

Abstract
Background and Purpose
Homocysteine is an intermediate product formed during the metabolism of methionine, and is increased in cells with folate deficiency. Patients with hyperhomocysteinemia tend to develop cardiovascular disease. Here, we have examined the molecular mechanisms underlying the anti-proliferative and anti-migratory effects of folate on homocysteine-challenged rat aortic smooth muscle cells (RASMCs).
Experimental Approach
Cultures of RASMC were challenged with homocysteine and then incubated with folate added. Changes in p21/p27, AKT and RhoA were followed by RT-PCR, Western blotting and immunocytochemistry. Transfection and anti-sense techniques were also used. Cell viability, growth and migration were measured.
Key Results
Folate up-regulated p21/p27 through a Src/ERK-dependent mechanism that accounted for its anti-proliferative effects on RASMC. Folate protected RASMC from the effects of homocysteine by reducing AKT1, focal adhesion kinase (FAK), paxillin, and p190RhoGAP activation/phosphorylation, along with cytosolic levels of p21 and p27, and increasing RhoA activation. Overexpression of AKT1, but not of AKT2, induced p21/p27 phosphorylation and increased cytosolic p21/p27 levels, as did homocysteine treatment. By contrast, and similarly to folate treatment, transfection with dominant negative (DN) AKT1 counteracted these effects. Additionally, AKT was shown to be an upstream target of FAK activation. In RASMC overexpressing constitutively active RhoA, activation of RhoA mediated the anti-migratory effects of folate. Addition of Y27632 (a RhoA inhibitor) and DNRhoA counteracted the anti-migratory effects, confirming RhoA involvement.
Conclusion and implications
Folate was anti-proliferative and anti-migratory in homocysteine-challenged RASMC. Mechanisms underlying folate-mediated protection against the proatherosclerotic effects of homocysteine have been delineated.
Keywords: folate, homocysteine, p21/cip1, p27/kip1, RhoA, AKT, p190RhoGAP, cell migration
Introduction
Homocysteine is a sulfur-containing intermediate product formed during the metabolism of methionine. It inhibits endothelial function, and thus represents a risk factor for atherothrombosis. Folate is involved in the metabolic pathway of homocysteine, and its supplementation reduces plasma homocysteine levels (Brattstrom et al., 1988; Wald et al., 2002). Folate also improves endothelial function in patients with cardiovascular disease, through mechanisms that act independently of its effects on homocysteine (Doshi et al., 2002). For example, 5-methyltetrahydrofolate, an active metabolite of folate, acts to stabilize and regenerate production of tetrahydrobiopterin (Verhaar et al., 1998; Miller, 2008), which is an essential cofactor for eNOS. It can rescue endothelial cells with homocysteine-mediated injury and can increase eNOS activity (Stroes et al., 2000; Title et al., 2000; Sasaki et al., 2003).
Homocysteine treatment induces growth-promoting effects in vascular smooth muscle cells (Tsai et al., 1994; 1996; Buemi et al., 2001). However, homocysteine treatment also inhibits endothelial cell growth in a dose-dependent manner (Wang et al., 2002); thus, patients with hyperhomocysteinemia might be susceptible to developing atherosclerosis. Studies have extensively investigated the effects of folate on the survival and proliferation of various cell types. For example, folate regulates trophoblast invasion and placental development in normal early human pregnancy (Williams et al., 2011). It also increases the proliferation of rat embryonic neural stem cells via the notch signalling pathway (Jia et al., 2008; Liu et al., 2010). However, folate reduces the proliferation of glioblastoma cells by increasing DNA methylation (Hervouet et al., 2009). The differential effects of homocysteine and folate on cell proliferation might therefore be tissue dependent.
Cell-cycle progression is mediated by the activation of cyclins and cyclin-dependent kinases (CDKs), which initiate progression from the G1 and G2 phases of the cell cycle to the S-phase and mitosis respectively. During cell cycle progression, CDK inhibitors, such as p21/cip1 and p27/kip1, regulate the formation of cyclin-CDK complexes and prevent abnormal proliferation by inhibiting their catalytic activities (Sherr and Roberts, 1999). Because of a high level of homology in their primary structures, p21 and p27 are thought to inhibit their targets through similar mechanisms. Researchers have become increasingly interested in the cell cycle-independent role of p21/p27 in regulating cell migration, a process that appears to occur through cytoplasmic sequestration of p21/p27 from the nuclei. Protein kinase B (AKT) induces resistance to cytokine-mediated G1 arrest because of its phosphorylation of p27. This impairs nuclear import and leads to the accumulation of cytoplasmic p27 (Liang et al., 2002). In addition, AKT1 raises cell proliferation by increasing the cytosolic fraction of p21/p27, whereas AKT2 causes cell cycle arrest by binding to p21 and preventing its phosphorylation by AKT1 (Heron-Milhavet et al., 2006).
Studies have shown that p27 regulates Cell migration is regulated by p27 as shown by the markedly reduced motility in a variety of cell types with p27 knock-down, compared with that in wild-type (WT) cells (McAllister et al., 2003; Wu et al., 2006). Conversely, ectopic overexpression of the p27Kip1 gene stimulates cell migration (Nagahara et al., 1998; McAllister et al., 2003). Additionally, re-expression of WT p27 (kip1) and p27CK (-), a mutant p27 that cannot bind to cyclins and CDKs, rescued the migrating ability of p27-/- fibroblasts. These cells displayed increased numbers of actin stress fibres and focal adhesions, comparable to those in cells in which the Rho pathway is activated. Cells lacking p27 displayed increased levels of active RhoA. Inhibition of Rho kinase (ROCK), a downstream effector of Rho, also rescued the migrating ability of p27-/- cells in response to growth factors. p27 was shown to bind to RhoA, thereby inhibiting RhoA activation by interfering with the interaction between RhoA and its activators. RhoA activators are referred to as guanine-nucleotide exchange factors (GEFs) (Besson et al., 2004). Also, cytoplasmic p21/Cip1 directly inhibits ROCK, resulting in increased neurite outgrowth (Tanaka et al., 2002; Lee and Helfman, 2004). Overall, these findings suggest the involvement of p21/p27 in the regulation of cell migration through modulating the Rho pathway.
Furthermore, tumour cells with mislocalized p21 and p27 in the cytosol showed poor prognosis, possibly associated with the de-inhibition of cell cycle arrest by p21 and p27 and a concomitant increase in cell migration (Rosen et al., 2005; Denicourt et al., 2007). The levels of active AKT1 and its short-term and long-term activation states can also regulate various signalling pathways to modulate the balance between pro-migratory and anti-migratory factors in vascular endothelial cells (Somanath et al., 2006). However, the isoform of AKT responsible for p21 and p27 cytosolic sequestration and the exact role of cytosolic p21 and p27 in anti-migration in rat aortic smooth muscle cell (RASMC) have yet to be fully elucidated.
Evidence of the effects of homocysteine on vascular smooth muscle cell migration is accumulating. Very high concentrations of homocysteine (500 μM) promoted vascular smooth cell migration in a co-culture system through the effects of adipocyte-derived resistin. However, the physiological concentration of homocysteine is less than 100 μM and the up-regulation of resistin has been linked to several disease conditions, possibly through an inflammatory mechanism; these diseases included metabolic syndrome, insulin resistance and atherosclerotic cardiovascular disease (Jiang et al., 2009). By contrast, another study showed that folate treatment reduced chemokine release from peripheral blood mononuclear cells in hyperhomocysteinemic patients (Holven et al., 2002). Studies investigating the use of folate to reduce homocysteine levels have not shown consistent protection from homocysteine-mediated cardiovascular disease or stroke (Ambrosi et al., 1999; Miller et al., 2010). However, Bloor et al. (2010) showed that folate reduced neointimal thickness and superoxide formation and increased microvessel density in diabetic animals. In addition to folate's protective effects on vascular endothelial cells, this evidence suggests its beneficial effects on vascular smooth muscle cells and a possible role in the prevention of atherosclerosis.
Folate supplementation restores ischaemia-induced angiogenesis in rats with hypercholesterolemia (Sasaki et al., 2003). However, the anti-migratory effects of folate on homocysteine-challenged RASMC have yet to be addressed. We have investigated the molecular mechanisms underlying the protective effects of folate on homocysteine-treated RASMC, focusing on its anti-proliferative and anti-migratory effects.
Methods
Isolation and primary culture of RASMC
All animal care and experimental treatments complied with the protocols of the Animal Center at Taipei Medical University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of six animals were used in the experiments described here. RASMCs were isolated from the thoracic aorta of male Sprague–Dawley rats (275–325 g) using an explant technique (Fisher-Dzoga et al., 1973). In brief, after removal of the endothelium and adventitia, the aortic explants were cultured in DMEM supplemented with the following: 10% FBS (Invitrogen, Carlsbad, CA, USA), penicillin (100 U·mL−1), streptomycin (100 μg·mL−1), and 25 mM HEPES (pH 7.4). After 2 weeks, cells that had migrated out of the explants were removed using trypsinization and were subcultured. Cell identity was confirmed using immunostaining with an antibody specific to smooth muscle cell α-actin. Cells from passages 5–12 were used in the experiments.
Cell viability and 5-bromo-2-deoxyuridine (BrdU) assays
The viability of cells was determined by examining the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma) to formazan by mitochondrial dehydrogenase. In brief, cells were plated in 24-well plates at a density of 1 × 104 cells per well and treated with DMSO or various concentrations of folate or homocysteine for 24 h. Thereafter, MTT was added, and the culture mixtures were incubated for a further 4 h. Formazan that formed was dissolved by overnight incubation with 10 mM HCl containing 10% SDS, and the results were then measured at a wavelength of 570 nm. To monitor DNA synthesis, RASMC cultured on 0.17 mm 0.1% gelatin-coated coverslips were pretreated with homocysteine for 30 min; this was followed by 4 h of folate treatment. Before harvesting, cells were pulsed with BrdU (a thymidine analogue; 75 mg·mL−1), to monitor DNA replication for 14 h. Cells were fixed in 3.7% formaldehyde and permeabilized with 0.2% Triton X-100 in a PBS. To detect the extent of BrdU incorporation by cells subjected to various treatments, we incubated RASMC with a Texas red-conjugated anti-BrdU (1:100) at 4°C overnight. Subsequent procedures were conducted as described previously (Chang et al., 2009).
We designed two types of negative control: the complete immunohistochemistry procedure performed on cells without BrdU administration and the complete procedure omitting the primary antibody. We used DAPI (Invitrogen) to stain DNA and reveal cell nuclei.
Transfection of various RhoA and AKT constructs and antisense oligonucleotides of p21 and p27
The RhoA cDNAs (dominant negative, DN; and constitutive active, CA) in pUSEamp were purchased from Millipore (Bedford, MA, USA). Dr Jin Q. Cheng kindly donated myr-HA-AKT1, -AKT2 and –DNAKT1 in pcDNA (Molecular Oncology, H. Lee Moffitt Cancer Center). The pcDNA overexpression variants (2 μg in a 3.5 cm Petri dish) were transfected into RASMCs using jetPEI™ (Polyplus-transfection, San Marcos, CA, USA). The sense and antisense oligonucleotides of p21 and p27 have been described previously (Sue et al., 2009). Post transfection, the cells were plated in a DMEM containing a 10% FBS, and the levels of RhoA variants, AKT1/2 and p21/p27 were analysed using Western blotting.
Cell migration
For the cell migration assay, an IBIDI culture insert (IBIDI GmbH) consisting of two reservoirs separated by a 500-μm-thick wall was placed into 1 well of a 12-well plate and gently pressed on the top to ensure tight adhesion. An equal number of control and experimental RASMC (70 μL; 7 × 105 cells mL−1) were added to the two reservoirs of the same insert and incubated at 37°C with 5% CO2. After 16 h, the insert was gently removed to create a gap of approximately 500 μm. Cells pre-challenged with homocysteine for 30 min were subjected to folate treatment for 18 h. Cells migrating from the leading edge were photographed at 0 and 18 h using a CCD camera (Olympus SC-5, Melville, NY, USA) attached to a microscope system (BX51, Olympus) and cells were counted. The migration of treated cells was normalized to the migration of untreated control cells. Data are expressed as mean ± SD from five randomly selected fields.
Analysis of gene expression using reverse-transcription (RT)-PCR and Western blot analysis
The method used to obtain the total RNA for RT-PCR analysis has been described previously (Sue et al., 2009). Protein expression was examined after subcellular fractionation. To prepare the nuclear protein extracts, RASMCs in 10 cm2 dishes were treated with NE-PER™ nuclear extraction reagents (Pierce, Rockford, IL, USA) for the indicated times. Protease inhibitors were added after treatment with 20 μM folate or 50 μM homocysteine. The subsequent procedures for nuclear protein extraction were performed according to the manufacturer's instructions. The fraction containing nuclear protein was stored at −80°C until use. To prepare the cytosolic-membrane extracts, cells were collected and incubated in 0.1 mL of a hypotonic buffer (10 mM Tris, pH 7.5; 0.5 mM EDTA; 2 mM PMSF) at 4°C for 30 min. After centrifugation, the supernatant (cytosolic fraction) was collected, and the pellet was resuspended in 0.1 mL of a RIPA buffer and incubated at 4°C for 30 min. The resulting fractions were sheared through an insulin syringe using a 29 G needle 100 times. After centrifugation, the supernatant (membrane fraction) was collected for analysis. Western blotting was conducted using the following antibodies: p21, p27, phospho-ERK1/2 and phospho-190RhoGAP from BD Biosciences (San Jose, CA, USA); AKT1/AKT2, phospho-focal adhesion kinase (FAK), phospho-p27(Ser10), phospho-p21(Ser146), RhoA, GAPDH, Lamin A/C and pan-cadherin from Santa Cruz Biotechnology (Santa Cruz, CA, USA); FAK, phospho-paxillin, paxillin and phosphoserine from Millipore; Src (Sigma); and phospho-AKT1 from GeneTex (Irvine, CA, USA). Cell lysates (50 μg) were electrophoresed on an 8–15% SDS-PAGE and then transblotted onto a Hybond-P membrane (GE Health care, Hong Kong, China). The remaining procedures were performed as described elsewhere (Lin et al., 2008).
Affinity precipitation of cellular GTP-RhoA and co-immunoprecipitation (CoIP)
RASMCs that had received the indicated treatments were washed with ice-cold Tris-buffered saline and lysed in the RIPA buffer. Cell lysates were clarified by centrifugation at 13 000× g at 4°C for 10 min. Thereafter, equal volumes of lysates were incubated with GST–rhotekin-binding domain (RBD) (20 μg) beads at 4°C for 1 h and were then centrifuged, washed and eluted by boiling in an SDS-loading buffer containing 5% β-mercaptoethanol for 10 min. RhoA was detected by Western blotting using an anti-RhoA antibody (Santa Cruz Biotechnology, Inc.). The level of RhoA activity in each sample was determined by normalizing the amount of RBD-bound RhoA to the total amount of RhoA in cell lysates.
In addition, cells were challenged with homocysteine for 30 min, then folate was added and the cells incubated for a further 6 h. The cells were then analysed for interactions among cell motility-related molecules. In brief, cell lysates were pre-cleared by adding protein A agarose beads for 60 min at 4°C, and then the protein concentrations of the samples were evaluated. Cell proteins (200 μg) were incubated with an anti-RhoA antibody (2 μg) and protein A plus G agarose beads (20 μg) at 4°C overnight. The precipitates were washed five times with a lysis buffer and once with PBS. The pellet was then resuspended in a sample buffer (50 mM Tris, pH 6.8; 100 mM bromophenol blue; 10% glycerol) and incubated at 90°C for 10 min. It was then analysed by electrophoresis, to release the proteins from the beads, on 10.0% SDS-PAGE gels and transferred to a PVDF membrane for Western blotting with specific antibodies.
Immunofluorescence
Cells cultured overnight on glass coverslips were treated as indicated, and then washed once with cold PBS and fixed for 10 min in 4% paraformaldehyde. Cells were permeabilized using a solution of 0.1% Triton X-100 and 0.05% Tween-20 in PBS. Coverslips were blocked using a 10% goat serum at room temperature for 60 min. Thereafter, the coverslips were either (i) stained with rhodamine-phalloidin (ICN Immunobiologicals, Costa Mesa, CA, USA; 1:100) or (ii) were incubated with anti-p21 or anti-p27 antibodies (1:100) at 4°C overnight, followed by further incubation with Texas red-conjugated goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA; 1:100) for 60 min at room temperature. Coverslips were mounted on slides using Vectashield anti-fade (Vector Laboratories, Inc., Burlingame, CA, USA) diluted 1:1 with PBS and were visualized using a DMI 6000B CS laser confocal microscope (Leica, Heidelberg, Germany) with an HCX PL APO l-blue 63 × /1.40-0.60 NA oil-immersion objective lens. Images were acquired using a CM350 CCD camera (Applied Precision, Issaquah, WA, USA) and TCS SP5 confocal spectral microscopy imaging system software (Leica). The images were processed using Photoshop 7.0 software (Adobe System, San Diego, CA, USA).
Data analysis
Values are expressed as the mean ± SD for data from at least three experiments. The P-values for the differences in means between experimental and control groups were calculated using Student's t-test or one-way anova with Bonferroni's method as a post hoc test. A P-value of less than 0.05 was considered statistically significant.
Materials
Homocysteine, folate, wortmannin (PI3K inhibitor) and YS-49 (PI3K activator) were purchased from Sigma Chemical (St. Louis, MO, USA). The compounds UO126 (ERK inhibitor) and Ly294002 (PI3K inhibitor) were obtained from Tocris Bioscience (Bristol, UK). The Src inhibitor 4-amino-5-(4-chloro-phenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine (PP2) was purchased from A.G. Scientific Inc. (San Diego, CA, USA).
Results
Folate reversed homocysteine-mediated increases in RASMC migration and proliferation
An earlier study showed that a very high concentration of homocysteine (500 μM) induced vascular smooth muscle cell migration (Jiang et al., 2009). In this study, we evaluated a range of moderate homocysteine concentrations (50–100 μM) and folate concentrations (20 nM–20 μM) for their effects on RASMC migration. Homocysteine increased cell migration in a concentration-dependent manner (Figure 1A). Low doses of folate (20 nM–2 μM) did not show anti-migratory effects in a wound-healing assay, whereas 20 μM of folate was effective (Figure 1A). Similarly, the addition of folate (20 μM) blocked the pro-migratory effects of homocysteine in RASMC. No apparent cell proliferation was noticed in cells cultured at 10% FBS medium within 0–18 h of folate or homocysteine treatment by an MTT assay. Therefore, we excluded the possibility that cell proliferation contributed to the pro-migratory effect of an 18 h homocysteine treatment. At a concentration of 50 μM, homocysteine increased cell proliferation significantly, as shown by the MTT and BrdU incorporation assays (Figure 1B and 1C respectively). The addition of folate to these cultures suppressed RASMC proliferation. These results suggested that folate blocked both the homocysteine-mediated increases in cell proliferation and in migration in RASMC.
Figure 1.
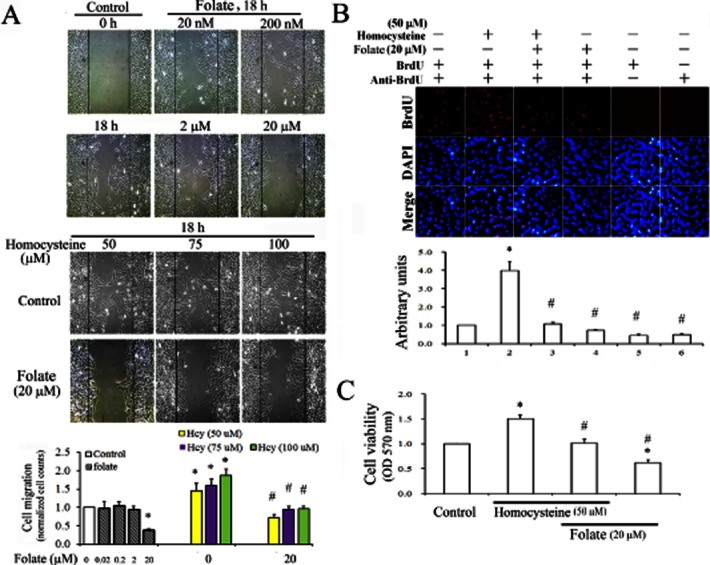
Anti-proliferative and anti-migratory effects of folate on homocysteine-treated RASMC. (A) Cells cultured in IBIDI culture inserts were treated with 2 nM–20 μM of folate alone to determine optimal concentrations for its anti-migratory effect in RASMC. Thus, cells were challenged with 50–100 μM homocysteine for 30 min, followed by 20 μM folate treatment for 18 h. Cell migration was observed using a microscope with a CCD camera attached. Magnification 100×. (B) Following various treatments, DNA synthesis in RASMC was detected using a BrdU incorporation assay. The BrdU-labelled nuclei are shown as bright red spots, and DAPI staining in the same fields revealed the positions of cell nuclei. We recorded micrographs of representative fields. Magnification 400×. Results in (A) and (B) are representative of three independent experiments. (C) Cells plated in 24-well plates (5 × 105 cells per well) or cultured on coverslips were challenged with 50 μM homocysteine for 30 min, then incubated with 20 μM folate for 24 h. The viability of cells was determined using an MTT assay. Data shown are the mean ± SD of five independent experiments. *P < 0.05 vs. the control; #P < 0.05 vs. the homocysteine-challenged group.
Involvement of p21/p27 in the anti-proliferative effects of folate, and their up-regulation through a Src/ERK-dependent pathway in RASMC
We evaluated the anti-proliferative effects of folate in homocysteine-treated RASMC by examining the expression of the endogenous CDK inhibitors, p21 and p27. We treated RASMC with 20 μM folate for 1–9 h, and then harvested the cells and analysed them using RT-PCR and Western blotting. Folate treatment up-regulated p21 and p27 in a time-dependent manner (Figure 2A). To examine the underlying signalling pathway(s), we maintained RASMC in a quiescent state in a DMEM containing 0.5% FBS for 48 h, and then treated the cells with 20 μM folate for 2–10 min in a medium containing 10% FBS. Western blot analysis showed that the activated forms of Src and ERK1/2 were increased at the time points shown in Figure 2B.
Figure 2.
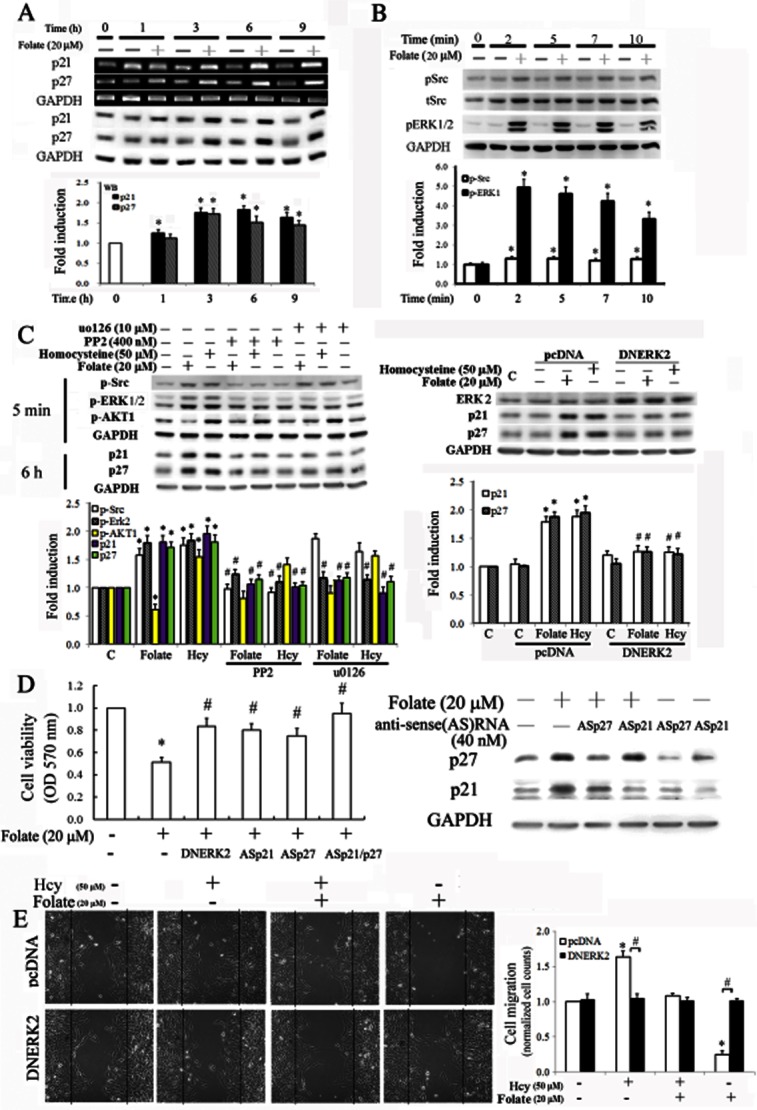
Molecular mechanisms underlying the effects of folate on p21/p27 up-regulation and the resulting anti-proliferative effect on RASMC. Cells were treated with folate for the indicated time points. The expression of p21/p27 was determined (A), and related signalling pathways of Src and Erk1/2 were analysed (B) using GAPDH as an internal control in RT-PCR or Western blot analysis. (C) Cells were pretreated with PP2 (a Src inhibitor) or U0126 (an ERK inhibitor) for 1 h, or transfected with DNERK2 overnight, followed by the addition of folate or homocysteine (Hcy). The indicated molecules were analysed by Western blots. A representative result for three separate experiments is shown and values are presented as the mean ± SD (*P < 0.05 vs. the control; #P < 0.05 vs. the folate- or homocysteine-treated group). (D) Cells were transfected with antisense (AS) p21/p27 oligonucleotides (40 nM) overnight and then treated with 20 μM folate for an additional 24 h. Effects of p21 and p27 AS oligonucleotides and DNERK2 overexpression in inducing the anti-proliferative effect of folate in RASMC were examined using an MTT assay. Six samples were analysed in each group, and values are presented as the mean ± SD. (E) Cells were transfected with DNERK2 plasmid overnight to reduce the levels of p21 and p27; thereafter, cells cultured at 0.5% FBS medium were challenged with 50 μM homocysteine (Hcy) for 30 min, followed by 20 μM folate treatment for 18 h. Cell migration was observed using a microscope with a CCD camera attached. Magnification 100×; graphs on the right indicate the migration normalized relative to the cell counts of the control. Data are expressed as mean ± SD from five randomly selected fields. *P < 0.05 vs. the control; #P < 0.05 vs. pCDNA alone.
To further elucidate the involvement of Src and ERK in p21/p27 up-regulation, we pretreated cells with their corresponding inhibitors PP2 and U0126, or overexpressed dominant negative (DN) ERK2, before treatment with folate or homocysteine. As shown in Figure 2C, PP2 blocked the folate-mediated ERK phosphorylation and concomitantly inhibited p21 and p27 up-regulation, suggesting that Src was upstream of ERK activation in folate-treated RASMC. Additional U0126 treatment or DNERK2 overexpression rescued folate-mediated p21/p27 up-regulation. We also observed that homocysteine increased p21 and p27 through a similar pathway of Src/Erk. However, further analysis showed that homocysteine-mediated increases in p21 and p27 occurred mainly in the cytosol and were involved in a pro-migratory effect in RASMC (Figure 4A).
Figure 4.
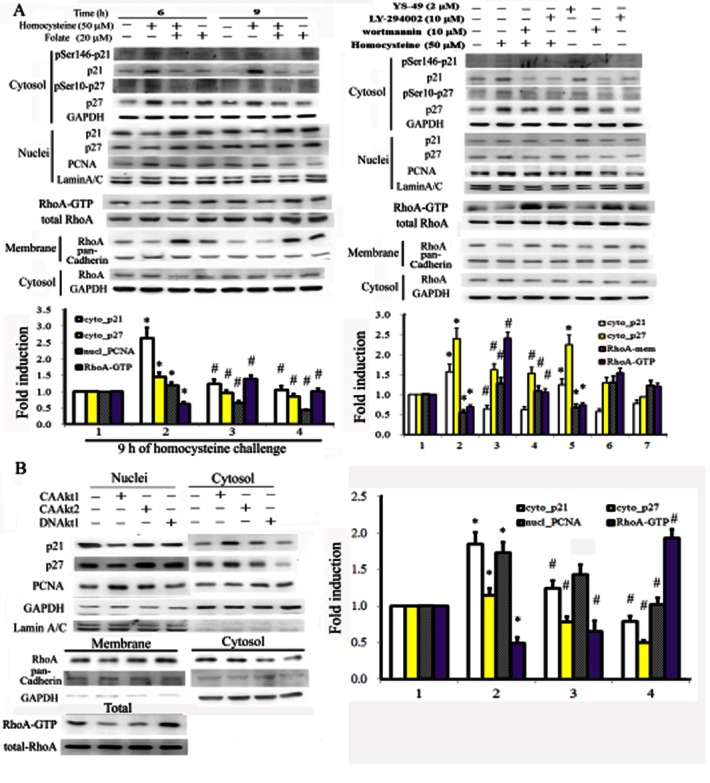
Folate prevented homocysteine-mediated increases in p21/p27 cytosolic and RhoA localization by inactivating AKT1. Cells were treated as described for Figure 3A, but with an increased duration (6 or 9 h) of homocysteine challenge (A), or were transfected with AKT constructs as described in Figure 3B. The resulting cell lysates were fractionated and analysed for the cytosolic-nuclear distribution of p21 and p27 and cytosolic-membrane RhoA by Western blotting. Data were derived from three independent experiments and are presented as the mean ± SD *P < 0.05 vs. the control; #P < 0.05 vs. the homocysteine-challenged or CAAKT1 group.
We transfected cells with anti-sense for p21 or for p27 or DNERK2 to evaluate the involvement of p21 and p27 in the anti-proliferative effects of folate by using an MTT assay. As shown in Figure 2D, silencing p21 or p27 partially reversed the folate-induced reduction of cell proliferation, whereas silencing both p21 and p27 or overexpressing DNERK2 additively prevented folate's inhibitory effects on cell proliferation. The sense oligonucleotides of p21 and p27 showed non-significant effects on the folate-mediated reduction in cell proliferation (data not shown). We then examined the involvement of p21 and p27 in the migratory effects of homocysteine and folate in RASMC using a wound-healing assay. In contrast with the group transfected with pcDNA alone, cells with DNERK2 overexpression not only blocked the pro-migratory effect of homocysteine, but also reduced the anti-migratory effect of folate in homocysteine-treated RASMC (Figure 2E), suggesting the importance of p21/p27, induced via the ERK pathway, in the migratory effects of homocysteine and folate.
Folate blocked homocysteine-mediated phosphorylation of FAK/paxillin/p190RhoGAP by inactivating AKT1
To evaluate the protective effect of folate on homocysteine-challenged RASMC, we assessed the levels of a number of signalling molecules involved in cell proliferation and migration, after treatment. Homocysteine treatment increased the activated forms of AKT1, FAK, paxillin, p190RhoGAP and PCNA in RASMC, whereas folate induced the opposite effect (Figure 3A). To confirm the essential role of AKT1 in homocysteine-mediated effects, we treated RASMC with compounds known to inhibit or activate PI3K. The addition of wortmannin and LY294002 prevented the effects of homocysteine, whereas YS-49, a PI3K activator, exerted the same effects as homocysteine. We then tested the effects of overexpressing (gain of function) or DN forms (loss of function) of AKT1/2 in RASMC. Cells overexpressing constitutively active (CA) AKT1 displayed characteristics similar to those of homocysteine-treated cells, such as increased FAK/paxillin/190RhoGAP phosphorylation (Figure 3B). In contrast, cells overexpressing DNAKT1 showed characteristics similar to those of folate-treated cells, such as dephosphorylation of FAK/paxillin/190RhoGAP. These results suggested that AKT1 might be the upstream regulator of the FAK/paxillin/190RhoGAP pathway. Protein kinase B (AKT) is a signalling molecule known to be involved in cell survival. As shown in Figure 3B, cells overexpressing AKT1 displayed increased nuclear levels of PCNA and those overexpressing DNAKT1 showed reduced nuclear PCNA. These findings suggested that homocysteine's effects on increased cell proliferation were derived at least in part from the activation of AKT1.
Figure 3.
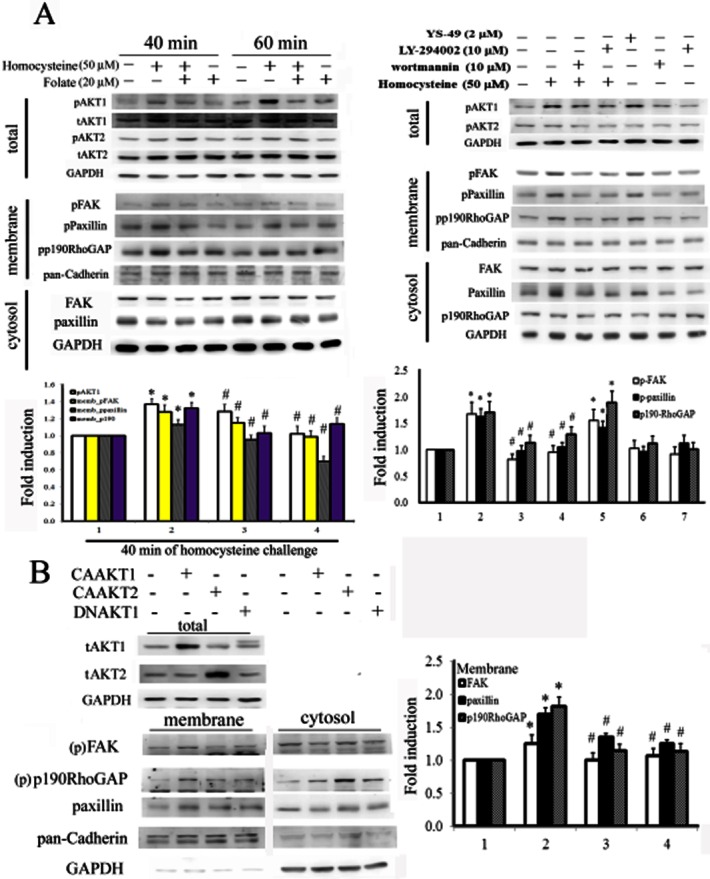
Folate treatment inactivated AKT1, thereby preventing homocysteine-mediated increases in phosphorylation of AKT1/FAK/paxillin/p190RhoGAP. (A) Cells were challenged with homocysteine for 30 min, followed by folate co-treatment for another 40 or 60 min. The resulting cell lysates were partitioned into nuclear and cytosolic fractions, which were probed with the indicated molecules and analysed by Western blotting. Representative results of three independent experiments are shown as the mean ± SD. *P < 0.05 vs. the control; #P < 0.05 vs. the homocysteine-challenged group. To evaluate the role of AKT activation in inducing the effect of folate, cells were pretreated with PI3K/AKT activators or inhibitors for 1 h, before challenge with homocysteine for 30 min. (B) Cells were transfected with CAAKT1, CAAKT2 or DNAKT1 (2 μg·mL−1) for 16 h. The resulting cell lysates were analysed and partitioned into cytosolic-nuclear and cytosolic-membrane fractions at the indicated time points. The activated forms of AKT1, FAK, paxillin and p190RhoGAP were analysed by Western blotting. *P < 0.05 vs. the control; #P < 0.05 vs. the CAAKT1 group.
Folate-mediated inactivation of AKT1 prevented homocysteine-mediated up-regulation of cytosolic p21/p27 and inactivation of RhoA
We investigated whether the opposing effects of folate and homocysteine on AKT phosphorylation were responsible for the differential downstream effects on p21, p27 and RhoA subcellular localization. Cell lysates treated with folate or homocysteine were partitioned into cytosolic-nuclei or cytosolic-membrane fractions for Western blot analysis. Homocysteine treatment was shown to increase the cytosolic sequestration and phosphorylation of p21 and p27, and to reduce the levels of the activated form of RhoA in membranes and RhoA-GTP. By contrast, folate treatment prevented these effects (Figure 4A). We then evaluated the involvement of AKT in the differential effects of homocysteine and folate on p21, p27, and RhoA subcellular localization using the overexpression of CAAKT1, CAAKT2 or DNAKT2. As shown in Figure 4B, RASMC in which AKT1 was overexpressed displayed characteristics similar to those of homocysteine-treated cells, such as increased cytosolic p21 and p27 levels, and reduced membrane-bound RhoA and RhoA-GTP. Cells overexpressing DNAKT1, however, showed characteristics similar to those of folate-treated cells, which were the opposite of patterns shown by homocysteine-treated cells.
Folate increased stress fibre formation associated with decreased cytosolic levels of p21 and p27, and decreased interaction among p21, p27, RhoA and p190RhoGAP
We used immunochemical staining to evaluate the effects of folate on the subcellular localization of p21, p27 and RhoA. The addition of folate to RASMC reversed the effect of homocysteine on the cytosolic sequestration of p21 and p27 (Figure 5A). The results of CoIP assays showed that these increased cytosolic levels of p21 and p27 led to increased recruitment of p190RhoGAP and RhoA to form a tetra-protein complex (Figure 5B). Adding folate abolished the formation of this complex (Figure 5B) and further inhibited p190RhoGAP phosphorylation (Figure 3A). Previous studies have shown that RhoA activation is primarily responsible for the formation of actin stress fibres (Ridley and Hall, 1992; Chang et al., 2009). These fibres potentially exert a contractile force and are necessary for the maintenance of sufficient adhesion to substrates. We further assessed the effects of folate on the activation of RhoA and stress fibre formation using rhodamine-phalloidin staining. Results from confocal microscopy indicated that folate treatment prevented the homocysteine-mediated inhibition of stress fibre formation (Figure 5C).
Figure 5.
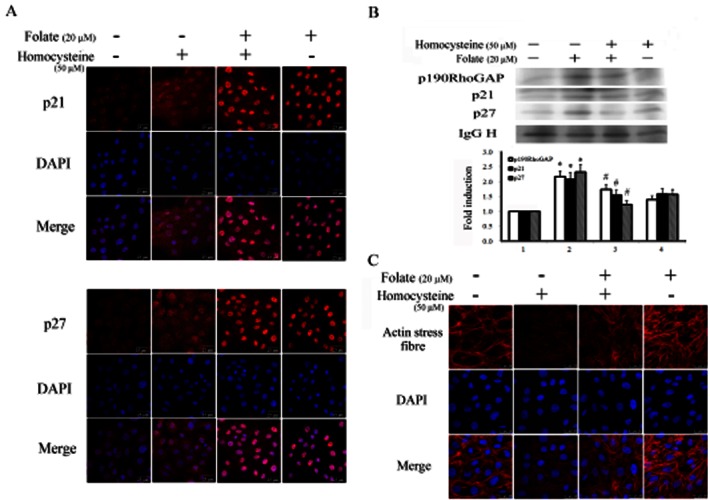
Folate-induced activation of RhoA correlated with decreased cytosolic distribution of p21 and p27, and interactions between p21, p27, RhoA and p190RhoGAP in RASMC. (A) Cells cultured on coverslips were challenged with homocysteine for 30 min followed by folate treatment for 6 h. Cells were then immunostained using an anti-p21 or anti-p27 antibody, and were then incubated with a second antibody conjugated with Texas red. Red spots represent p21- or p27-positive staining in the cytosol or nuclei. Fields stained with p21 or p27 were also stained with DAPI to reveal the positions of cell nuclei. Micrographs of representative fields were recorded. (B) Cells receiving similar treatments were immunoprecipitated using an anti-RhoA antibody. The pulled-down complex was detected using anti-p21, anti-p27 and anti-p190RhoGAP antibodies; an IgG heavy chain was used as an internal control for normalization. Bar charts show the band intensities of the indicated molecules according to densitometry. Data were derived from three independent experiments and are presented as the mean ± SD. *P < 0.05 vs. the control; #P < 0.05 vs. the homocysteine-challenged group. (C) Cells were pre-challenged with 50 μM homocysteine for 30 min, followed by incubation with 20 μM folic acid for 6 h. Cell morphology was captured using a fluorescent confocal microscope with a CCD camera attached. Red filaments represent stress fibre formation. Fields stained for the formation of stress fibres were also stained using DAPI to reveal the positions of cell nuclei. Magnification 630×.
Contributions of FAK silencing and RhoA activation to the anti-migratory effects of folate in RASMC
Tomar et al. (2009) showed that FAK modulated p190RhoGAP through phosphorylation. To evaluate the effects of folate-induced FAK inactivation on the signalling pathway for RhoA activation, we silenced FAK using the corresponding siRNA to mimic the effects of folate on RASMC. As with folate-treated cells, RASMC with FAK knock-down displayed decreased paxillin and p190RhoGAP phosphorylation and increased membrane-bound RhoA levels and RhoA-GTP (Figure 6A). In addition, the silencing of FAK resulted in decreased levels of PCNA, a cell proliferation marker. This finding suggested that FAK, a signalling molecule downstream of AKT, was an essential component of cell survival signalling in folate-treated RASMC. We confirmed the involvement of RhoA activation in inducing the anti-migratory effects of folate in homocysteine-treated RASMC using pharmacological and genetic approaches. As shown in Figure 6B, treatment with Y27632, a ROCK inhibitor, prevented folate-mediated inhibition of cell migration. In addition, CARhoA-overexpressing cells displayed reduced migration in a manner similar to that observed in folate-treated cells. This finding suggested the involvement of RhoA activation in the anti-migratory effects of folate in RASMC. By contrast, DNRhoA-overexpressing cells resembled homocysteine-treated cells and displayed increased cell migration.
Figure 6.
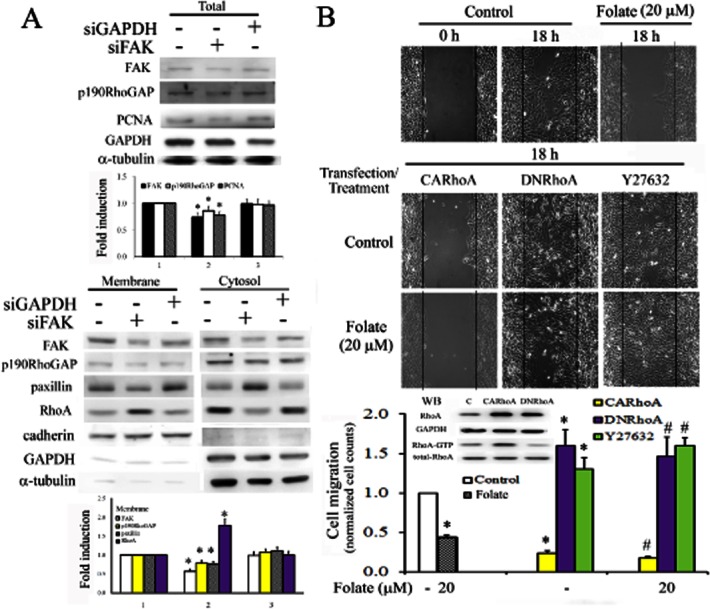
Relationship between FAK and RhoA in inducing the anti-migratory effect of folate in homocysteine-treated RASMC. (A) Cells were transfected with scrambled siRNA, FAK or GAPDH siRNA for 2 days. Total cell lysates were analysed for the expression of FAK, p190RhoGAP, PCNA and GAPDH. Partitioned cytosolic-membrane fractions were analysed for the subcellular localization of FAK, p190RhoGAP, paxillin and RhoA. α-tubulin was used as an internal control for total lysates or cytosolic fractions, and cadherin was used as an internal control for membrane fractions, to confirm equivalent loading. Bar charts on the right show the band intensities of indicated molecules according to densitometry. Data were derived from three independent experiments and are presented as the mean ± SD. *P < 0.05 vs. the control. (B) Cells were transfected with CARhoA or DNRhoA overnight or were pretreated with Y27632 for 1 h, and were then incubated with folate for 18 h. Cells cultured in IBIDI culture inserts overnight were removed and replaced with a 10% FBS medium to assess the extent of cell migration into the cell-free gap, 18 h after treatment. Images were captured using an Olympus SC-35 attached to an inverted microscope (Nikon, Tokyo, Japan) at a magnification of 100×. Data are normalized to the cell counts of the control and are presented as the mean ± SD of three independent experiments. *P < 0.05 vs. the control; #P < 0.05 vs. folate alone.
Discussion
The results of this study showed that folate inhibited homocysteine-mediated increases in RASMC proliferation and migration by inactivating AKT1. The inactivation of AKT1 inhibited homocysteine-induced phosphorylation of FAK, paxillin, and p190RhoGAP, and cytoplasmic sequestration of p21/p27. Folate treatment elicited RhoA activation by reducing the phosphorylation of p190RhoGAP (a RhoA inhibitor) and by decreasing the interactions between cytosolic p21/p27, RhoA and p190RhoGAP. Our results confirmed that RhoA activation contributed to the anti-migratory effects of folate in homocysteine-challenged RASMC (Figure 7).
Figure 7.
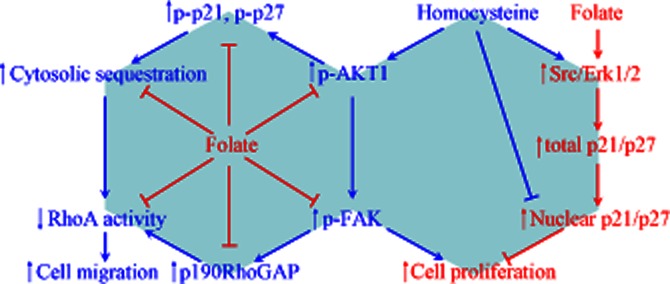
Schematic representation of the signalling pathways and molecular mechanisms involved in the anti-proliferative and anti-migratory effects of folate on homocysteine-treated RASMC. Both homocysteine and folate treatment increased p21 and p27 induction through a Src-Erk-dependent pathway, whereas their differential effects on AKT1 (de)phosphorylation caused different outcomes in the cytosol-nuclear distribution of p21 and p27. The increased nuclear fraction of p21 and p27 induced by AKT1 inactivation resulting from folate treatment contributed to the anti-proliferative effect in RASMC. By contrast, activation of AKT1 following homocysteine treatment increased p21 and p27 phosphorylation, resulting in their increased cytosolic sequestration and increased phosphorylation of p190RhoGAP. This contributed to RhoA inactivation, leading to the pro-migratory effect of homocysteine on RASMC. Folate treatment prevented the effects of homocysteine by activating RhoA through the inhibition of AKT1-mediated signalling pathways.
In addition to confirming folate's anti-migratory effects on RASMC, we demonstrated that folate's anti-proliferative effects on RASMC were attributable to the up-regulation of p21 and p27. Homocysteine also up-regulated p21 and p27 through a similar pathway of Src and ERK phosphorylation. However, in contrast to the anti-proliferative effects of folate, homocysteine increased cell proliferation in RASMC, as shown by MTT and BrdU assays (Figure 1C). These findings were in agreement with those obtained by Buemi et al. (2001), which showing that homocysteine increased cell proliferation. They tested concentrations similar to those used in our study (10–15 μM), although cell proliferation was also concomitant with cell deaths by apoptosis and necrosis (Buemi et al., 2001). Furthermore, our results showed that homocysteine caused apoptosis through caspase-3 activation, which might have resulted from the inflammatory effect of homocysteine in RASMC (Y. Chou, unpubl. data). However, the relationships and balance between cell proliferation, cell death and cell continuity require further investigation.
Folate-mediated up-regulation of p21 and p27 occurred primarily in the nuclei of cells. Homocysteine-mediated increases in p21 and p27, however, were predominantly localized to the cytosol. This led to further RhoA inactivation, resulting in the pro-migratory effect of homocysteine in RASMC. Our findings also showed that activation of the AKT1-FAK pathway was involved in the effect of homocysteine in promoting cell proliferation. The cell proliferation marker PCNA increased in AKT1-overexpressing RASMC, whereas it decreased in DNAKT1-overexpressing or FAK knock-down RASMC (Figures 4 and 6).
An increasing body of evidence shows that phosphorylation of p27 at various sites induces differential subcellular localization regulatory or protein degradation responses (Nacusi and Sheaff, 2006). For example, phosphorylation at Thr187 in p27 increased proteasome degradation, whereas phosphorylation at Ser10 by AKT1 activation increased cytosolic sequestration. In agreement with these findings, our results showed that homocysteine increased p27 phosphorylation at Ser10 through AKT1 phosphorylation, resulting in increased p27 cytosolic levels in RASMC. Similarly, we observed that activation of AKT1 by homocysteine increased p21 phosphorylation at Ser146 (Figure 4A). This result confirmed a previous report that the activation of AKT by extracellular growth signal caused phosphorylation at Thr145 and/or Ser146 and resulted in cytoplasmic localization and stabilization (Li et al., 2002).
FAK is widely considered to act upstream of AKT (Xia et al., 2004). Our study showed that AKT1 phosphorylation induced by homocysteine activated FAK in RASMC. However, cells with FAK knock-down displayed non-significant changes in levels of AKT (data not shown), suggesting that AKT1 occurs upstream of FAK phosphorylation. These results provided support for the findings of Wang et al. (2002) in which AKT1 was shown to regulate FAK through direct association, and implicating serine phosphorylation in pressure-induced colon cancer metastasis. Our results also showed that homocysteine increased paxillin and p190RhoGAP phosphorylation, and concomitantly reduced activation of RhoA. Similarly, Tsubouchi et al. (2002) showed that the binding of phosphorylated paxillin to p120RasGAP increased the free form of p190RhoGAP, thereby inhibiting RhoA activation in NMuMG cells.
Several studies have investigated the effects of p27 on RhoA activation. Besson et al. (2004) showed that p27 binds to RhoA and inhibits RhoA activation by interfering with interactions between RhoA activators, the GEFs. The current study was the first to show that increased cytosolic levels of p21 and p27 in response to homocysteine treatment raised the recruitment of p190RhoGAP and RhoA to form a tetra-protein complex, and that the addition of folate inhibited this effect (Figure 5B). Inactivation of RhoA and pro-migratory effects in RASMC can be attributed to an increased interaction between p190RhoGAP and RhoA (caused by homocysteine-induced increases in cytosolic p21 and p27) together with increased p190RhoGAP phosphorylation. This mechanism suggests the involvement of p21 and p27 in regulating cell migration by modulating the Rho pathway (Besson et al., 2004).
This study showed that folate inhibited RASMC migration by increasing RhoA activation through the suppression of pathways of AKT/p21/p27 cytosolic localization and AKT/paxillin/p190RhoGAP. We confirmed the involvement of RhoA in folate's anti-migratory effects using a RhoA inhibitor (Y27632) and by overexpressing CARhoA or DNRhoA (Figure 6). Our previous study showed that RhoA activation suppressed cell migration. In human umbilical vein endothelial cells, inactivation of FAK and p190RhoGAP by aryl hydrocarbon receptor activation led to RhoA activation, which was followed by anti-angiogenesis (Chang et al., 2009). The pathological influence of RhoA activation has been demonstrated in numerous biological systems. For example, increased RhoA activity was responsible for a thrombin-induced increase in endothelial permeability and this effect was caused by increased contraction and could be rescued using FAK to increase p190RhoGAP phosphorylation (Holinstat et al., 2006). Similarly, the results of our study showed that RhoA activation by folate was caused by inactivation of FAK and p190RhoGAP, which in turn was induced through AKT1 inactivation in RASMC.
Overall, folate treatment increased RhoA activation by suppressing the activation of AKT1-FAK-paxillin-p190RhoGAP and AKT1-mediated decreases in the cytosolic levels of p21 and p27, leading to the inhibition of RASMC migration. Folate up-regulated p21 and p27 expression and inactivated homocysteine-mediated AKT1 phosphorylation, resulting in anti-proliferative effects in homocysteine-challenged RASMC. This study was the first to show that folate exerts a protective effect against homocysteine challenge in vitro through anti-migration and anti-proliferation. The results of earlier studies on the protective effects of folate against homocysteine-mediated cardiovascular disease or stroke in vivo have been inconsistent. Our findings have elucidated some of the signalling pathways and molecular mechanisms involved in the anti-proliferative and anti-migratory effects of folate on RASMC and the resultant protection against the atherosclerosis-inducing effects of homocysteine.
Acknowledgments
This study was supported by a grant (99TMU-SHH-02-2) from Taipei Medical University-Shuang-Ho Hospital, Taipei, Taiwan.
Glossary
- ASp21
antisense p21
- ASp27
antisense p27
- CARhoA
constitutive active RhoA
- DNRhoA
dominant negative RhoA
- FAK
focal adhesion kinase
- PP2
4-amino-5-(4-chloro-phenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine
- RASMC
rat aortic smooth muscle cell
Conflict of interest
The authors state no conflict of interest.
References
- Ambrosi P, Rolland PH, Bodard H, Barlatier A, Charpiot P, Guisgand G, et al. Effects of folate supplementation in hyperhomocysteinemic pigs. J Am Coll Cardiol. 1999;34:274–279. doi: 10.1016/s0735-1097(99)00144-8. [DOI] [PubMed] [Google Scholar]
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloor J, Shukla N, Smith FC, Angelini GD, Jeremy JY. Folic acid administration reduces neointimal thickening, augments neo-vasa vasorum formation and reduces oxidative stress in saphenous vein grafts from pigs used as a model of diabetes. Diabetologia. 2010;53:980–988. doi: 10.1007/s00125-010-1680-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brattstrom LE, Israelsson B, Jeppsson JO, Hultberg BL. Folic acid – an innocuous means to reduce plasma homocysteine. Scand J Clin Lab Invest. 1988;48:215–221. doi: 10.3109/00365518809167487. [DOI] [PubMed] [Google Scholar]
- Buemi M, Marino D, Di Pasquale G, Floccari F, Ruello A, Aloisi C, et al. Effects of homocysteine on proliferation, necrosis, and apoptosis of vascular smooth muscle cells in culture and influence of folic acid. Thromb Res. 2001;104:207–213. doi: 10.1016/s0049-3848(01)00363-2. [DOI] [PubMed] [Google Scholar]
- Chang CC, Tsai SY, Lin H, Li HF, Lee YH, Chou Y, et al. Aryl-hydrocarbon receptor-dependent alteration of FAK/RhoA in the inhibition of HUVEC motility by 3-methylcholanthrene. Cell Mol Life Sci. 2009;66:3193–3205. doi: 10.1007/s00018-009-0102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res. 2007;67:9238–9243. doi: 10.1158/0008-5472.CAN-07-1375. [DOI] [PubMed] [Google Scholar]
- Doshi SN, McDowell IF, Moat SJ, Payne N, Durrant HJ, Lewis MJ, et al. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation. 2002;105:22–26. doi: 10.1161/hc0102.101388. [DOI] [PubMed] [Google Scholar]
- Fisher-Dzoga K, Jones RM, Vesselinovitch D, Wissler RW. Ultrastructural and immunohistochemical studies of primary cultures of aortic medial cells. Exp Mol Pathol. 1973;18:162–176. doi: 10.1016/0014-4800(73)90014-2. [DOI] [PubMed] [Google Scholar]
- Heron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, et al. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol Cell Biol. 2006;26:8267–8280. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervouet E, Debien E, Campion L, Charbord J, Menanteau J, Vallette FM, et al. Folate supplementation limits the aggressiveness of glioma via the remethylation of DNA repeats element and genes governing apoptosis and proliferation. Clin Cancer Res. 2009;15:3519–3529. doi: 10.1158/1078-0432.CCR-08-2062. [DOI] [PubMed] [Google Scholar]
- Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. The Journal of Biological Chemistry. 2006;281:2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- Holven KB, Aukrust P, Holm T, Ose L, Nenseter MS. Folic acid treatment reduces chemokine release from peripheral blood mononuclear cells in hyperhomocysteinemic subjects. Arterioscler Thromb Vasc Biol. 2002;22:699–703. doi: 10.1161/01.atv.0000013288.35930.90. [DOI] [PubMed] [Google Scholar]
- Jia DY, Liu HJ, Wang FW, Liu SM, Ling EA, Liu K, et al. Folic acid supplementation affects apoptosis and differentiation of embryonic neural stem cells exposed to high glucose. Neurosci Lett. 2008;440:27–31. doi: 10.1016/j.neulet.2008.05.053. [DOI] [PubMed] [Google Scholar]
- Jiang C, Zhang H, Zhang W, Kong W, Zhu Y, Xu Q, et al. Homocysteine promotes vascular smooth muscle cell migration by induction of the adipokine resistin. Am J Physiol Cell Physiol. 2009;297:C1466–C1476. doi: 10.1152/ajpcell.00304.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem. 2004;279:1885–1891. doi: 10.1074/jbc.M306968200. [DOI] [PubMed] [Google Scholar]
- Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002;277:11352–11361. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Lin H, Lee JL, Hou HH, Chung CP, Hsu SP, Juan SH. Molecular mechanisms of the antiproliferative effect of beraprost, a prostacyclin agonist, in murine vascular smooth muscle cells. J Cell Physiol. 2008;214:434–441. doi: 10.1002/jcp.21214. [DOI] [PubMed] [Google Scholar]
- Liu H, Huang GW, Zhang XM, Ren DL, X Wilson J. Folic Acid supplementation stimulates notch signaling and cell proliferation in embryonic neural stem cells. J Clin Biochem Nutr. 2010;47:174–180. doi: 10.3164/jcbn.10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AL. The methylation, neurotransmitter, and antioxidant connections between folate and depression. Altern Med Rev. 2008;13:216–226. [PubMed] [Google Scholar]
- Miller ER, 3rd, Juraschek S, Pastor-Barriuso R, Bazzano LA, Appel LJ, Guallar E. Meta-analysis of folic acid supplementation trials on risk of cardiovascular disease and risk interaction with baseline homocysteine levels. Am J Cardiol. 2010;106:517–527. doi: 10.1016/j.amjcard.2010.03.064. [DOI] [PubMed] [Google Scholar]
- Nacusi LP, Sheaff RJ. Akt1 sequentially phosphorylates p27kip1 within a conserved but non-canonical region. Cell Div. 2006;1:11. doi: 10.1186/1747-1028-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, et al. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Rosen DG, Yang G, Cai KQ, Bast RC, Jr, Gershenson DM, Silva EG, et al. Subcellular localization of p27kip1 expression predicts poor prognosis in human ovarian cancer. Clin Cancer Res. 2005;11(2 Pt 1):632–637. [PubMed] [Google Scholar]
- Sasaki K, Duan J, Murohara T, Ikeda H, Shintani S, Shimada T, et al. Rescue of hypercholesterolemia-related impairment of angiogenesis by oral folate supplementation. J Am Coll Cardiol. 2003;42:364–372. doi: 10.1016/s0735-1097(03)00629-6. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Somanath PR, Razorenova OV, Chen J, Byzova TV. Akt1 in endothelial cell and angiogenesis. Cell Cycle. 2006;5:512–518. doi: 10.4161/cc.5.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroes ES, van Faassen EE, Yo M, Martasek P, Boer P, Govers R, et al. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ Res. 2000;86:1129–1134. doi: 10.1161/01.res.86.11.1129. [DOI] [PubMed] [Google Scholar]
- Sue YM, Chung CP, Lin H, Chou Y, Jen CY, Li HF, et al. PPARdelta-mediated p21/p27 induction via increased CREB-binding protein nuclear translocation in beraprost-induced antiproliferation of murine aortic smooth muscle cells. Am J Physiol Cell Physiol. 2009;297:C321–C329. doi: 10.1152/ajpcell.00069.2009. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yamashita T, Asada M, Mizutani S, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) regulates neurite remodeling by inhibiting Rho-kinase activity. J Cell Biol. 2002;158:321–329. doi: 10.1083/jcb.200202071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Title LM, Cummings PM, Giddens K, Genest JJ, Jr, Nassar BA. Effect of folic acid and antioxidant vitamins on endothelial dysfunction in patients with coronary artery disease. J Am Coll Cardiol. 2000;36:758–765. doi: 10.1016/s0735-1097(00)00809-3. [DOI] [PubMed] [Google Scholar]
- Tomar A, Lim ST, Lim Y, Schlaepfer DD. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J Cell Sci. 2009;122(Pt 11):1852–1862. doi: 10.1242/jcs.046870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Perrella MA, Yoshizumi M, Hsieh CM, Haber E, Schlegel R, et al. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci U S A. 1994;91:6369–6373. doi: 10.1073/pnas.91.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Wang H, Perrella MA, Yoshizumi M, Sibinga NE, Tan LC, et al. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. J Clin Invest. 1996;97:146–153. doi: 10.1172/JCI118383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi A, Sakakura J, Yagi R, Mazaki Y, Schaefer E, Yano H, et al. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J Cell Biol. 2002;159:673–683. doi: 10.1083/jcb.200202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaar MC, Wever RM, Kastelein JJ, van Dam T, Koomans HA, Rabelink TJ. 5-methyltetrahydrofolate, the active form of folic acid, restores endothelial function in familial hypercholesterolemia. Circulation. 1998;97:237–241. doi: 10.1161/01.cir.97.3.237. [DOI] [PubMed] [Google Scholar]
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002;325:1202. doi: 10.1136/bmj.325.7374.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Jiang X, Yang F, Chapman GB, Durante W, Sibinga NE, et al. Cyclin A transcriptional suppression is the major mechanism mediating homocysteine-induced endothelial cell growth inhibition. Blood. 2002;99:939–945. [PMC free article] [PubMed] [Google Scholar]
- Williams PJ, Bulmer JN, Innes BA, Broughton Pipkin F. Possible roles for folic acid in the regulation of trophoblast invasion and placental development in normal early human pregnancy. Biol Reprod. 2011;84:1148–1153. doi: 10.1095/biolreprod.110.088351. [DOI] [PubMed] [Google Scholar]
- Wu FY, Wang SE, Sanders ME, Shin I, Rojo F, Baselga J, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66:2162–2172. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279:33024–33034. doi: 10.1074/jbc.M313265200. [DOI] [PubMed] [Google Scholar]