.
Reaction of NO with radical species
Unpaired electrons can impart high reactivity to many radical species in that they can be electron poor and, therefore, oxidants. The resistance of NO to dimerization is highly unusual for a radical, but is critical to its function as a signalling agent. NO does react rapidly with other radicals, as shown in Reactions 04 and 09. In these reactions, two radicals combine to form a diamagnetic species (formally via nitrosylation). Since O2 has two unpaired electrons, two NO molecules react with it (Reaction 06). Another important radical to consider with NO is superoxide (O2−). Although reduction of O2 is unfavourable, interaction of O2 with strong reductants can result in production of O2−. In particular, O2− can be produced by the respiratory chain necessitating a high concentration of superoxide dismutase in mitochondria. However, immune cells have evolved to utilize O2− in the control of invading microorganisms. Since the immune system also produces NO, the interaction of NO and O2− has received substantial attention both as a scavenger of NO and in the formation ONOO− (Reaction 16).
| (16) |
Peroxynitrite itself is not a radical species (i.e. it does not have any unpaired electrons). However, proton dependent decomposition of ONOO− has the potential to generate potent odd electron oxidants NO2/HO· as fleeting intermediates contained in a solvent cage in a reaction pathway that gives predominantly nitrate (NO3−) (Reaction 17) (e.g. Gunaydin and Houk, 2008).
| (17) |
The biochemical fate of ONOO− is highly dependent on its environment. Under normal biological conditions, reaction of ONOO− with CO2 is likely the major reaction giving nitrosoperoxycarbonate (Reaction 18). Further decomposition of nitrosoperoxycarbonate (via possible homolytic cleavage of the O-O bond, analogous to Reaction 17 above), generates potent oxidants, NO2/CO3·−, as fleeting intermediates on a reaction path that eventually forms nitrocarbonate (which will further decompose to give NO3−) (Reaction 19).
| (18) |
| (19) |
Thus peroxynitrite is not itself a nitrosating agent. However, the intermediates in H+- or CO2-mediated decomposition can produce powerful oxidants (NO2, HO· or CO3·−). Yet, in excess NO, these oxidants can be converted to a variety of species including the nitrosating agent N2O3 (Reactions 20, 21, 22). This balance between oxidative chemistry and nitrosative chemistry may provide an important interface between these two chemical conditions.
| (20) |
| (21) |
| (22) |
The biochemical consequences of ONOO− formation and degradation will be discussed further later.
In addition to small radicals such as NO2 or O2−, NO can react with carbon-centred radicals (R·) to give the corresponding C-nitroso compound (Reaction 23).
| (23) |
Most alkyl nitroso compounds are unstable with respect to tautomerization to the corresponding oxime (Reaction 24).
| (24) |
Such reactivity (i.e. tautomerization to an oxime) is not available to tertiary or phenyl nitroso compounds.
In aerobic systems, carbon-centred radicals can be converted into oxy- and peroxy-radicals. This can be particularly deleterious since these radicals can initiate and/or propagate chain reactions. That is, a single radical initiating event can lead to the destruction or modification of numerous molecules via propagation by chain-carrying and O2-derived reactive intermediates. One of the most important examples of this is lipid peroxidation. Production of a one-electron oxidant (e.g. HO·, NO2, HOO·, etc.) in a membrane lipid environment can lead to the generation of a carbon-centred radical via one-electron oxidation of an unsaturated fatty acid (typically by hydrogen-atom abstraction or addition to an unsaturation). In the presence of polyunsaturated fatty acids (PUFA), membranes are more susceptible to lipid peroxidation due to the fact that PUFAs are more easily oxidized compared to saturated or monounsaturated fatty acids. In an aerobic environment, the fatty acid radical will react with O2, but only one of the two unpaired electrons of O2 will be paired by this process. The resulting alkyl peroxyl radical (LOO·) is a reasonable one-electron oxidant that can propagate the chain reaction by oxidizing another fatty acid. This process is schematically depicted in Figure 3 using linoleic acid as an example.
Figure 3.
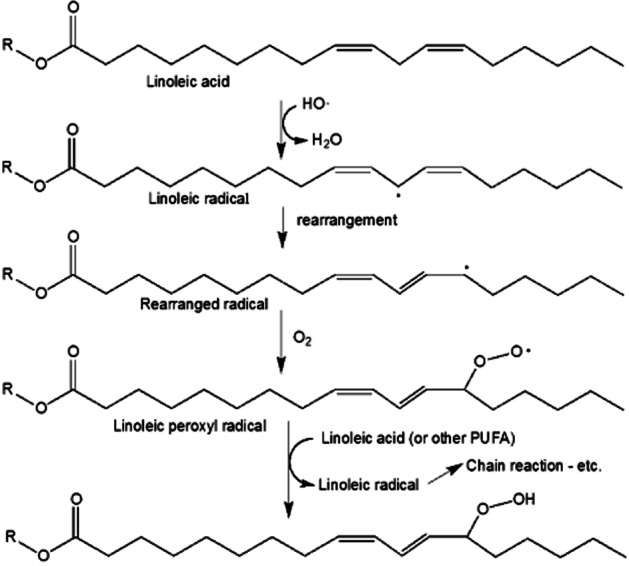
Lipid peroxidation mechanism.
Lipid peroxidation can be initiated during the inflammatory process and can ultimately lead to cell death by disrupting cell membranes (e.g. Higdon et al., 2012). There are multiple radical intermediates in the lipid peroxidation process (HO·, lipid radical, lipid peroxyl radical), all of which can react with NO (e.g. Reaction 25).
| (25) |
This quenching by NO of the radical nature of intermediates involved in the initiation or propagation of lipid peroxidation leads to termination of the process (e.g. Padmaja and Huie, 1993; Rubbo et al., 1995; Wink et al., 1995). This protective function of NO has been observed in both endothelial (Struck et al., 1995) and macrophage cells (Hogg et al., 1995). Clearly, the utility of NO in this regard will be highly dependent on having NO generated in proximity to these radical events and having it at high enough concentrations. Importantly, NO will partition favourably into lipid environments (Liu et al., 1998).
The build up of cholesterol in arteries produces atherosclerotic plaques, which are the cause of most coronary artery disease and strokes. Such plaques are initiated by accumulation of oxidized lipoproteins produced by the inflammatory response. Donors of NO have been found to prevent the macrophage-dependent oxidation of low-density lipoproteins by termination of lipid peroxidation (Hogg et al., 1993). This study indicates that NO may provide endogenous protection against plaque development. In fact, NO has been shown to be a more potent inhibitor of lipid peroxidation than the antioxidant α-tocopherol (vitamin E) (O'Donnell et al., 1997).
NO has also been designated as a pro-oxidant, capable of initiating radical chain chemistry. Such effects are not likely due to NO itself since it is such a poor oxidant (one-electron reduction of NO is highly unfavourable). Rather, NO2 and other strong oxidants, derived from the reaction of NO for instance with O2 and O2−, are likely responsible. Again, association of NO with such oxidants will lead to radical quenching and thus will limit lipid oxidation.
Lipid peroxidation can also be initiated indirectly by oxidation of metal centres. In biological systems, FeII and FeIII are the most common oxidation states. However, more oxidized species can form transiently, for example, due to reaction of H2O2 (Reaction 26).
| (26) |
Such highly reactive species [i.e. Fe(IV)=O or HO·, Reaction 1988). NO can inhibit this chemistry by reducing these oxidized centres to a normal valence state (Reaction 27) (Kanner et al., 1991; Wink et al., 1994), providing a protective mechanism against peroxide-mediated cytotoxicity (Wink et al., 1993c).
| (27) |
Oxygen-based radicals are also produced during normal metabolism, most notably the tyrosyl radical. Both ribonucleotide reductase and cyclooxygenase/prostaglandin H-synthase contain a catalytically important tyrosyl radical. Reaction with NO results in enzyme inhibition (for a review, see Guittet et al., 1999). The resulting suppression of DNA or prostaglandin synthesis is a possible deleterious function of NO. However, the mammalian immune system utilizes this inhibitory effect to combat pathogens.
As discussed immediately above, the anti- and pro-oxidative properties of NO are highly dependent on the concentration of NO as well as the biochemical environment. Thus, it is often difficult to predict the effects NO will have on a system without an intimate understanding of these issues.
Nitration via NO-derived species
In a formal sense, the term nitration is analogous to that of nitrosation, referring specifically to chemical reactions involving the addition of a nitronium ion (NO2+) to a nucleophilic group. However, nitration has been used extensively, particularly by organic chemists, as a general term to denote formation of a nitro compound without implying any specific mechanism of X-NO2 formation (X representing an atom to which an NO2 group has been added). As with NO+, the highly electrophilic NO2+ is only generated under highly acidic conditions (e.g. nitric acid in sulfuric acid). Such conditions have been used extensively to nitrate organic compounds but are not relevant to biological conditions. In contrast to nitrosation, a physiologically relevant NO2+ transfer agent has not been established. Thus, the likely and primary mechanism for nitration in cellular systems involves association of NO2 to existing radicals (comparable to nitrosylation). However, a unique term has not been defined for this process.
As already described, NO2 can be formed from the autoxidation of NO (Reaction 06) and from the reaction of NO with O2− (Reaction 16). In the first case, as discussed earlier, the second order dependence of NO on autoxidation to give NO2 limits its prevalence in the aqueous environment of cells but to a lesser extent in lipophilic regions. Also, NO2 generated from NO autoxidation will be kept at low levels due to further reaction with NO leading to N2O3 formation (thus not allowing accumulation NO2) (Reaction 04). Significantly, NO2 can also be formed in the absence of NO from oxidation of NO2− by H2O2 in the presence of metals (e.g. Burner et al., 2000; Monzani et al., 2004) (Reaction 28).
| (28) |
Again, the metal centres function as peroxidases as shown here for FeIII systems. In the first step, H2O2 leads to production of a more oxidized species (compound I; Reaction 29; please note that the metal oxidation states are shown primarily as a means of electron accounting and may not accurately represent the actual oxidizing species).
| (29) |
This oxidized species can participate in two sequential one-electron oxidations of NO2−, ultimately regenerating the FeIII starting complex (Reactions 30 and 31).
| (30) |
| (31) |
There has been significant speculation that biological nitration primarily follows this pathway. Nitrite is an end-product of NO biosynthesis, and thus can lead to NO2 formation by Reactions 30 and 31. Importantly, the possible temporal separation of NO biosynthesis from metal-mediated NO2− oxidation reduces the likelihood of reaction of NO2 with NO, which would lead to nitrosation rather than nitration (vide supra). Nitrite can also be ingested in the diet, which completely eliminates the dependence on NO biosynthesis.
In biological systems, typically an oxygen, carbon or other main group element is nitrated. It is well established that nitrated adducts, including those of tyrosine, tryptophan, fatty acids and guanosine, are prevalent biological species under conditions of NO-generation (e.g. Akuta et al., 2006; Souza et al., 2008; Nuriel et al., 2011; Schopfer et al., 2011) (Figure 4). Unlike many of the chemical processes involving nitrogen oxides, as described above, nitration is currently considered to be a biologically irreversible modification. That is, biologically mediated conversion of a nitrated species back to the original molecule is not reported to happen (although nitro group reduction has been reported).
Figure 4.
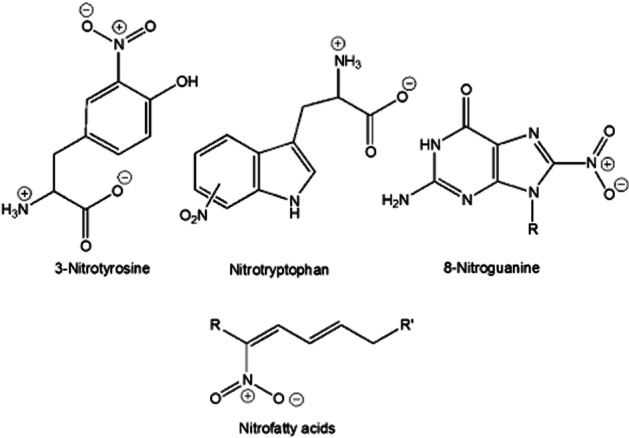
Examples of nitrated biological species.
The term ‘nitrative stress’ is often used to indicate conditions associated with nitration events on, for example, proteins or nucleotides leading to pathologies (e.g. Zaki et al., 2005). It is important to note the distinction between the terms ‘nitrosative stress’ and ‘nitrative stress’. As discussed earlier, nitrosative stress results from the nitrosation of biological molecules (e.g. protein cysteines) while nitrative stress is associated with nitration of biological molecules (e.g. protein tyrosines). ‘Nitrative stress’ has been characterized primarily via the detection of nitrated proteins. For example, nitration of tyrosines (3-nitrotyrosine formation) detected by antibodies is often used to characterize/indicate a nitrative stress. However, several important questions regarding this phenomenon remain. For example, is nitration merely a dosimeter of a chemical/biochemical condition? Are there any significant functional consequences of nitration? To date, answers to these questions are not well established. A further complication is that nitration of tyrosine may result from oxidative or nitrosative conditions (vide infra) indicating that nitration does not represent or result from a unique biochemical condition.
One mechanism of 3-nitrotyrosine formation from interaction with ONOO− and derived species has been determined to involve first oxidation of tyrosine by the carbonate radical anion (CO3·−) generated from the nitrosoperoxycarbonate intermediate (vide supra), and then reaction of the tyrosyl radical with NO2 (Reactions 17, 18, 19, 32, 33) (e.g. Gunaydin and Houk, 2009).
| (32) |
| (33) |
It should be noted that the yield of 3-nitrotyrosine by this chemistry is highly dependent on the relative concentrations and fluxes of NO and O2− (the precursors to ONOO−) with the highest yield occurring at equal fluxes. Moreover, even under ideal conditions a less than 40% yield is realized under purely chemical conditions (Goldstein et al., 2000).
Since 3-nitrotyrosine is formed by the reaction of the tyrosyl radical with NO2 (Reaction 33), any oxidant capable of removing an electron from tyrosine in the presence of NO2 can lead to 3-nitrotyrosine production. Importantly, NO2 itself is capable of oxidizing tyrosine to Tyr-O·, and thus any system that generates NO2 can lead to the formation of 3-nitrotyrosine.
The reaction of the tyrosyl radical with NO is also facile. Interestingly, the product 3-nitrosotyrosine has been suggested to further oxidize to give 3-nitrotyrosine via an intermediate iminoxyl radical (Gunther et al., 2002). In this pathway, an existing tyrosyl radical reacts with NO to give a ‘weak complex’ that tautomerizes to 3-nitrosotyrosine. Subsequent tautomerization to an oxime can be followed by one-electron oxidation generating an iminoxyl radical intermediate that is further oxidized to give an electrophilic species capable of reacting with water to give 3-nitrotyrosine (Figure 5).
Figure 5.
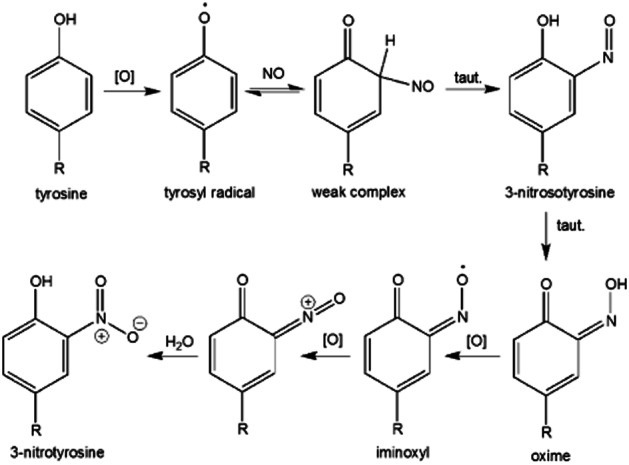
Possible mechanism of 3-nitrotyrosine formation from tyrosyl radical and NO.
Conclusion and summary of terms
The biological chemistry of NO is dominated by its ability to coordinate to metals and to react with other radical species. The subsequent chemistry that is initiated by the formation of metal nitrosyls and NO-radical adducts is important to the ultimate fate and chemical biology of NO. Importantly, much of the lexicon of nitrogen oxide chemistry was established prior to the relatively recent explosion of biological interest/application. Unfortunately, a lack of adherence to the established chemical terminology can lead to problems or misunderstandings regarding the intimate chemical mechanisms and processes being described. Herein described are some of the biologically relevant chemistry of NO (and derived species) along with what we believe to be the proper terminology.
To be sure, not all biological/biochemical terms are as chemically rigorous or descriptive as would be preferred. For example (and particularly relevant to this discussion and mentioned above) is the term ‘phosphorylation’. In biochemistry textbooks, the term phosphorylation is used to describe the transfer of a phosphate from the atom of one biomolecule to another (typically from one oxygen atom to another). ‘Phosphoryl’ groups are derivatives of phosphoric acid (H3PO4). Therefore, ‘phosphorylation’, as used in biochemistry, is a term that describes the products of the reaction but not the mechanism. However, it needs to be noted that biochemical phosphorylation occurs via attack of a nucleophilic atom of a biomolecule at an electrophilic phosphorous atom of the phosporyl donor (to our knowledge, there are no other known reaction pathways) thus precluding a need for numerous mechanistically distinct terms.
Since formation of an X-NO bond can occur by two distinct mechanisms, (described above), it is preferable to use a mechanistically defined term when possible. The question then arises as to the proper nomenclature when the mechanism is neither known nor implied. Both nitrosation and nitrosylation are often used in the literature to denote the general (mechanistically ill-defined) formation of X-NO compounds. However, given that both terms inherently describe a mechanism of formation, neither term is appropriate in cases where the mechanism is unknown or ambiguous. Rather, a more generic description such as thiol modification or S-nitrosothiol production is recommended (in the case of thiol targets). Currently, there is the tendency for many to view ‘nitrosation’ as a chemical term that specifically denotes NO+-mediated modification of a nucleophilic species (such as a thiol) and ‘nitrosylation’ as a biological term that denotes the mere formation of an X-NO species (X = S, N, etc.) without regard to mechanism. However, it needs to be stressed that nitrosylation is also a chemical term with mechanistic implications (vide supra) and use of nitrosylation to merely draw analogy to, for example, phosphorylation seems unwarranted at this time.
Below is a summary of the terms discussed in this review.
Nitrosation: refers to the addition of a nitrosonium ion (NO+) to a nucleophilic centre (e.g. a thiol or amine) either directly or by transfer from an NO+ donor (e.g. N2O3 or FeIINO+).
Transnitrosation: the transfer of NO+ from one nucleophilic centre to another.
Nitrosative stress: a biological stress caused by nitrosation of biological molecules.
Nitrosylation: refers to the direct formation of a nitrosyl species (X-NO; X = metal centre or radical species) via direct reaction with NO.
Transnitrosylation: technically, this term would refer to the transfer of NO from one molecule to another. This process will likely involve NO dissociation from one system before binding to another. Use of transnitrosylation to indicate transfer of NO+ is incorrect and should be discontinued.
Nitration: any process that leads to the generation of an X-NO2 species (nitro group formation). This term is not mechanistically specific.
Nitrative stress: refers to a biological stress associated with nitration of biological molecules.
Glossary
- PUFA
polyunsaturated fatty acids
- sGC
soluble guanylate cyclase
Conflict of interest
There are no potential conflicts of interest.
References
- Akuta T, Zaki MH, Yoshitake J, Okamoto T, Akaiake T. Nitrative stress through formation of 8-nitroguanosine: insights into microbial pathogenesis. Nitric Oxide. 2006;14:101–108. doi: 10.1016/j.niox.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med. 2012;90:233–244. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch H, Ohshima H, Shuker DE, Pignatelli B, Calmels S. Exposure of humans to endogenous N-nitroso compounds: implications in cancer etiology. Mutat Res. 1990;238:255–267. doi: 10.1016/0165-1110(90)90017-6. [DOI] [PubMed] [Google Scholar]
- Brouwer M, Chamultrat W, Ferruzzi G, Sauls DL, Weinberg JB. Nitric oxide interactions with cobalamins: biochemical and functional consequences. Blood. 1996;88:1857–1864. [PubMed] [Google Scholar]
- Burner U, Furtmuller PG, Kettle AJ, Koppenol WH, Obinger C. Mechanism of reaction of myeloperoxidase with nitrite. J Biol Chem. 2000;275:20597–20601. doi: 10.1074/jbc.M000181200. [DOI] [PubMed] [Google Scholar]
- Castro CE, Bartnicki EW. The interconversion of nucleic acid bases by iron(III) porphyrins and nitric oxide. J Org Chem. 1994;59:4051–4052. [Google Scholar]
- Caulfield JL, Wishnok JS, Tannenbaum SR. Nitric oxide-induced deamination of cytosine and guanine in deoxynucleosides and oligonucleotides. J Biol Chem. 1998;273:12689–12695. doi: 10.1074/jbc.273.21.12689. [DOI] [PubMed] [Google Scholar]
- Drapier J-C, Bouton C. Modulation by nitric oxide of metalloprotein regulatory activities. Bioessays. 1996;18:549–556. doi: 10.1002/bies.950180706. [DOI] [PubMed] [Google Scholar]
- Eich RF, Li T, Lemon DD, Doherty DH, Curry SR, Aitken JF, et al. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- Farias-Eisner R, Chaudhuri G, Aeberhardt E, Fukuto JM. The chemistry and tumoricidal activity of nitric oxide/hydrogen peroxide and the implications to cell resistance/susceptibility. J Biol Chem. 1996;271:6144–6151. doi: 10.1074/jbc.271.11.6144. [DOI] [PubMed] [Google Scholar]
- Ford PC. Reactions of NO and nitrite with heme models and proteins. Inorg Chem. 2010;49:6226–6239. doi: 10.1021/ic902073z. [DOI] [PubMed] [Google Scholar]
- Ford PC, Fernandez BO, Lim MD. Mechanisms of reductive nitrosylation in iron and copper models relevant to biological systems. Chem Rev. 2005;105:2439–2455. doi: 10.1021/cr0307289. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA, et al. Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide and their derived species. Chem Res Toxicol. 2012;25:769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn SA, Boersma BJ, Dorsey TH, Yi M, Yfantis HG, Ridnour LA, et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J Clin Invest. 2010;120:3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S, Czapski G, Lind J, Merenyi G. Tyrosine nitration by simultaneous generation of NO and O2− under physiological conditions. J Biol Chem. 2000;275:3031–3036. doi: 10.1074/jbc.275.5.3031. [DOI] [PubMed] [Google Scholar]
- Guittet O, Roy B, Lepoivre M. Nitric oxide: a radical molecule in quest of free radicals in proteins. Cell Mol Life Sci. 1999;55:1054–1067. doi: 10.1007/s000180050356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin H, Houk KN. Molecular dynamics simulation of the HOONO decomposition and the HO·/NO2· caged radical pair in water. J Am Chem Soc. 2008;130:10036–10037. doi: 10.1021/ja711365e. [DOI] [PubMed] [Google Scholar]
- Gunaydin H, Houk KN. Mechanisms of peroxynitrite-mediated nitration of tyrosine. Chem Res Toxicol. 2009;22:894–898. doi: 10.1021/tx800463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther MR, Sturgeon BE, Mason RP. Nitric oxide trapping of the tyrosyl radical-chemistry and biochemistry. Toxicology. 2002;177:1–9. doi: 10.1016/s0300-483x(02)00191-9. [DOI] [PubMed] [Google Scholar]
- Gwost D, Caulton KG. Reductive nitrosylation of group VIIIb metals. Inorg Chem. 1973;12:2095–2099. [Google Scholar]
- Hibbs JB., Jr Synthesis of nitric oxide from L-arginine: a recently discovered pathway induced by cytokines with antitumor and antimicrobial activity. Res Immunol. 1991;142:565–569. doi: 10.1016/0923-2494(91)90103-p. [DOI] [PubMed] [Google Scholar]
- Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. Cell signaling by reactive lipid species: new concepts and molecular mechanisms. Biochem J. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S. Inhibition of low-density lipoprotein oxidation by nitric oxide. Potential role in atherogenesis. FEBS Lett. 1993;334:170–174. doi: 10.1016/0014-5793(93)81706-6. [DOI] [PubMed] [Google Scholar]
- Hogg N, Struck A, Goss SPA, Santanam N, Joseph J, Parthasarathy S, et al. Inhibition of macrophage-dependent low density lipoprotein oxidation by nitric-oxide donors. J Lipid Res. 1995;36:1756–1762. [PubMed] [Google Scholar]
- Hoshino M, Ozawa K, Seki H, Ford PC. Photochemistry of nitric oxide adducts of water-soluble iron(III) porphyrin and ferrihemoproteins studies by nanosecond laser photolysis. J Am Chem Soc. 1993;115:9568–9575. [Google Scholar]
- Houk KN, Hietbrink N, Bartberger MD, McCarren PR, Choi BY, Voyksner RD, et al. Nitroxyl disulfides, novel intermediates in transnitrosation reactions. J Am Chem Soc. 2003;125:6972–6976. doi: 10.1021/ja029655l. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. Nitric oxide: a unique endogenous signaling molecule in vascular biology. Biosci Rep. 1999;19:51–71. doi: 10.1023/a:1020150124721. [DOI] [PubMed] [Google Scholar]
- Jourd'heuil D, Jourd'Heuil FL, Feelisch M. Oxidation and nitrosation of thiols at low micromolar exposure to nitric oxide. J Biol Chem. 2003;278:15720–15726. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- Kanner J, Harel S, Granit R. Nitric oxide as an antioxidant. Arch Biochem Biophys. 1991;289:130–136. doi: 10.1016/0003-9861(91)90452-o. [DOI] [PubMed] [Google Scholar]
- Kapil V, Webb AJ, Ahluwalia A. Inorganic nitrate and the cardiovascular system. Heart. 2010;96:1703–1709. doi: 10.1136/hrt.2009.180372. [DOI] [PubMed] [Google Scholar]
- Keshive M, Singh S, Wishnok JS, Tannenbaum SR, Deen WM. Kinetics of S-nitrosation of thiols in nitric oxide solutions. Chem Res Toxicol. 1996;9:988–993. doi: 10.1021/tx960036y. [DOI] [PubMed] [Google Scholar]
- Khatsenko OG, Gross SS, Rifkind AB, Vane JR. Nitric oxide is a mediator of the decrease in cytochrome P450-dependent metabolism caused by immunostimulants. Proc Natl Acad Sci U S A. 1993;90:11147–11151. doi: 10.1073/pnas.90.23.11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiishi I, Takajo T, Tsuchida K. Regulation of S-thiolation and S-nitrosylation in the thiol/nitric oxide system by radical scavengers. Nitric Oxide. 2007;16:356–361. doi: 10.1016/j.niox.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Lim MD, Lorkovic IM, Ford PC. NO and NOx interaction with group 8 metalloporphyrins. J Inorg Biochem. 2005;99:151–165. doi: 10.1016/j.jinorgbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Liu X, Miller MJS, Joshi MS, Thomas DD, Lancaster JR., Jr Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci U S A. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marletta MA. Mammalian synthesis of nitrite, nitrate, nitric oxide and N-nitrosating agents. Chem Res Toxicol. 1988;1:249–257. doi: 10.1021/tx00005a001. [DOI] [PubMed] [Google Scholar]
- Marozkina NV, Gaston B. S-nitrosylation signaling regulates cellular protein interactions. Biochim Biophys Acta. 2012;1820:722–729. doi: 10.1016/j.bbagen.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc Res. 2007;75:220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- Miwa M, Stuehr DJ, Marletta MA, Wishnok JS, Tannenbaum SR. Nitrosation of amines by stimulated macrophages. Carcinogenesis. 1987;8:955–958. doi: 10.1093/carcin/8.7.955. [DOI] [PubMed] [Google Scholar]
- Monzani E, Roncone R, Galliano M, Koppenol WH, Casella L. Mechanistic insight into the peroxidase catalyzed nitration of tyrosine derivatives by nitrite and hydrogen peroxide. Eur J Biochem. 2004;271:895–906. doi: 10.1111/j.1432-1033.2004.03992.x. [DOI] [PubMed] [Google Scholar]
- Moran EE, Timerghazin QK, Kwong E, English AM. Kinetics and mechanism of S-Nitrosothiol acid-catalyzed hydrolysis: sulfur activation promotes facile NO+ release. J Phys Chem. 2011;115:3112–3126. doi: 10.1021/jp1035597. [DOI] [PubMed] [Google Scholar]
- Nuriel T, Hansler A, Gross SS. Protein nitrotryptophan: formation, significance and identification. J Proteomics. 2011;74:2300–2312. doi: 10.1016/j.jprot.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell VB, Chumley PH, Hogg N, Bloodsworth A, Darley-Usmar VM, Freeman BA. Nitric oxide inhibition of lipid peroxidation: kinetics of reaction with lipid peroxyl radicals and comparison with α-tocopherol. Biochemistry. 1997;36:15216–15223. doi: 10.1021/bi971891z. [DOI] [PubMed] [Google Scholar]
- Padmaja S, Huie RE. The reaction of nitric oxide with organic peroxyl radicals. Biochem Biophys Res Commun. 1993;195:539–544. doi: 10.1006/bbrc.1993.2079. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Church DF, Govindan CK, Crank G. Oxidation of thiols by nitric oxide and nitrogen dioxide: synthetic utility and toxicological implications. J Org Chem. 1982;47:156–159. [Google Scholar]
- Puppo A, Halliwell B. Formation of hydroxyl radicals from hydrogen peroxide in the presence of iron. Is hemoglobin a biological fenton reagent. Biochem J. 1988;249:185–190. doi: 10.1042/bj2490185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridnour LA, Barasch KM, Windhausen AN, Dorsey TH, Lizardo MM, Yfantis HG, et al. Nitric oxide synthase and breast cancer: role of TIMP-1 in NO-mediated Akt activation. PLoS ONE. 2012;7:e44081. doi: 10.1371/journal.pone.0044081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbo H, Parthsarathy S, Barnes S, Kirk M, Kalyanaraman B, Freeman BA. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch Biochem Biophys. 1995;324:15–25. doi: 10.1006/abbi.1995.9935. [DOI] [PubMed] [Google Scholar]
- Schopfer FJ, Cipollina C, Freeman BA. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinyashiki M, Pan CJG, Switzer CH, Fukuto JM. Mechanisms of nitric oxide-mediated disruption of metalloprotein function: an examination of the copper-responsive yeast transcription factor ace1. Chem Res Toxicol. 2001;14:1584–1589. doi: 10.1021/tx010102i. [DOI] [PubMed] [Google Scholar]
- Singh RJ, Hogg N, Joseph J, Kalyanaraman B. Mechanism of nitric oxide release from S-nitrosothiols. J Biol Chem. 1996;271:18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- Souza JM, Peluffo G, Radi R. Protein tyrosine nitration – functional alteration or just a biomarker. Free Radic Biol Med. 2008;45:357–366. doi: 10.1016/j.freeradbiomed.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Stadler J, Trockfeld J, Schmalix WA, Brill T, Siewert R, Greim H, et al. Inhibition of cytochromes P4501A by nitric oxide. Proc Natl Acad Sci U S A. 1994;91:3559–3563. doi: 10.1073/pnas.91.9.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struck AT, Hogg N, Thomas JP, Kalyanaraman B. Nitric oxide donor compounds inhibit the toxicity of oxidized low-density lipoprotein to endothelial cells. FEBS Lett. 1995;361:291–294. doi: 10.1016/0014-5793(95)00178-c. [DOI] [PubMed] [Google Scholar]
- Stubbe J, van der Donk WA. Protein radicals in enzyme catalysis. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Marletta MA. Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc Natl Acad Sci U S A. 1985;82:7738–7742. doi: 10.1073/pnas.82.22.7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr DJ, Nathan CF. Nitric oxide: macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzer CH, Glynn SA, Cheng RY, Ridnour LA, Ambs S, Wink DA. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol Cancer Res. 2012;10:1203–1215. doi: 10.1158/1541-7786.MCR-12-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JC, Jr, Augusto O. Connecting the chemical and biological properties of nitric oxide. Chem Res Toxicol. 2012;25:979–985. doi: 10.1021/tx300042g. [DOI] [PubMed] [Google Scholar]
- Traylor TG, Sharma VS. Why NO? Biochemistry. 1992;31:2847–2848. doi: 10.1021/bi00126a001. [DOI] [PubMed] [Google Scholar]
- Wade RS, Castro CE. Redox reactivity of iron(III) porphyrins and heme proteins with nitric oxide. Nitrosyl transfer to carbon, oxygen, nitrogen and sulfur. Chem Res Toxicol. 1990;3:289–291. doi: 10.1021/tx00016a002. [DOI] [PubMed] [Google Scholar]
- Wayland BB, Olson LW. Nitric oxide complexes of iron(II) and iron(III) porphyrins. J Chem Soc Chem Commun. 1973:897–898. doi: 10.1021/ja00826a013. [DOI] [PubMed] [Google Scholar]
- Williams DLH. Nitrosation mechanisms. Adv Phys Org Chem. 1983;19:381–429. [Google Scholar]
- Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into regulatory, cytotoxic and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Wink DA, Darbyshire JF, Nims RW, Saavedra JE, Ford PC. Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media: determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem Res Toxicol. 1993a;6:23–27. doi: 10.1021/tx00031a003. [DOI] [PubMed] [Google Scholar]
- Wink DA, Osawa Y, Darbyshire JF, Jones CR, Eshenaur SC, Nims RW. Inhibition of cytochromes P450 by nitric oxide and a nitric oxide-releasing agent. Arch Biochem Biophys. 1993b;300:115–123. doi: 10.1006/abbi.1993.1016. [DOI] [PubMed] [Google Scholar]
- Wink DA, Hanbauer I, Krishna MC, DeGraff W, Gamson J, Mitchell JB. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc Natl Acad Sci U S A. 1993c;90:9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wink DA, Hanbauer I, Laval F, Cook JA, Krishna MC, Mitchell JB. Nitric oxide protects against the cytotoxic effects of reactive oxygen species. Ann N Y Acad Sci. 1994;738:265–278. doi: 10.1111/j.1749-6632.1994.tb21812.x. [DOI] [PubMed] [Google Scholar]
- Wink DA, Cook JA, Pacelli R, Liebmann J, Krishna MC, Mitchell JB. Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol Lett. 1995;82/83:221–226. doi: 10.1016/0378-4274(95)03557-5. [DOI] [PubMed] [Google Scholar]
- Wong PSY, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, et al. Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- Zaki MH, Akuta T, Akaike T. Nitric oxide-induced nitrative stress involved in microbial pathogenesis. J Pharmacol Sci. 2005;98:117–129. doi: 10.1254/jphs.crj05004x. [DOI] [PubMed] [Google Scholar]