

Abstract
Background and Purpose
Perivascular adipose tissue (PVAT) releases adipocyte-derived hyperpolarizing factors (ADHFs) that may partly act by opening myocyte K+ channels. The present study in rat and mouse mesenteric arteries aimed to identify the myocyte K+ channel activated by PVAT and to determine whether adiponectin contributed to the hyperpolarizing effects of PVAT.
Experimental Approach
Myocyte membrane potential was recorded from de-endothelialized, non-contracted rat and mouse mesenteric arteries in the presence and absence of PVAT.
Key Results
The β3-adrenoceptor agonist, CL-316,243 (10 μM), generated PVAT-dependent, iberiotoxin-sensitive myocyte hyperpolarizations resulting from BKCa channel opening and which were partially blocked by L-NMMA (100 μM). Adiponectin (5 μg·mL−1) also produced iberiotoxin-sensitive hyperpolarizations in PVAT-denuded arterioles. Activation of myocyte AMP-activated protein kinase (AMPK) using 5 μM A-769662 also induced BKCa-mediated hyperpolarizations. Dorsomorphin abolished hyperpolarizations to CL-316,243, adiponectin and A-769662. In vessels from Adipo−/− mice, hyperpolarizations to CL-316,243 were absent whereas those to A-769662 and adiponectin were normal. In rat vessels, adipocyte-dependent hyperpolarizations were blocked by glibenclamide and clotrimazole but those to NS1619 (33 μM) were unaltered.
Conclusions and Implications
Under basal, non-contracted conditions, β3-adrenoceptor stimulation of PVAT releases an ADHF, which is probably adiponectin. This activates AMPK to open myocyte BKCa channels indirectly and additionally liberates NO, which also contributes to the observed PVAT-dependent myocyte hyperpolarizations. Clotrimazole and glibenclamide each reversed hyperpolarizations to adiponectin and A-769662, suggesting the involvement of myocyte TRPM4 channels in the ADHF-induced myocyte electrical changes mediated via the opening of BKCa channels.
Keywords: BKCa channels, adipose tissue, vascular myocytes, membrane potential, CL-316, 243, iberiotoxin, AMP kinase, adipocyte-derived hyperpolarizing factor, adiponectin, β3-adrenoceptors
Introduction
Perivascular adipose tissue (PVAT) exerts anti-contractile influences on the arteries that it surrounds (Dubrovska et al., 2004; Verlohren et al., 2004; Fésüs et al., 2007; Gao et al., 2007; Greenstein et al., 2009). Several reports have concluded that some of the anti-contractile effects of PVAT result from the opening of myocyte K+ channels (Löhn et al., 2002; Verlohren et al., 2004; Fésüs et al., 2007; Gao et al., 2007; Schleifenbaum et al., 2010), and in this paper the hypothetical factor(s) involved is/are designated adipocyte-derived hyperpolarizing factor(s) – ADHF(s).
The possible involvement of any ADHF-induced K+ channel opening has largely been inferred from myograph experiments in which vessels surrounded by PVAT were simply contracted in the presence or absence of K+ channel blockers without any measurement of myocyte membrane potential. Because receptors for the contractile agonists used exist not only on myocytes but also on adipocytes (Germack et al., 1997; Kinoshita et al., 2010; Stunes et al., 2011), these agonists could themselves have modified the release of any such ADHFs.
Adiponectin is a peptide secreted almost exclusively by adipocytes (Scherer et al., 1995). Myograph studies using 4-aminopyridine suggested that adiponectin can relax mesenteric artery rings by opening KV channels (Fésüs et al., 2007), and Greenstein et al. (2009) concluded that adiponectin was the anti-contractile adipokine released from rat mesenteric artery PVAT. In vivo, serum adiponectin levels are raised by CL-316,243 (Oana et al., 2005; Fu et al., 2008), a selective β3-adrenoceptor agonist (Bloom et al., 1992; Dolan et al., 1994; Michel et al., 2010) that has been used to investigate their role on adipose tissue (Collins et al., 1997). β3-adrenoceptors exist on rat adipocytes but are not functionally present on either mesenteric artery myocytes or endothelial cells (Kozlowska et al., 2003; Briones et al., 2005).
Using an electrophysiological approach in the present study, evidence was obtained for the β3-adrenoceptor-mediated release from PVAT of an ADHF which opens myocyte BKCa channels in both rat and mouse mesenteric arteries. Authentic adiponectin hyperpolarized mesenteric artery myocytes by opening BKCa channels by activation of AMP-activated protein kinase (AMPK). Although CL-316,243-stimulated release of adiponectin itself from PVAT could not be detected, this agonist generated PVAT-dependent myocyte hyperpolarizations in vessels from control mice yet had no effect on myocytes from Adipo−/− mice which were unable to synthesize adiponectin.
A preliminary account of some of these findings has been published (Egner et al., 2011).
Methods
Animals
Experiments were performed on second- or third-order mesenteric artery branches dissected from male Sprague–Dawley rats (approximately 250–300 g body weight; bred in-house, The University of Manchester) or on first- or second-order branches from C57BL/6J mice (control; Harlan, UK) or adiponectin-deficient (Adipo−/−) mice (B6.129-adipoqtm1Chan/J [originally generated by Chan and co-workers (Ma et al., 2002) but purchased from Jackson Laboratory Repository, Bar Harbor, ME, USA], each 10–11 weeks old. The homozygous Adipo−/− mice were confirmed by Jackson Laboratories to lack adiponectin mRNA (adipose tissue) and protein (serum). All animals were killed by cervical dislocation in compliance with Schedule 1 of the UK Animals (Scientific Procedures) Act, 1986. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Electrophysiology
Segments of artery (length 2–3 mm) that, unless otherwise stated, had been de-endothelialized by introducing deionized water into the vessel lumen for approximately 10–15 s were pinned to the Sylgard base of a thermostatically controlled bath and superfused (10 mL·min−1) with a Krebs solution (pH 7.5; 37°C; bubbled with 5% CO2 in air). Myocytes were impaled through the adventitial surface using 3 M KCl-filled microelectrodes (resistance 40–80 MΩ). About 50 Hz interference at the high impedance amplifier (Intra 767; WPI, Hitchin, UK) output was selectively removed (Humbug; Digitimer, Welwyn Garden City, UK). Signals were digitized and analysed using Chart software (Mac version 5.5.6) in conjunction with a PowerLab 4/SP acquisition system (ADInstruments, Chalgrove, UK).
Adiponectin assay
Segments of rat mesenteric bed (comprising artery, vein and PVAT) were weighed and then incubated in 5 mL Krebs solution (37°C; gassed with 5% CO2 : 95% air) in the absence (control) or presence of 10 μM CL-316,243. After 30 min of pre-incubation, the total adiponectin incubate concentration was determined (ALPCO Diagnostics Rat Adiponectin Immunoassay Kit; Stratech Scientific Ltd, Newmarket, UK). Reactions were performed in a 96-well plate and absorbance read at 450 nm. The adiponectin concentration was calculated from an adiponectin standard curve.
Statistical analysis
Results were analysed by two-way repeated-measures anova (with Bonferroni post hoc test) or Student's t-test as appropriate and are expressed as means ± SEM; n represents both the number of replications and the number of different animals from which the artery segments were obtained. A P-value of ≤0.05 was considered significant.
Drugs, reagents and solutions
A-769662 (4-hydroxy-3-(2′-hydroxybiphenyl-4-yl)-6-oxo-6,7-dihydrothieno[2,3-b]pyridine-5-carbonitrile; Tocris, Bristol, UK), glibenclamide and levcromakalim were dissolved in ethanol. ACh chloride, CL-316,243 (5-[(2R)-2-[[(2R)-2-(3-chlorophenyl)-2-hydroxyethyl]amino]-propyl]-1,3-benzodioxole-2,2-dicarboxylic acid disodium salt; Tocris), dorsomorphin (Tocris; prepared freshly each day) and iberiotoxin (Latoxan, 26000 Valence, France) were each dissolved in deionized water. Recombinant (human) adiponectin (BioVendor, Modrice, Czech Republic) and L-NMMA (NG-monomethyl-L-arginine; Tocris) were prepared in Krebs solution and used immediately. Clotrimazole and NS1619 (1-(2′-hydroxy-5′ trifluoromethylphenyl)-5-trifluoromethyl-2(3H)benzimidazolone; Research Biochemicals International, St. Albans, UK) were dissolved in dimethyl sulfoxide. Unless otherwise stated, all drugs and reagents were obtained from Sigma-Aldrich (Poole, Dorset, UK).
ACh, NS1619 and levcromakalim were each added as bolus injections directly into the recording bath in quantities calculated to obtain (transiently) the concentrations indicated. Unless otherwise stated, A-769662, CL-316,243, clotrimazole, dorsomorphin and iberiotoxin were added to the reservoir of Krebs solution superfusing the bath. Due to the expense of adiponectin, Krebs flow was stopped and the peptide was added directly into the recording chamber.
The composition of the Krebs solution was (in mM): NaCl, 118; KCl, 3.4; CaCl2, 1.0; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 25; glucose, 11.
Results
Hyperpolarizing effects of PVAT stimulation in rat mesenteric arteries using CL-316,243: comparison with adiponectin and involvement of myocyte BKCa channels
In endothelium-denuded vessels (no response to 10 μM ACh) and in the presence of PVAT, the selective β3-adrenoceptor agonist, CL-316,243 (10 μM), produced myocyte hyperpolarizations of 11.2 ± 1.0 mV (Figure 1A, n = 4), which were abolished (0.2 ± 0.1 mV, n = 4) by the selective BKCa channel inhibitor, iberiotoxin (100 nM). Hyperpolarizations to the KATP opener, levcromakalim, were similar before (24.6 ± 0.4 mV, n = 4) and during exposure to iberiotoxin (28.6 ± 1.5 mV, n = 4) (see Figure 1A).
Figure 1.
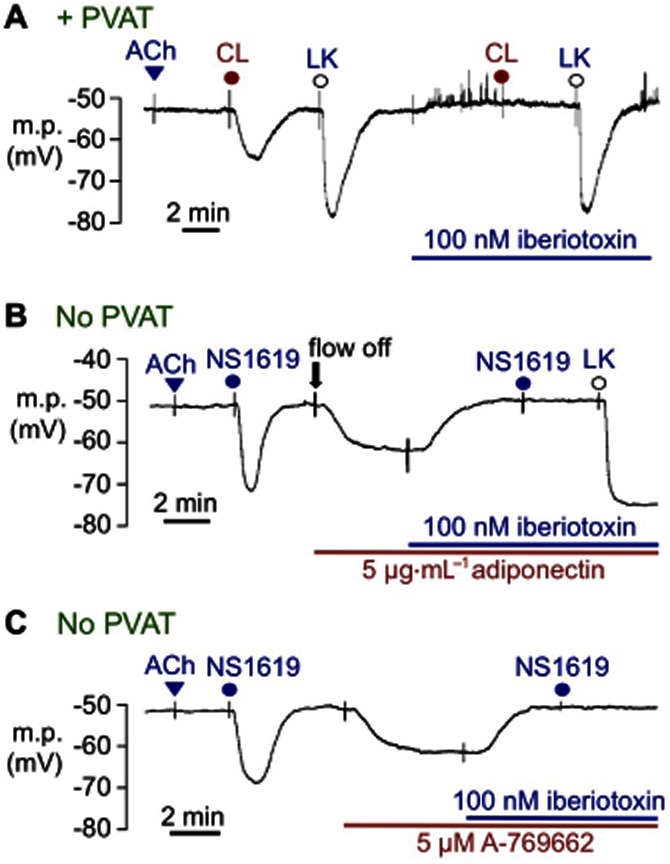
Hyperpolarizing effects of PVAT in rat mesenteric artery segments, all in the absence of the vascular endothelium (no response to 10 μM ACh). (A) In the presence of PVAT (+PVAT), hyperpolarizations induced by 10 μM CL-316,243 (CL) were abolished by prior exposure to 100 nM iberiotoxin, which had no effect on 10 μM levcromakalim (LK)-induced hyperpolarizations. (B) In the absence of PVAT (no PVAT), transient exposure to 33 μM NS1619 produced myocyte hyperpolarizations. After washout, the flow of Krebs was stopped (flow off); exposure to adiponectin hyperpolarized the myocytes, a response that was reversed by 100 nM iberiotoxin. Subsequent exposure to 33 μM NS1619 was without effect whereas 10 μM LK still hyperpolarized myocytes to close to EK in the continuing presence of iberiotoxin. (C) In the absence of PVAT, the AMPK activator A-769662 evoked hyperpolarizations, which were reversed by 100 nM iberiotoxin. Subsequent exposure to 33 μM NS1619 was also without effect, confirming iberiotoxin-induced blockade of myocyte BKCa channels.
In separate experiments, 10 μM CL-316,243-induced hyperpolarizations (13.4 ± 2.6 mV, n = 4) in the presence of the vascular endothelium were not different (P < 0.05) from those in endothelium-denuded vessels (11.2 ± 1.0 mV, n = 4) but were completely absent after PVAT removal (n = 3; data not shown). Subsequent experiments were therefore conducted in vessels denuded of their vascular endothelium.
Authentic adiponectin (5 μg·mL−1), like CL-316,243, also evoked iberiotoxin-sensitive myocyte hyperpolarizations in the absence of both PVAT and endothelium (10.3 ± 0.24 mV, n = 4; Figure 1B). Because of the expense of adiponectin, the flow of Krebs over the vessel was stopped immediately before adiponectin exposure (flow off, Figure 1B). In these experiments, hyperpolarizations to the BKCa channel opener, NS1619 (33 μM; 16.2 ± 2.1 mV, n = 4), were abolished in the presence of 100 nM iberiotoxin, whereas those of the KATP opener, levcromakalim (10 μM), were still present, indicating both the effectiveness and selectivity of iberiotoxin as a BKCa channel blocker.
Comparison of hyperpolarizing effects of the AMPK activator, A-769662 with those of CL-316,243 and adiponectin
AMPK is a downstream effector of the actions of adiponectin (Yamauchi et al., 2002; Kadowaki and Yamauchi, 2005). To clarify whether adiponectin-induced myocyte hyperpolarizations resulted from myocyte AMPK activation, the effects of a selective AMPK activator, A-769662 (Cool et al., 2006; Göransson et al., 2007), on membrane potential in the absence of both PVAT and endothelium were investigated. A-769662 (5 μM) hyperpolarized the myocytes by 9.2 ± 0.6 mV (n = 4), an effect reversed by 100 nM iberiotoxin (which also prevented the effects of subsequent exposure to NS1619; Figure 1C).
Hyperpolarizing effects of PVAT stimulation using CL-316,243 are absent in Adipo−/− mice
Although exogenous adiponectin mimicked the hyperpolarizing effects of CL-316,423, the rate of release of adiponectin from PVAT in the presence of CL-316,423 (0.64 ± 0.13 ng·mg·PVAT·h−1; n = 6) was similar to that in its absence (0.63 ± 0.15 ng·mg·PVAT·h−1; n = 6). However, commercially available kits for the quantification of rat adiponectin do not distinguish between the various adiponectin aggregates, which differ in their biological activity (see Brochu-Gaudreau et al., 2010). Thus, our inability to detect any CL-316,243-induced increase in a biologically relevant form of adiponectin could have been due to masking by unimportant aggregates.
Vessels from homozygous, adiponectin-deficient (Adipo−/−) mice (B6.129-adipoqtm1Chan/J) which lacked adiponectin mRNA and protein were therefore utilized. Hyperpolarizations to 10 μM CL-316,243 (10.7 ± 0.4 mV, n = 4) in control (C57BL/6J) mouse mesenteric arteries in the presence of PVAT were similar in magnitude (Figure 2A,C) to those in rat vessels under similar conditions (see Figure 1). Mouse myocyte membrane potentials were also increased by 5 μM A-769662 (6.4 ± 0.5 mV; n = 4) and by 5 μg·mL−1 adiponectin (9.7 ± 0.6 mV; n = 4). In arteries from Adipo−/− mice (Figure 2B,C), the hyperpolarizations to A-769662 (6.2 ± 0.4 mV; n = 4) and to 5 μg·mL−1 adiponectin (9.9 ± 1.1 mV; n = 4) were also not significantly different from those of controls. In marked contrast, however, 10 μM CL-316,243 completely failed to hyperpolarize vessels from Adipo−/− mice in the presence of PVAT (0.3 ± 0.2 mV; n = 4) (Figure 2B,C).
Figure 2.
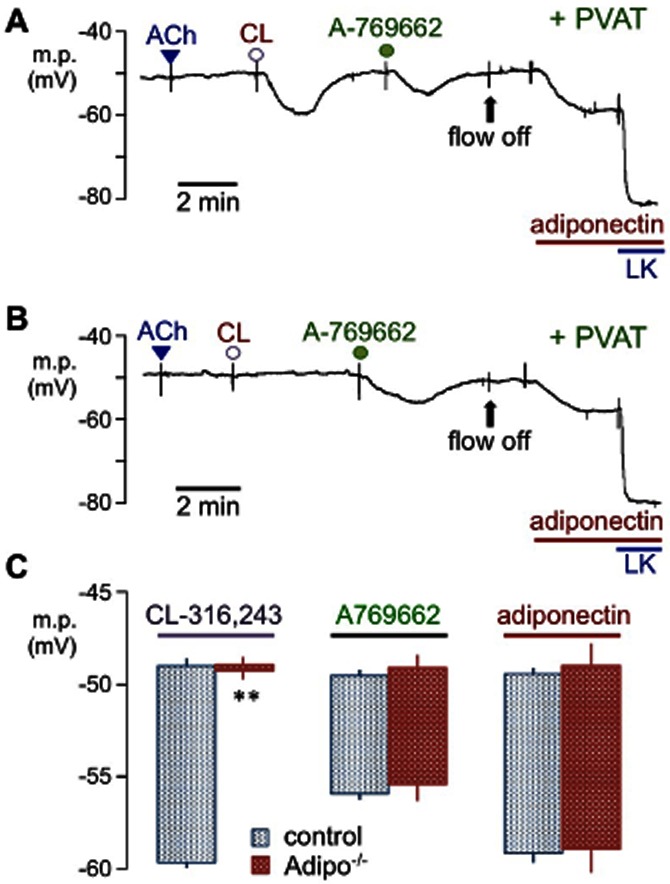
Effects of adiponectin deficiency on responses to CL-316,243, A-769662 and adiponectin in mouse mesenteric arteries. Typical traces showing the hyperpolarizations induced by 10 μM CL-316,243 (CL), 5 μM A-769662 and 5 μg·mL−1 adiponectin in PVAT-intact, endothelium-denuded artery segments (no response to 10 μM ACh) from (A) control (wild-type C57BL/6J) or (B) adiponectin-deficient (Adipo−/−; B6.129-adipoqtm1Chan/J) mice. In (A) and (B), bolus doses of ACh, CL-316,243 or A-769662 were added to produce, transiently, concentrations of 10, 10 and 5 μM respectively. Flow was stopped (flow off) to minimize the quantity of adiponectin required. Note the complete absence of CL-316,243-induced hyperpolarizations in vessels from Adipo−/− mice whereas authentic adiponectin (5 μg·mL−1) still hyperpolarized the myocytes. Levcromakalim (LK 10 μM, an opener of myocyte ATP-sensitive K+ channels) was added to establish the K+ equilibrium potential. (C) Histograms comparing the mean membrane potential in artery segments from wild-type and adiponectin-deficient (Adipo−/−) mice before and after exposure to CL-316,243, A-769662 and adiponectin. Each point represents the mean ± SEM (n = 4).
Effects of AMPK inhibition in rat vessels using dorsomorphin
Following 30 min of pre-incubation with 1 μM dorsomorphin (an AMPK inhibitor; Zhou et al., 2001) and in its continued presence, rat myocytes became depolarized (-47.9 ± 0.5 mV, n = 4) relative to controls (–50.3 ± 0.4 mV, n = 8; P = 0.0033, unpaired Student's t-test) and often exhibited spontaneous spike discharges (Figure 3A). In the presence of dorsomorphin, mean hyperpolarizations to 5 μg·mL−1 adiponectin (2.6 ± 0.5 mV, n = 4) and to 5 μM A-769662 (2.4 ± 0.3 mV, n = 4) were markedly reduced (P < 0.001; compare Figures 1B,C & 3A), although in the same preparations the hyperpolarizations to 33 μM NS1619 (16.7 ± 0.9 mV, n = 4) were not significantly changed (see Figure 1C and related text above). Responses to 10 μM CL-316,243 in the presence of PVAT (2.0 ± 0.3 mV, n = 4) were similarly reduced by dorsomorphin (P < 0.001; Figure 3B).
Figure 3.
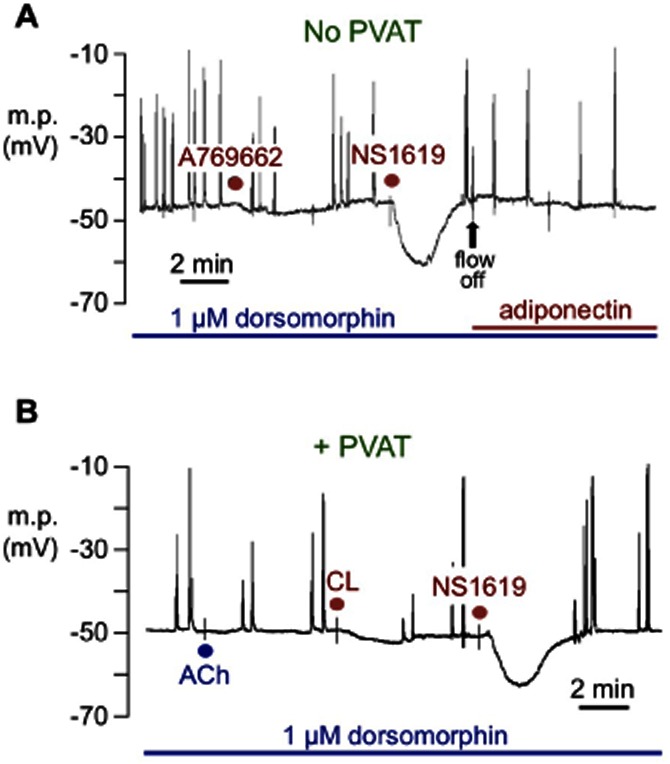
Typical recordings showing the effect of the AMPK inhibitor dorsomorphin on membrane potential in rat mesenteric arteries in the absence of the endothelium (no response to 10 μM ACh). (A) In the absence of PVAT (no PVAT), 30 min of prior exposure to 1 μM dorsomorphin inhibited responses both to 5 μM A-769662 and to 5 mg·mL−1 adiponectin, but not those to 33 μM NS1619. (B) In the presence of PVAT (+ PVAT), dorsomorphin inhibited hyperpolarizations to 10 μM CL-316,243 (CL) but not those to the BKCa channel opener NS1619. In (A), flow was stopped (flow off) to minimize the quantity of adiponectin required; in (B) the tissue was constantly superfused with Krebs to which bolus doses of the drugs were added as previously described.
Does stimulation of PVAT β3-adrenoceptors also release NO?
AMPK activates NOS in endothelial cells (Morrow et al., 2003). We therefore speculated that AMPK might enhance NO production from adipocyte NOS and contribute to the hyperpolarizing effects of CL-316,243.
L-NMMA (100 μM) did not change myocyte resting membrane potentials (control, −51.0 ± 0.9 mV, n = 4; L-NMMA, −50.1 ± 0.8 mV, n = 4) or NS1619-induced hyperpolarizations (control, 18.1 ± 0.9 mV, n = 4; presence of L-NMMA, 17.5 ± 0.9 mV, n = 4) but partially reduced those generated by CL-316,243 in the presence of PVAT (P < 0.0001, two-way anova; Figure 4). Subsequent addition of dorsomorphin in the presence of L-NMMA abolished the residual hyperpolarizations to 10 μM CL-316,243 and repolarized the membrane to control levels (–49.4 ± 0.4 mV, n = 4).
Figure 4.
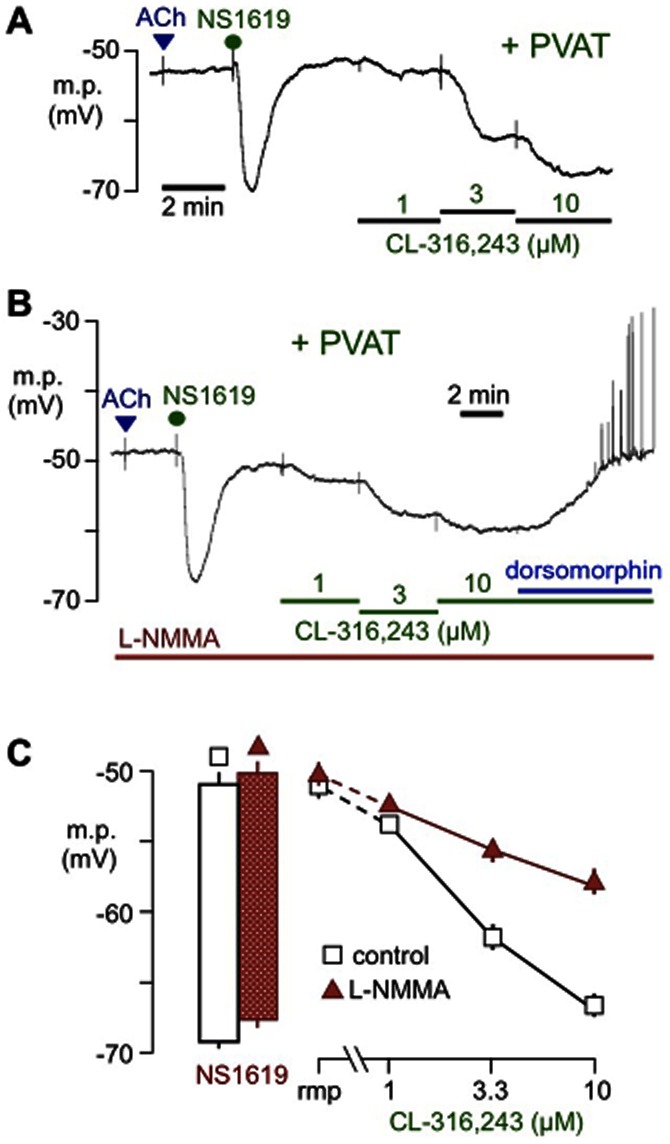
Effects of the NOS inhibitor (L-NMMA) on hyperpolarizations to CL-316,243. Typical traces showing sharp microelectrode recordings from rat mesenteric artery myocytes in the absence of the endothelium (no response to 10 μM ACh) but in the presence of PVAT (+PVAT). The concentration-dependent hyperpolarizations produced by CL-316,243 (A) were reduced by 100 μM L-NMMA (B), although the inhibitor had no effect on hyperpolarizations to 33 μM NS1619. In the presence of L-NMMA, hyperpolarizations to CL-316,243 were reversed by 1 μM dorsomorphin (see text for more details). The line graph in (C) indicates the mean resting membrane potential (rmp) and hyperpolarizations to each concentration of CL-316,243 (increased cumulatively) in the absence (control, open symbols) or presence (filled symbols) of L-NMMA. The histograms to the left depict the membrane potential before and after exposure to 33 μM NS1619 in the absence or presence of L-NMMA. Each point represents the mean ± SEM (n = 4).
Inhibitory effects of clotrimazole and glibenclamide
The present findings that BKCa channels in native myocytes were opened by myocyte AMPK activation conflicted with the results of Ross et al. (2011), who reported that the AMPK activator, aminoimidazole-4-carboxamide-ribonucleoside (AICAR), either had no effect or even inhibited BKCa channels heterologously expressed in HEK293 cells.
To clarify possible mechanisms involved in BKCa activation by AMPK in native myocytes following exposure to adiponectin or A-769662, we investigated the effects of clotrimazole, an inhibitor of several types of vascular, non-selective cation channels which could have acted as myocyte calcium-entry pathways. Myocyte hyperpolarizations to CL-316,243 adiponectin and to A-769662 were always reversed by 33 μM clotrimazole (Figure 5A–C). This agent inhibits Na+- and K+-permeable TRPM4 channels (Vennekens and Nilius, 2007) that are present in vascular myocytes (Morita et al., 2007; Zholos et al., 2011), an action shared with glibenclamide (Demion et al., 2007), the sulphonylurea previously reported to inhibit the anti-contractile effects of PVAT in rat aorta (Löhn et al., 2002; Fang et al., 2009).
Figure 5.
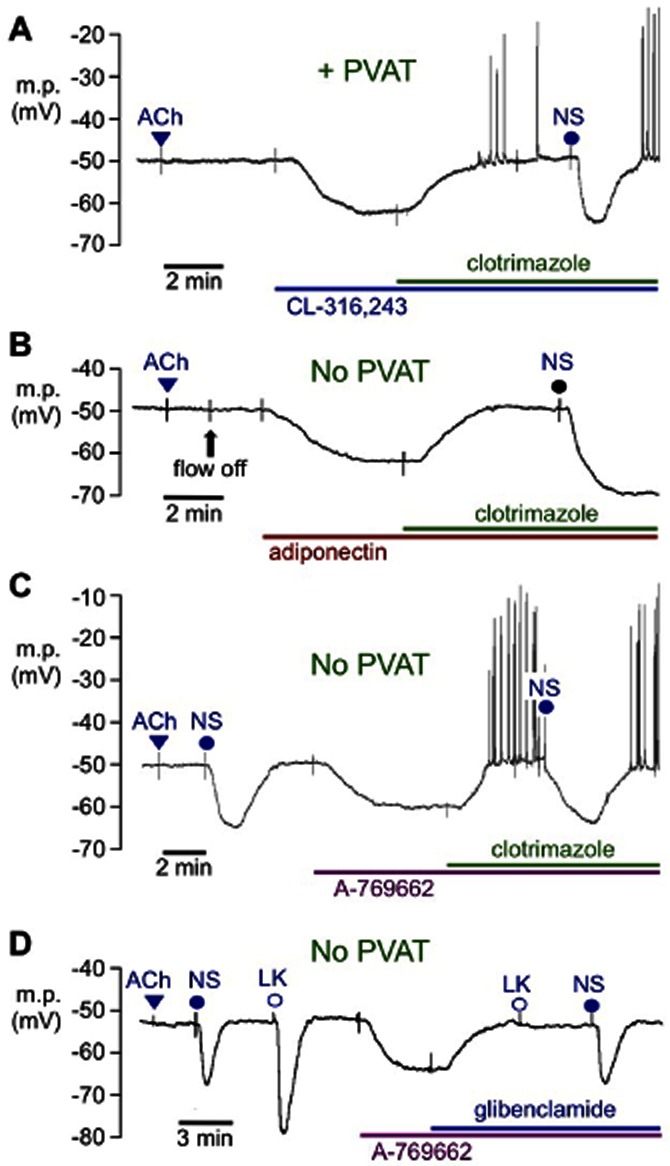
Typical traces showing the inhibitory effects of clotrimazole and glibenclamide on myocyte hyperpolarizations in endothelium-denuded segments (no response to 10 μM ACh) of rat mesenteric artery. In the presence (A; +PVAT) or absence (B and C; no PVAT) of PVAT, hyperpolarizations induced by (A) 10 μM CL-316,243, (B) 5 μM A-769662 or (C) 5 μg·mL−1 adiponectin were reversed by 33 μM clotrimazole although 33 μM NS1619 (NS) was subsequently able to induce hyperpolarization. (D) In the absence of PVAT, hyperpolarizations to 5 μM A-769662 were reversed by 10 μM glibenclamide. The hyperpolarizations induced by 10 μM levcromakalim (LK) but not by 33 μM NS1619 were inhibited by glibenclamide. Note that in (A), (C) and (D), the tissue was constantly superfused with Krebs solution to which bolus doses of ACh, NS1619 or LK were added to produce transient responses. Other modulators were added to the superfusate to produce sustained responses; in (B), flow was stopped as indicated (flow off) to minimize the quantity of adiponectin required.
Although 10 μM glibenclamide essentially abolished hyperpolarizations to 5 μM A-769662 (absence: 11.8 ± 1.1 mV; presence: 2.2 ± 0.5 mV, each n = 4), those due to direct opening of BKCa with 33 μM NS1619 were unaffected (absence: 14.6 ± 0.5 mV; presence: 14.3 ± 1.1 mV, each n = 4; Figure 5D), an indication that these actions of glibenclamide were not due to BKCa blockade.
Discussion
Experimental approach: simplify the experimental system
The clarity of the present findings was made possible by simplifying the experimental system in marked contrast to earlier studies. Thus, the vascular endothelium was removed in most experiments and microelectrode techniques allowed assessment of myocyte responses in the absence of spasmogens. Their presence is a powerful modulator of many ion channels, pumps and transporters, and greatly complicates analysis of the results obtained. Consistent with the use of a simplified system, the selective β3-adrenoceptor agonist CL-316,243 (Bloom et al., 1992; Dolan et al., 1994; Michel et al., 2010) was employed to stimulate adipocytes to release ADHF because these receptors are not functionally present on either rat mesenteric artery myocytes or endothelial cells (Kozlowska et al., 2003; Briones et al., 2005).
The results clearly show that the PVAT surrounding both rat and mouse mesenteric arteries can be stimulated to release an ADHF, which opens myocyte BKCa channels. The hyperpolarizing effects of the endogenous ADHF on myocyte BKCa were mimicked by adiponectin, the 30 kDa peptide secreted almost exclusively by adipocytes (Scherer et al., 1995), and proposed by Greenstein et al. (2009) to be an adipocyte-derived relaxing factor in rat and human vessels. Powerful evidence was also obtained for the involvement of myocyte AMP kinase, an enzyme known to be activated by adiponectin (Yamauchi et al., 2002; Kadowaki and Yamauchi, 2005) (Figure 6).
Figure 6.
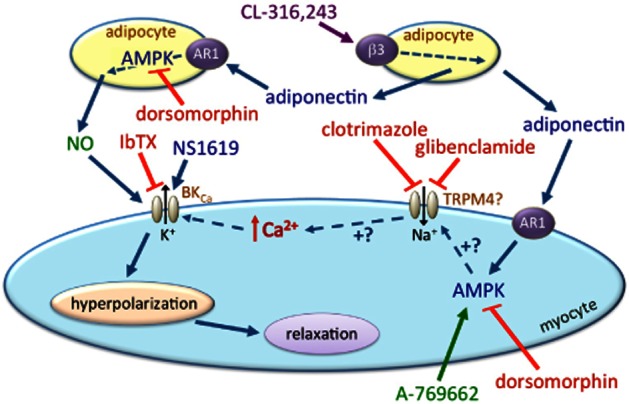
Working model for the stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery PVAT and involvement of adiponectin and myocyte BKCa channels. CL-316,243 stimulates the release of adiponectin, which acts through adiponectin receptors (AR1) and AMPK activation both in adipocytes (to release NO) and in myocytes (to open BKCa channels and induce hyperpolarization sensitive to iberiotoxin – IbTX). All effects of CL-316,243 are blocked by the AMPK inhibitor dorsomorphin. In the absence of PVAT, the hyperpolarizing effects of exogenous adiponectin are mimicked by A-769662, the AMPK activator. Both clotrimazole and glibenclamide (known inhibitors of TRPM4 channels) block the AMPK-induced activation of BKCa (by A-769662, adiponectin and CL-316,243) but do not modify direct BKCa activation by NS1619. We speculate that AMPK activation indirectly opens BKCa by first activating TRPM4 channels; the link between this putative event and BKCa activation requires further study.
Involvement of myocyte BKCa channels
CL-316,243 always hyperpolarized myocyte in artery segments surrounded by PVAT. Because these electrical changes were abolished by iberiotoxin (a selective BKCa blocker; Garcia et al., 1991), they were clearly generated by myocyte BKCa channel opening with no evidence that a voltage-gated K+ channel such as KV (as proposed by Verlohren et al., 2004; Fésüs et al., 2007; Schleifenbaum et al., 2010) or indeed any K+ channel other than BKCa was involved. In the absence of PVAT, CL-316,243 had no effect on myocyte membrane potentials in segments from the same mesenteric branch (even in vessels with an intact endothelium), confirming that the β3 agonist exerted its effects on the adipose tissue (Figure 6).
Is adiponectin the CL-316,243-induced ADHF?
In vivo, serum adiponectin levels in obese diabetic mice (db/db and KKAy) are raised by CL-316,243 (Oana et al., 2005; Fu et al., 2008). Exposure of rat mesenteric arteries to authentic adiponectin always hyperpolarized the myocytes in the absence of PVAT or the endothelium, reaching a maximum effect within a few minutes. This action, like the hyperpolarizations induced by CL-316,243 in the presence of PVAT, was also blocked by iberiotoxin, indicating the adiponectin-induced opening of myocyte BKCa channels (Figure 6).
Although attempts to measure CL-316,243-induced release of adiponectin from PVAT in vitro were unsuccessful, adiponectin exists predominantly in trimeric, hexameric and multimeric (>18 adiponectin molecules) aggregates, which differ in their biological activities (see Brochu-Gaudreau et al., 2010). Because commercially available kits for quantification of rat adiponectin do not distinguish between these forms, their presence could have masked any CL-316,243-induced release of a biologically relevant form of the peptide. Thus, although no direct evidence of stimulated adiponectin release was obtained, these experiments collectively suggest that the interaction of CL-316,243 with adipocyte β3-adrenoceptors is likely to release adiponectin which then hyperpolarizes the mesenteric vessel myocytes by activating BKCa channels (Figure 6).
A component of CL-316,243-induced hyperpolarizations was blocked by L-NMMA. If adiponectin is the ADHF involved, we propose that this peptide can activate adipocyte NOS to release NO. All the effects of CL-316,243 were dorsomorphin sensitive, an indication that the postulated adipocyte NO pathways also involve AMPK activation (Figure 6).
Experiments with vessels from Adipo−/− mice: supportive evidence of adiponectin release from PVAT
Exposure of mouse control (C57BL/6J) mesenteric vessels to CL-316,243 in the presence of PVAT generated iberiotoxin-sensitive hyperpolarizations, which were similar to those observed in rat vessels. Such changes were, however, absent in vessels from Adipo−/− mice, the strain (B6.129-Adipoqtm1Chan/J; Ma et al., 2002) engineered to be adiponectin deficient. This strongly suggests that CL-316,243-induced activation of β3-adrenoceptors on mouse (and by extrapolation, rat) PVAT can generate adiponectin-mediated myocyte hyperpolarization involving BKCa activation. This is strongly supported by observations that adiponectin-induced hyperpolarizations are absent from Slo−/− mice mesenteric artery myocytes, which lack BKCa pore-forming α-subunits (Lynch et al., 2013).
Our findings suggest that adiponectin can fully explain the hyperpolarizing effect of PVAT stimulation in non-contracted vessels. However, in a previous study, Fésüs et al. (2007) found that 4-aminopyridine (a relatively non-selective blocker of delayed rectifier K+ channels, KV) produced a PVAT-dependent increase in perfusion pressure in mouse-isolated mesenteric bed preparations in the presence or absence of a spasmogen (5-hydroxytryptamine). This effect, which was not modified by adiponectin gene deletion, was interpreted as an indication that the relaxant factor released by PVAT acted by opening myocyte KV channels. However, little consideration was paid to the possibility that the enhanced contractile effect of 4-aminopyridine following PVAT removal would have occurred irrespective of the mechanism of action of the PVAT-derived factor. It is well established that the vascular endothelium releases several relaxant factors in response to a single stimulus (reviewed by Triggle et al., 2010), and Gollasch (2012) has summarized the various agents, in addition to adiponectin, which can be released from PVAT. Our inability to detect evidence of a role for factors other than adiponectin in our electrophysiological experiments in non-contracted vessels suggests that such additional factors are released from PVAT by relatively high spasmogen concentrations and/or that their actions are not exerted through myocyte hyperpolarization.
The AMP kinase pathway and ADHF
To obtain further information about the possible role of adiponectin as the mediator of PVAT-dependent myocyte hyperpolarizations, experiments were conducted to determine whether the hyperpolarizing effect of CL-316,243 in the presence of PVAT was associated with modulation of AMPK, the major downstream signalling target activated by adiponectin following its interaction with AdipoR1 receptors (Yamauchi et al., 2007; reviewed by Xu et al., 2010). AMPK activity was first stimulated using A-769662 that, like AMP itself, acts both as an allosteric activator and inhibits the dephosphorylation and consequent inactivation of the enzyme (Göransson et al., 2007). However, unlike other AMPK activators such as the widely employed AICAR, A-769662 does not mimic AMP. Therefore, it neither stimulates glycogen phosphorylase nor inhibits fructose-1,6-bisphosphatase (Göransson et al., 2007), and so is a more selective AMPK activator than AICAR. Although off-target effects for A-769662 have been reported (Moreno et al., 2008; Scott et al., 2008; Treebak et al., 2009), these are associated with concentrations (100 μM–1 mM) that are significantly higher than that (5 μM) employed in the present study.
In strong support of a role for myocyte AMPK in the BKCa-opening actions of both CL-316,243 and exogenous adiponectin, A-769662 itself generated marked myocyte hyperpolarizations (in the absence of PVAT), which were inhibited not only by iberiotoxin but also by the AMPK inhibitor, dorsomorphin. Furthermore, this inhibitor also abolished the myocyte-hyperpolarizing, BKCa-opening effects of both CL-316,243 and authentic adiponectin (Figure 6).
Inhibitory actions of glibenclamide and clotrimazole – some speculations
A striking finding was the pivotal involvement of myocyte BKCa channels in the hyperpolarizing actions of adiponectin and A-769662 and in those of the ADHF liberated from PVAT by CL-316,243. Previous myograph studies (Löhn et al., 2002; Fang et al., 2009) have simplistically concluded that ‘ADHF’ was an opener of myocyte KATP channels because the anti-contractile effects of PVAT in rat aorta were inhibited by glibenclamide, the widely used blocker of KATP. However, direct measurements of membrane potential in the present study have clearly shown that ADHF opens BKCa channels.
Because 10 μM glibenclamide, the concentration employed by Löhn et al. (2002) and Fang et al. (2009), also inhibits the cation channel TRPM4 (Demion et al., 2007), we reasoned that myocyte AMPK activation (by an endogenous ADHF) might allow Na+ entry through TRPM4 channels. As reviewed by Guinamard et al. (2010), TRPM4 activation provides an important driving force for Ca2+ entry into a variety of cell types, an event that would increase the open probability of a Ca2+-sensitive K+ channel like BKCa. If correct, iberiotoxin should block BKCa opening induced by both A-769662 and NS1619, whereas glibenclamide should only inhibit any ‘indirect’ effect of A-769662 mediated by hypothetical AMPK-induced activation of Ca2+ entry via TRPM4. Consistent with this hypothesis, glibenclamide did inhibit iberiotoxin-sensitive hyperpolarizations induced by the AMPK activator, A-769662, without affecting direct BKCa activation by NS1619.
In addition to their glibenclamide sensitivity (Demion et al., 2007), TRPM4 cation channels are also inhibited by clotrimazole (Vennekens and Nilius, 2007), which has no effect on NS1619-induced BKCa currents (Edwards et al., 1996). In the present study, clotrimazole abolished PVAT-dependent hyperpolarizations not only to CL-316,243 but also to A-769664 and adiponectin, with no effect on responses to NS1619 (Figure 6).
Collectively, therefore, the hyperpolarizations generated by ADHF (i.e. by CL-316,243 in the presence of PVAT), A-769664 and adiponectin are consistent with their ability to activate myocyte BKCa channels indirectly. Experiments to investigate these possibilities further and to explore how the opening of a non-selective, poorly Ca2+-permeable cation channel like TRPM4 might be involved in the activation of myocyte BKCa channels are currently in progress.
Conclusions
Activation of adipocyte β3-adrenoceptors on PVAT releases an ADHF, which is likely to be adiponectin. In non-contracted vessels, this adipokine indirectly opens myocyte BKCa channels, an effect that involves AMPK since it can be mimicked by the AMPK activator, A-769662, and blocked by the kinase inhibitor, dorsomorphin (Figure 6). The ability of glibenclamide and clotrimazole to block adipocyte-dependent myocyte hyperpolarizations but not responses to a BKCa channel opener, NS1619, suggests that AMPK does not activate BKCa directly but may involve a cation channel such as TRPM4. Some of the effects of the liberated ADHF are L-NMMA sensitive, a possible indication of adipocyte NO-synthase activation also involving an AMPK-dependent pathway (Figure 6). The PVAT-dependent, BKCa-mediated hyperpolarizing effects of adiponectin on vascular myocytes may have a variety of long-term consequences in vivo, and form the basis of the complex role of adiponectin in cardiovascular disease, including the effects of this adipokine on systolic blood pressure (Avery et al., 2011).
Acknowledgments
This study was funded by the British Heart Foundation Grants FS/07/043 (E. L. P.), FS/09/055/28034 (Y. D.) and BBSRC CASE Studentship, BB/D526561/1 (I. E.).
Glossary
- A-769662
4-hydroxy-3-(2′-hydroxybiphenyl-4-yl)-6-oxo-6,7-dihydrothieno[2,3-b]pyridine-5-carbonitrile
- ADHF
adipocyte-derived hyperpolarizing factor
- AMPK
AMP-activated kinase
- BKCa
large-conductance Ca2+-activated K+ channels
- CL-316,243
5-[(2R)-2-[[(2R)-2-(3-chlorophenyl)-2-hydroxyethyl]amino]-propyl]-1,3-benzodioxole-2,2-dicarboxylic acid
- KATP
ATP-sensitive K+ channel
- NS1619
1-(2′-hydroxy-5′ trifluoromethylphenyl)-5-trifluoromethyl-2(3H)benzimidazolone
- PVAT
perivascular adipose tissue
- TRPM4
transient receptor potential melastatin channel subtype 4
Conflicts of interest
None.
References
- Avery PJ, Patel SK, Ibrahim IM, Walker M, Keavney BD. Common variation in the adiponectin gene has an effect on systolic blood pressure. J Hum Hypertens. 2011;25:719–724. doi: 10.1038/jhh.2010.122. [DOI] [PubMed] [Google Scholar]
- Bloom JD, Dutia MD, Johnson BD, Wissner A, Burns MG, Largis EE, et al. Disodium (R,R)-5-[2-[[2-(3-chlorophenyl)-2-hydroxyethyl]-amino] propyl]-1,3-benzodioxole-2,2-dicarboxylate (CL 316,243). A potent β-adrenergic agonist virtually specific for β3 receptors. A promising antidiabetic and antiobesity agent. J Med Chem. 1992;35:3081–3084. doi: 10.1021/jm00094a025. [DOI] [PubMed] [Google Scholar]
- Briones AM, Daly CJ, Jimenez-Altayo F, Martinez-Revelles S, Gonzalez JM, McGrath JC, et al. Direct demonstration of β1- and evidence against β2- and β3-adrenoceptors, in smooth muscle cells of rat small mesenteric arteries. Br J Pharmacol. 2005;146:679–691. doi: 10.1038/sj.bjp.0706369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochu-Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, Palin M-F. Adiponectin action from head to toe. Endocr. 2010;37:11–32. doi: 10.1007/s12020-009-9278-8. [DOI] [PubMed] [Google Scholar]
- Collins S, Daniel KW, Petro AE, Surwit RS. Strain-specific response to β3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. 1997;138:405–413. doi: 10.1210/endo.138.1.4829. [DOI] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc Res. 2007;73:531–538. doi: 10.1016/j.cardiores.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Dolan JA, Muenkel HA, Burns MG, Pellegrino SM, Fraser CM, Pietri F, et al. Beta-3 adrenoceptor selectivity of the dioxolane dicarboxylate phenethanolamines. J Pharmacol Exp Ther. 1994;269:1000–1006. [PubMed] [Google Scholar]
- Dubrovska G, Verlohren S, Luft FC, Gollasch M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol. 2004;286:H1107–H1113. doi: 10.1152/ajpheart.00656.2003. [DOI] [PubMed] [Google Scholar]
- Edwards G, Zygmunt P, Högestätt E, Weston AH. Effects of cytochrome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. Br J Pharmacol. 1996;119:691–701. doi: 10.1111/j.1476-5381.1996.tb15728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner I, Weston AH, Porter EL, Edwards G. Hyperpolarizing effect of perivascular adipose tissue in rat mesenteric artery myocytes: stimulation by β3 adrenoceptor activation. 2011. Proceedings of the British Pharmacological Society. Available at: http://www.pA2online.org/abstracts/Vol8Issue1abst141P.pdf (accessed 02/04/2013)
- Fang L, Zhao J, Chen Y, Ma T, Xu G, Tang C, et al. Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens. 2009;27:2174–2185. doi: 10.1097/HJH.0b013e328330a900. [DOI] [PubMed] [Google Scholar]
- Fésüs G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, et al. Adiponectin is a novel humoral vasodilator. Cardiovasc Res. 2007;75:719–727. doi: 10.1016/j.cardiores.2007.05.025. [DOI] [PubMed] [Google Scholar]
- Fu L, Isobe K, Zeng Q, Suzukawa K, Takekoshi K, Kawakami Y. The effects of β3-adrenoceptor agonist CL-316,243 on adiponectin, adiponectin receptors and tumor necrosis factor-α expressions in adipose tissues of obese diabetic KKAy mice. Eur J Pharmacol. 2008;584:202–206. doi: 10.1016/j.ejphar.2008.01.028. [DOI] [PubMed] [Google Scholar]
- Gao Y-J, Lu C, Su L-Y, Sharma AM, Lee RMKW. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151:323–331. doi: 10.1038/sj.bjp.0707228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia ML, Galvez A, Garcia-Calvo M, King VF, Vazquez J, Kaczorowski GJ. Use of toxins to study potassium channels. J Bioenerg Biomembr. 1991;23:615–646. doi: 10.1007/BF00785814. [DOI] [PubMed] [Google Scholar]
- Germack R, Starzec AB, Vassy R, Perret GY. Beta-adrenoceptor subtype expression and function in rat white adipocytes. Br J Pharmacol. 1997;120:201–210. doi: 10.1038/sj.bjp.0700885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollasch M. Vasodilator signals from perivascular adipose tissue. Br J Pharmacol. 2012;165:633–642. doi: 10.1111/j.1476-5381.2011.01430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119:1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Demion M, Launay P. Physiological roles of the TRPM4 channel extracted from background currents. Physiology. 2010;25:155–164. doi: 10.1152/physiol.00004.2010. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Ono K, Horie T, Nagao K, Nishi H, Kuwabara Y, et al. Regulation of adipocyte differentiation by activation of serotonin (5-HT) receptors 5-HT2AR and 5-HT2CR and involvement of microRNA-448-mediated repression of KLF5. Mol Endocrinol. 2010;24:1978–1987. doi: 10.1210/me.2010-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowska H, Szymska U, Schlicker E, Malinowska B. Atypical β-adrenoceptors, different from β3-adrenoceptors and probably from the low-affinity state of β1-adrenoceptors, relax the rat isolated mesenteric artery. Br J Pharmacol. 2003;140:3–12. doi: 10.1038/sj.bjp.0705421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16:1057–1063. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH, et al. Perivascular adipose tissue-derived adiponectin activates BKCa channels to induce anticontractile responses. Am J Physiol. 2013;304:H786–H795. doi: 10.1152/ajpheart.00697.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Cabrero A, Saha PK, Kojima H, Li L, Chang BH-J, et al. Increased β-oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem. 2002;277:34658–34661. doi: 10.1074/jbc.C200362200. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Ochodnicky P, Summers RJ. Tissue functions mediated by β3-adrenoceptors – findings and challenges. Naunyn-Schmied Arch Pharmacol. 2010;382:103–108. doi: 10.1007/s00210-010-0529-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno D, Knecht E, Viollet B, Sanz P. A-769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008;582:2650–2654. doi: 10.1016/j.febslet.2008.06.044. [DOI] [PubMed] [Google Scholar]
- Morita H, Honda A, Inoue R, Ito Y, Abe K, Nelson MT, et al. Membrane stretch-induced activation of a TRPM4-like nonselective cation channel in cerebral artery myocytes. J Pharmacol Sci. 2007;103:417–426. doi: 10.1254/jphs.fp0061332. [DOI] [PubMed] [Google Scholar]
- Morrow VA, Foufelle F, Connell JMC, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- Oana F, Takeda H, Matsuzawa A, Akahane S, Isaji M, Akahane M. Adiponectin receptor 2 expression in liver and insulin resistance in db/db mice given a β3-adrenoceptor agonist. Eur J Pharmacol. 2005;518:71–76. doi: 10.1016/j.ejphar.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Ross FA, Rafferty JN, Dallas ML, Ogunbayo O, Ikematsu N, McClafferty H, et al. Selective expression in carotid body type I cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by AMP-activated protein kinase. J Biol Chem. 2011;286:11929–11936. doi: 10.1074/jbc.M110.189779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Köhn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010;28:1875–1882. doi: 10.1097/HJH.0b013e32833c20d5. [DOI] [PubMed] [Google Scholar]
- Scott JW, van Denderen BJW, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase β1-containing complexes. Chem Biol. 2008;15:1220–1230. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Stunes AK, Reseland JE, Hauso O, Kidd M, Tømmerås K, Waldum HL, et al. Adipocytes express a functional system for serotonin synthesis, reuptake and receptor activation. Diabetes Obes Metab. 2011;13:551–558. doi: 10.1111/j.1463-1326.2011.01378.x. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JFP. A-769662 activates AMPK β1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol. 2009;297:C1041–C1052. doi: 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- Triggle CR, Samuel SM, Ravishankar S, Marei I, Arunachalam G, Ding H. The endothelium: influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol. 2010;90:713–738. doi: 10.1139/y2012-073. [DOI] [PubMed] [Google Scholar]
- Vennekens R, Nilius B. Insights into TRPM4 function, regulation and physiological role. Handb Exp Pharmacol. 2007;179:269–285. doi: 10.1007/978-3-540-34891-7_16. [DOI] [PubMed] [Google Scholar]
- Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y, et al. Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension. 2004;44:271–276. doi: 10.1161/01.HYP.0000140058.28994.ec. [DOI] [PubMed] [Google Scholar]
- Xu A, Wang Y, Lam KSL, Vanhoutte PM. Vascular actions of adipokines molecular mechanisms and therapeutic implications. Adv Pharmacol. 2010;60:229–255. doi: 10.1016/B978-0-12-385061-4.00008-8. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- Zholos A, Johnson C, Burdyga T, Melanaphy D. TRPM channels in the vasculature. Adv Exp Med Biol. 2011;704:707–729. doi: 10.1007/978-94-007-0265-3_37. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]