

Abstract
Background and Purpose
Our previous study demonstrated that 6-(pyrrolidin-1-yl)-2-(3-methoxyphenyl)quinazolin-4-one (HMJ38) was a potent anti-tubulin agent. Here, HMJ38 was used as a lead compound to develop more potent anti-cancer agents and to examine the anti-cancer mechanisms.
Experimental Approach
Using computer-aided drug design, 2-aryl-6-substituted quinazolinones (MJ compounds) were designed and synthesized by introducing substituents at C-2 and C-6 positions of HMJ38. The cytotoxicity of MJ compounds towards human cancer cells was examined by Trypan blue exclusion assay. Microtubule distribution was visualized using TubulinTracker™ Green reagent. Protein expression of cell cycle regulators and JNK was assessed by Western blot analysis.
Key Results
Compounds MJ65–70 exhibited strong anti-proliferative effects towards melanoma M21, lung squamous carcinoma CH27, lung non-small carcinoma H460, hepatoma Hep3B and oral cancer HSC-3 cells, with one compund MJ66 (6-(pyrrolidin-1-yl)-2-(naphthalen-1-yl)quinazolin-4-one) highly active against M21 cells (IC50 about 0.033 μM). Treatment of CH27 or HSC-3 cells with MJ65–70 resulted in significant mitotic arrest accompanied by increasing multiple asters of microtubules. JNK protein expression was involved in the MJ65–70-induced CH27 and M21 cell death. Consistent with the cell cycle arrest at G2/M phase, marked increases in cyclin B1 and Bcl-2 phosphorylation were also observed, after treatment with MJ65–70.
Conclusions and Implication
MJ65–70 are dual-targeted, tubulin- and JNK-binding, anti-cancer agents and induce cancer cell death through up-regulation of JNK and interfering in the dynamics of tubulin. Our work provides a new strategy and mechanism for developing dual-targeted anti-cancer drugs, contributing to clinical anti-cancer drug discovery and application.
Keywords: 2-aryl-6-substituted quinazolinones, c-Jun N-terminal kinase, computer-aided drug design, dual-targeted anti-cancer agents, tubulin
Introduction
Microtubules are polymers that are made up of consecutive dimers of α- and β-tubulin protein and play a crucial role in cell mitosis. In recent years, drugs that derange microtubule dynamics have been investigated and commonly used as chemotherapeutic agents for a variety of cancers (Burns et al., 2009; Choi et al., 2011). Several clinically used anti-cancer drugs, such as paclitaxel and vinblastine, bind to microtubules, thereby altering normal microtubule dynamics, which induces cells to arrest at the G2/M phase of the cell cycle and ultimately leads to apoptosis of cancer cells (Manfredi et al., 1982; Hadfield et al., 2003). Therefore, microtubules are a proven target for the development of new anti-cancer drugs because they are critical for mitotic spindle fibre formation and the separation of chromosomes at mitosis. JNK, also known as stress-activated protein kinase, is the member of the MAPK family. The activation of JNK has been demonstrated to be involved in apoptosis of cancer cells, induced by tubulin-binding agents (Sánchez-Perez et al., 1998; Ma et al., 2009). It has also been reported that the phosphorylation of Bcl-2 on Ser70 is not proportional to the extent of apoptosis (Furukawa et al., 2000), and Bcl-2 phosphorylation is only a marker of M-phase events (Ling et al., 1998). Furthermore, drugs affecting the integrity of microtubules could induce Bc1-2 phosphorylation (Haldar et al., 1997). Therefore, JNK should also be an important target for the development of new anti-tubulin drugs.
Cancer is the leading cause of death worldwide. Anti-tubulin drugs such as vinblastine (microtubule-destabilizing) and paclitaxel (microtubule-stabilizing) have been developed against a variety of cancers. However, development of drug resistance limits the effectiveness of paclitaxel and vinblastine (Gottesman et al., 2002; Doyle and Ross, 2003). Therefore, many investigators concentrated their efforts on understanding the mechanisms of drug resistance or identifying novel and highly specific anti-cancer drugs (Kartalou and Essigmann, 2001; Agarwal and Kaye, 2003). Furthermore, multi-target anti-cancer drugs were proposed to provide a new opportunity for treating paclitaxel- or vinblastine-resistant tumour cells.
In our earlier studies, substituted 2-phenyl-4-quinazolinones (PQZs) were identified as potent inhibitors of tubulin polymerization (Hour et al., 2000; Yang et al., 2004). We also concluded that the substituents at C-6 of PQZs were important to their anti-cancer activities. Compounds with electron-donating groups, such as methoxy group, N-alkyl group or a heterocyclic ring, at 6-position of PQZs displayed promising anti-cancer activities. Among them, 6-methoxy, 6-N,N-dimethyl, 6-(pyrrolidin-1-yl), 6-(piperidin-1-yl) and 6-(morpholin-1-yl) PQZs showed strong cytotoxicity with low IC50 values against a panel of human tumour cell lines. Notably, 6-(pyrrolidin-1-yl)-2-(3-methoxyphenyl)quinazolin-4-one (HMJ38) was the most potent anti-tubulin agent. Based on our previous study, the five substituents described earlier were selected as building blocks in our further design of more potent cytotoxic anti-tubulin agents.
Molecular docking techniques were also applied in this study to supplement the efficient and effective design of new analogues. Computer-aided drug design (CADD) system is a powerful tool in drug design process. Molecular docking methodology that predicts the best mode by which a given compound will fit into a binding site of a macromolecular target is of practical importance for the generation of the lead compound or optimizing the lead compound in drug discovery. Molecular docking also allows the rapid screening of many chemical structures and thus the identification of promising candidates for practical synthesis and further experimental processing (Bajorath, 2002; Booth and Zemmel, 2004; Chen et al., 2006). As the parent compound, HMJ38 was docked initially with tubulin to explore its ligand–protein binding mode and binding energy. Subsequently, new 2-aryl-6-substituted quinazolinones (MJ series compounds) were designed based on the docking results with HMJ38. Finally, the designed compounds were synthesized and evaluated for in vitro anti-tumour activity.
The major purpose of this study was to identify more potent anti-cancer compounds and to examine the underlying mechanisms. A better understanding of the cytotoxic mechanisms involved would assist in the development of potent derivatives with high therapeutic efficacy. In this study, the expression of JNK protein and microtubule assembly were found to be involved in the cytotoxicity and G2/M cell cycle arrest in HSC-3 and CH27 cells, induced by compounds MJ65–70. The results of docking simulations provided further support that these compounds were tubulin- and JNK-binding agents.
Methods
Molecular docking
The crystal structures of tubulin with paclitaxel (1jff) or vinblastine (1Z2b), respectively, and JNK protein (3e7o) recovered from the RCSB Protein Data Bank (http://www.rcsb.org/pdb) were used as the targets for molecular docking. The docking calculations of the designed compounds with tubulin and JNK were performed with LigandFit program within the software package Discovery Studio 2.5 (Accelrys, San Diego, CA, USA), which is an automated tool for ligand–protein docking and scoring. The prepared protein protocol was used to prepare 1z2b, 1jff and 3e7o protein structures including the following actions: standardize atom names, insert missing atoms in residues and remove alternate conformations, insert missing loop regions based on SEQRES data, optimize short and medium size loop regions with Looper Algorithm, minimize remaining loop regions, and calculate pK and protonate structure.
General procedure for synthesis of 2-aryl-6-substituted-4-quinazolinones (compounds 17–31)
The intermediate benzamides (2, 4, 5–13) were known compounds and their preparation has been published in a previous study (Hour et al., 2000). Sodium hydrogen sulfite (0.8 g, 7.5 mmol) was added to a solution of 5-methoxy-2-aminobenzamide (9) (1.2 g, 7.3 mmol) and 1-naphthaldehyde (14) (1.1 g, 7.3 mmol) in N,N-dimethylacetamide (20 mL). The mixture was heated with stirring at 150°C for 3 h and poured into ice water (200 mL). The precipitate was collected, washed with water and dried in vacuo. After purification by column chromatography (silica gel; chloroform) and followed by recrystallization from EtOH, 6-methoxy-2-(naphthalen-1-yl)-4-quinazolinone (17) was obtained (1.5 g, 70%) as pale yellow needles. The method used to prepare 17 was also applied with the appropriate substituted benzaldehyde and benzamide to afford 18–31. Yields ranged from 60 to 70%.
Synthesis of 6-methoxy-2-(naphthalen-1-yl)quinazolin-4-one (17, MJ70)
1.5 g from 5-methoxy-2-aminobenzamide (9) (1.2 g, 7.3 mmol) and 1-naphthaldehyde (14) (1.1 g, 7.3 mmol): pale yellow needles.
Synthesis of 6-methoxy-2-(1H-indol-3-yl)quinazolin-4-one (18, MJ74)
1.4 g from 5-methoxy-2-aminobenzamide (9) (1.2 g, 7.3 mmol) and indo-3-carboxaldehyde (15) (1.1 g, 7.3 mmol): brown crystals.
Synthesis of 6-methoxy-2-(benzo[b]thiophen-3-yl)quinazolin-4-one (19, MJ79)
1.3 g from 5-methoxy-2-aminobenzamide (9) (1.2 g, 7.3 mmol) and thianaphthene-3-carboxaldehyde (16) (1.2 g, 7.3 mmol): brown crystals.
Synthesis of 6-(N,N-dimethylamino)-2-(naphthalen-1-yl)quinazolin-4-one (20, MJ69)
1.5 g from 5-N,N-dimethyl-2-aminobenzamide (10) (1.3 g, 7.3 mmol) and 1-naphthaldehyde (14) (1.1 g, 7.3 mmol): yellow crystals.
Synthesis of 6-(N,N-dimethylamino)-2-(1H-indol-3-yl)quinazolin-4-one (21, MJ75)
1.3 g from 5-N,N-dimethyl-2-aminobenzamide (10) (1.3 g, 7.3 mmol) and indo-3-carboxaldehyde (15) (1.1 g, 7.3 mmol): yellow crystals.
Synthesis of 6-(N,N-dimethylamino)-2-(benzo[b]thiophen-3-yl)quinazolin-4-one (22, MJ80)
1.5 g from 5-N,N-dimethyl-2-aminobenzamide (10) (1.3 g, 7.3 mmol) and thianaphthene-3-carboxaldehyde (16) (1.2 g, 7.3 mmol): pale yellow needles.
Synthesis of 6-(pyrrolidin-1-yl)-2-(naphthalen-1-yl)quinazolin-4-one (23, MJ66)
1.6 g from 2-amino-5-(pyrrolidin-1-yl)benzamide (11) (1.5 g, 7.3 mmol) and 1-naphthaldehyde (14) (1.1 g, 7.3 mmol): pale yellow needles.
Synthesis of 6-(pyrrolidin-1-yl)-2-(1H-indol-3-yl)quinazolin-4-one (24, MJ65)
1.5 g from 2-amino-5-(pyrrolidin-1-yl)benzamide (11) (1.5 g, 7.3 mmol) and indo-3-carboxaldehyde (15) (1.1 g, 7.3 mmol): brown powder.
Synthesis of 6-(pyrrolidin-1-yl)-2-(benzo[b]thiophen-3-yl)quinazolin-4-one (25, MJ78)
1.5 g from 2-amino-5-(pyrrolidin-1-yl)benzamide (11) (1.5 g, 7.3 mmol) and thianaphthene-3-carboxaldehyde (16) (1.2 g, 7.3 mmol): brown powder.
Synthesis of 6-(piperidin-1-yl)-2-(naphthalen-1-yl)quinazolin-4-one (26, MJ68)
1.6 g from 2-amino-5-(piperidin-1-yl)benzamide (12) (1.6 g, 7.3 mmol) and 1-naphthaldehyde (14) (1.1 g, 7.3 mmol): brown needles.
Synthesis of 6-(piperidin-1-yl)-2-(1H-indol-3-yl)quinazolin-4-one (27, MJ72)
1.6 g from 2-amino-5-(piperidin-1-yl)benzamide (12) (1.6 g, 7.3 mmol) and indo-3-carboxaldehyde (15) (1.1 g, 7.3 mmol): brown crystals.
Synthesis of 6-(piperidin-1-yl)-2-(benzo[b]thiophen-3-yl)quinazolin-4-one (28, MJ73)
1.6 g from 2-amino-5-(piperidin-1-yl)benzamide (12) (1.6 g, 7.3 mmol) and thianaphthene-3-carboxaldehyde (16) (1.2 g, 7.3 mmol): pale yellow crystals.
Synthesis of 6-(morpholin-1-yl)-2-(naphthalen-1-yl)quinazolin-4-one (29, MJ67)
1.7 g from 2-amino-5-(morpholin-1-yl)benzamide (13) (1.6 g, 7.3 mmol) and 1-naphthaldehyde (14) (1.1 g, 7.3 mmol): yellow crystals.
Synthesis of 6-(morpholin-1-yl)-2-(1H-indol-3-yl)quinazolin-4-one (30, MJ76)
1.6 g from 2-amino-5-(morpholin-1-yl)benzamide (13) (1.6 g, 7.3 mmol) and indo-3-carboxaldehyde (15) (1.1 g, 7.3 mmol): brown crystals.
Synthesis of 6-(morpholin-1-yl)-2-(benzo[b]thiophen-3-yl)quinazolin-4-one (31, MJ77)
1.6 g from 2-amino-5-(morpholin-1-yl)benzamide (13) (1.6 g, 7.3 mmol) and thianaphthene-3-carboxaldehyde (16) (1.2 g, 7.3 mmol): brown crystals.
Cell culture
The M21 cells were grown in monolayer culture in RPMI medium 1640 (Invitrogen, Carlsbad, CA, USA) containing 10% FBS (HyClone Laboratories, Logan, UT, USA), 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin (Invitrogen) at 37°C in a humidified atmosphere comprising 95% air and 5% CO2. CH27, H460 and Hep3B cells were cultured in DMEM (Invitrogen) containing 5% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin and 2 mM glutamine (Merck). HSC-3 cells were cultured in DMEM/F12 (1:1) with l-glutamine and HEPES (HyClone) containing 5% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. When cells were treated with 2-aryl-6-substituted quinazolinones, culture medium containing 1% FBS was used.
Trypan blue exclusion assay
Cells were seeded at a density of 5 × 104 cells per well onto 12-well plates 48 h before being treated with drugs. The cells were incubated with various concentrations of MJ series compounds for 24 h. The control cultures were treated with 0.1% DMSO. The number of viable cells was determined by staining the cell population with Trypan blue. After treatment, cells were washed with PBS, trypsinized and the number of unstained (viable) cells was counted.
Localization of microtubules
Cells grown on coverslips were treated with 0.1% DMSO or with MJ65, 66, 67, 68, 69, 70 or 72 for 12 h. To detect β-tubulin, cells were incubated for 30 min at 37°C with 250 nM TubulinTracker™ Green reagent according to the manufacturer's instructions (Abcam, Cambridge, MA, USA). The specimens were observed by fluorescence microscopy (H600L, Nikon, Tokyo, Japan).
Cell cycle analysis
Cell cycle was determined as described previously (Wu et al., 2011). After treatment, cells were stained with propidium iodide (50 μg·mL−1) and analysed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). ModFit LT3.0 software (Verity Software House, Topsham, ME, USA) was employed for cell cycle distribution analysis.
Protein preparation and Western blot analysis
Protein preparation and Western blot analysis were performed as described by Lee et al.,(2010). The protein concentrations were estimated with the Bradford method. Equal amounts of protein (50 μg) were separated by various indicated concentrations of SDS-PAGE, and the SDS-separated proteins were electrotransferred to Immobilon-P Transfer Membranes (Millipore, Darmstadt, Germany). Membranes were probed with antibodies to β-actin (1:5000), Bcl-2 (1:1000), Bcl-2(pS70) (1:1000), Cdk1 (1:2500), cyclin B1 (1:500) and JNK (1:1000). Secondary antibody consisted of a 1:20 000 dilution of HRP-conjugated goat anti-mouse IgG (for β-actin, Bcl-2, cyclin B1, Cdk1 and JNK) or HRP-conjugated goat anti-rabbit IgG [for Bcl-2(pS70)]. The enhanced chemiluminescent detection system (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) was used for immunoblot protein detection.
Data analysis
Data are presented as means ±SEM and differences between means assessed by Student's t-test for paired data.
Materials
All of the solvents and reagents were obtained commercially and used without further purification. TLC plates and silica gel for column chromatography were purchased from Merck (Darmstadt, Germany). Melting points were determined with a Yanaco MP-500D (Kyoto, Japan) melting point apparatus and are uncorrected. NMR spectra were obtained on a Bruker Avance DPX-200 FT-NMR spectrometer (Billerica, MA, USA) in DMSO-d6. MS spectra were measured with a Finnigan/Thermo Quest MAT 95XL instrument (Bremen, Germany).
Antipain, aprotinin, DTT, EGTA, leupeptin, pepstatin, PMSF and Tris were purchased from Sigma (St. Louis, MO, USA). Antibodies to various proteins were obtained from the following sources: Bcl-2 and β-actin antibodies from Sigma; cyclin B1, Cdk1 and JNK from BD Biosciences (San Jose, CA, USA); Bcl-2(pS70) from (Abcam). HRP-conjugated goat anti-mouse and anti-rabbit IgG were from Abcam Inc.
Results
Computer-aided drug design
In order to efficiently optimize HMJ38 and obtain prospective potent candidates, the docking program LigandFit within the software package Discovery Studio 2.5 was applied in our design process. In our docking experiments, we found that HMJ38 could dock into 1z2b using the vinblastine domain as the binding site; moreover, there were more available space and amino acid residues encompassed in the binding pocket, which would allow us to introduce larger substituents into the structure of HMJ38. Based on our preliminary suggestions of structure–activity relationships (SAR) of PQZs, methoxy, N,N-dimethyl, pyrrolidin-1-yl, piperidin-1-yl and morpholin-1-yl substitutents were selected preferentially for incorporation at position 6 of PQZs. In addition, some aryl groups larger than phenyl, such as naphthalen-1-yl, 1H-indol-3-yl and benzo[b]thiophen-3-yl group, were introduced at the C-2 position of PQZs. The newly designed compounds were then docked into the tubulin active site to explore their binding positions and to calculate and rank their binding energies to predict if the best binding poses are promising in actual activity estimate. A virtual comparison of the new compound MJ66 with the lead compound HMJ38, in tubulin was shown in Figure 1A. We scored our models using several scoring functions, LigScore, PLP, PMF and DockScore (Table 1-1). The results showed that the designed 2-aryl-6-substituted quinazolinones (MJ series compounds) could bind adequately to tubulin. For example, as shown in Figure 1B, MJ66 can bind well to tubulin via hydrogen bonding and hydrophobic interactions. A hydrogen bond is present between the quinazoline ring (N3–H) of the ligand and Asp179. Complementary, van der Waals interactions are also formed between the ligand and Ser178, Asp179, Pro222, Thr223, Tyr224 and Ile332. These interactions caused MJ66 to bind readily to tubulin. The other MJ series compounds also bound well and in a similar mode. The vinblastine was re-docked into the tubulin–vinblastine domain to generate an RMSD (root mean square deviation) value of 1.9860 Å (Figure 1C).
Figure 1.
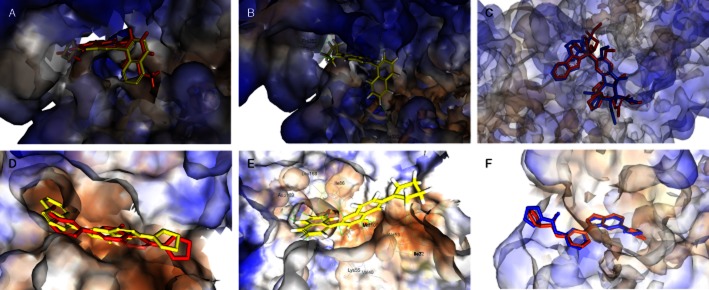
Molecular modelling. Virtual comparison of newly developed compound MJ66 with lead compound HMJ38 in tubulin (A) and JNK (B). MJ66 (yellow stick) docked well with tubulin (C) and JNK (D). The outputs of structural alignment show the superposition of vinblastine in tubulin–vinblastine domain (E) and 35F in JNK-35F domain (F) with RMSD values of 1.9860 and 0.8931 Å respectively. The binding amino acids are shown as lines and labelled. The carved surface representation of the pocket formed from MJ66 binding is shown as transparent blue-white-brown. Red and blue sticks represent the ligands co-crystallized within protein and re-docked into the protein respectively. The compound 35F is N-[3-[5-(1H-1,2,4-triazol-3-yl)-1H-indazol-3-yl]phenyl]furan-2-carboxamide.
Table 1-1.
The docking results of MJ65-80 and HMJ38 with tubulin (1z2b)
| Name | LigScore1 | LigScore2 | (-)PLP1 | (-)PLP2 | Jain | (-)PMF | Dock_Score |
|---|---|---|---|---|---|---|---|
| MJ65 | 3.34 | 4.90 | 55.50 | 49.92 | 2.70 | 32.51 | 53.028 |
| MJ66 | 3.21 | 5.13 | 63.69 | 55.36 | 1.66 | 16.46 | 56.452 |
| MJ67 | 3.58 | 5.26 | 61.38 | 54.38 | 2.75 | 31.28 | 55.67 |
| MJ68 | 3.20 | 5.16 | 69.37 | 60.84 | 1.64 | 21.00 | 57.04 |
| MJ69 | 2.82 | 4.90 | 61.31 | 54.03 | 2.12 | 22.51 | 53.97 |
| MJ70 | 3.21 | 4.83 | 63.33 | 52.82 | 1.09 | 19.4 | 49.31 |
| MJ72 | 2.00 | 4.79 | 64.24 | 57.23 | 2.03 | 31.52 | 50.14 |
| MJ73 | 1.61 | 4.48 | 54.57 | 47.51 | 0.85 | 16.00 | 53.39 |
| MJ74 | 3.29 | 4.70 | 55.89 | 49.63 | 2.58 | 32.11 | 47.96 |
| MJ75 | 3.30 | 4.72 | 50.17 | 44.98 | 2.76 | 30.15 | 52.10 |
| MJ76 | 3.55 | 4.81 | 61.65 | 50.36 | 2.36 | 39.76 | 51.06 |
| MJ77 | 3.34 | 4.97 | 58.94 | 53.4 | 2.71 | 33.11 | 56.54 |
| MJ78 | 2.76 | 4.69 | 58.42 | 53.55 | 3.09 | 34.41 | 55.72 |
| MJ79 | 2.54 | 4.34 | 46.62 | 40.91 | 1.56 | 21.28 | 52.01 |
| MJ80 | 2.67 | 4.49 | 46.96 | 41.46 | 1.57 | 21.66 | 55.04 |
| HMJ38 | 2.85 | 4.73 | 66.24 | 57.31 | 1.14 | 18.85 | 52.84 |
Chemistry
All of the compounds were synthesized according to Schemes 1–3. Structural elucidation of compounds was based on careful inspections of spectroscopic data. The physical and spectroscopic data of target compounds 17–31 are presented in Table 2.
Scheme 1.
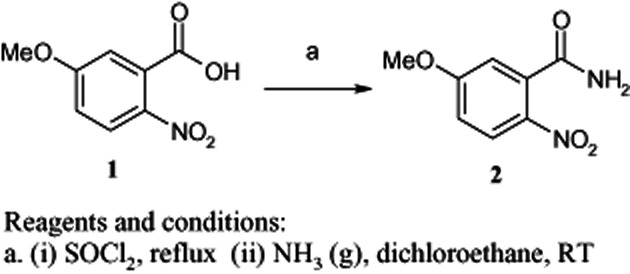
Synthesis of 5-methoxy-2-nitrobenzamide
Scheme 3.
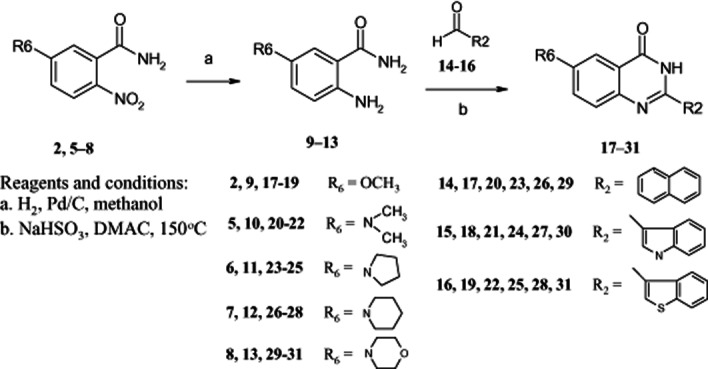
Synthesis of 2-aryl-6-substituted quinazolinones
Table 2.
Physical and spectroscopic data of compounds 17–31
| Compounds | Yield (%) | Mp (°C) | Formula | HRMS-EI, m/z (calcd.) | 1H NMR (DMSO-d6)δ |
|---|---|---|---|---|---|
| 17 (MJ70) | 70 | 174–175 | C19H14N2O2 | 302.1057 (302.1055) | 3.87 (3H, s, OCH3), 7.22–7.34 (2H, m, 2 × ArH), 7.53–7.72 (5H, m, 5 × ArH), 7.95–8.22 (3H, m, 3 × ArH), 12.62 (1H, br s, NH) |
| 18 (MJ74) | 68 | 311–312 | C17H13N3O2 | 291.1001 (291.1008) | 3.84 (3H, s, OCH3), 7.15–7.19 (2H, m, 2 × ArH), 7.35–7.49 (3H, m, 3 × ArH), 7.63–7.68 (1H, m, 1 × ArH), 8.45 (1H, s, 1 × ArH), 8.46–8.63 (1H, m, 1 × ArH), 11.76 (1H, s, NH), 12.11 (1H, br s, NH) |
| 19 (MJ79) | 60 | 289–290 | C17H12N2O2S | 308.0628 (308.0619) | 3.89 (3H, s, OCH3), 7.43–7.57 (4H, m, 4 × ArH), 7.76–7.80 (1H, m, 1 × ArH), 8.07–8.11 (1H, m, 1 × ArH), 8.73 (1H, s, 1 × ArH), 8.98–9.01 (1H, m, 1 × ArH), 12.49 (1H, br s, NH) |
| 20 (MJ69) | 65 | 254–255 | C20H17N3O | 315.1367 (315.1372) | 2.99 [6H, s, N(CH3)2], 7.22–7.34 (2H, m, 2 × ArH), 7.51–7.72 (5H, m, 5 × ArH), 7.96–8.23 (3H, m, 3 × ArH), 12.39 (1H, br s, NH) |
| 21 (MJ75) | 60 | >300 | C18H16N4O | 304.1316 (304.1324) | 2.99 [6H, s, N(CH3)2], 7.17–7.34 (4H, m, 4 × ArH), 7.43–7.47 (1H, m, 1 × ArH), 7.58–7.63 (1H, m, 1 × ArH), 8.44 (1H, s, 1 × ArH), 8.65–8.69 (1H, m, 1 × ArH), 11.71 (1H, s, NH), 11.92 (1H, br s, NH) |
| 22 (MJ80) | 62 | >300 | C18H15N3OS | 321.0941 (321.0936) | 2.99 [6H, s, N(CH3)2], 7.20–7.67 (5H, m, 5 × ArH), 8.02–8.05 (1H, m, 1 × ArH), 8.63 (1H, s, 1 × ArH), 8.97–9.01 (1H, m, 1 × ArH), 12.21 (1H, br s, NH) |
| 23 (MJ66) | 65 | 276–277 | C22H19N3O | 341.1523 (341.1528) | 1.99 (4H, m, CH2CH2NCH2CH2), 3.27 (4H, m, CH2NCH2), 7.10–7.21 (2H, m, 2 × ArH), 7.56–7.76 (5H, m, 5 × ArH), 8.01–8.21 (3H, m, 3 × ArH), 12.34 (1H, br s, NH) |
| 24 (MJ65) | 61 | 298 | C20H18N4O | 330.1488 (330.1481) | 1.94 (4H, m, CH2CH2NCH2CH2), 3.28 (4H, m, CH2NCH2), 6.97–7.20 (4H, m, 4 × ArH), 7.39–7.43 (1H, m, 1 × ArH), 7.53–7.58 (1H, m, 1 × ArH), 8.38 (1H, s, 1 × ArH), 8.61–8.65 (1H, m, 1 × ArH), 11.68 (1H, s, NH), 11.88 (1H, br s, NH) |
| 25 (MJ78) | 60 | >300 | C20H17N3OS | 347.1090 (347.1092) | 1.97 (4H, m, CH2CH2NCH2CH2), 3.47 (4H, m, CH2NCH2), 7.43–7.57 (4H, m, 4 × ArH), 7.76–7.80 (1H, m, 1 × ArH), 8.07–8.11 (1H, m, 1 × ArH), 8.73 (1H, s, 1 × ArH), 8.98–9.02 (1H, m, 1 × ArH), 12.51 (1H, br s, NH) |
| 26 (MJ68) | 65 | 233–235 | C23H21N3O | 355.1680 (355.1685) | 1.57–1.62 [6H, m, (CH2)2CH2NCH2CH2], 3.26–3.31 (4H, m, CH2NCH2), 7.48–7.65 (6H, m, 6 × ArH), 7.72–7.76 (1H, m, 1 × ArH), 8.00–8.13 (3H, m, 3 × ArH), 12.43 (1H, br s, NH) |
| 27 (MJ72) | 64 | >300 | C21H20N4O | 344.1638 (344.1637) | 1.59–1.62 [6H, m, (CH2)2CH2NCH2CH2], 3.20–3.24 (4H, m, CH2NCH2), 7.18–7.21 (2H, m, 2 × ArH), 7.41–7.48 (3H, m, 3 × ArH), 7.50–7.62 (1H, m, 1 × ArH), 8.46 (1H, s, 1 × ArH), 8.65–8.69 (1H, m, 1 × ArH), 11.73 (1H, s, NH), 11.96 (1H, br s, NH) |
| 28 (MJ73) | 60 | >300 | C21H19N3OS | 361.1240 (361.1249) | 1.57–1.59 [6H, m, (CH2)2CH2NCH2CH2], 3.22–3.25 (4H, m, CH2NCH2), 7.42–7.54 (4H, m, 4 × ArH), 7.63–7.67 (1H, m, 1 × ArH), 8.03–8.07 (1H, m, 1 × ArH), 8.66 (1H, s, 1 × ArH), 8.97–9.00 (1H, m, 1 × ArH), 12.30 (1H, br s, NH) |
| 29 (MJ67) | 64 | 291–293 | C22H19N3O2 | 357.1470 (357.1477) | 3.22 (4H, t, J = 4.7 Hz, CH2NCH2), 3.77 (4H, t, J = 4.7 Hz, CH2OCH2), 7.51–7.66 (6H, m, 6 × ArH), 7.73–7.76 (1H, m, 1 × ArH), 8.00–8.11 (3H, m, 3 × ArH), 12.48 (1H, br s, NH) |
| 30 (MJ76) | 65 | >300 | C20H18N4O2 | 346.1434 (346.1430) | 2.97 (4H, t, J = 4.7 Hz, CH2NCH2), 3.74 (4H, t, J = 4.7 Hz, CH2OCH2), 7.12–7.22 (2H, m, 2 × ArH), 7.41–7.63 (4H, m, 4 × ArH), 8.43 (1H, s, 1 × ArH), 8.60–8.64 (1H, m, 1 × ArH), 11.76 (1H, s, NH), 12.01 (1H, br s, NH) |
| 31 (MJ77) | 60 | >300 | C20H17N3O2S | 363.1034 (363.1041) | 2.47 (4H, t, J = 4.7 Hz, CH2NCH2), 3.75 (4H, t, J = 4.7 Hz, CH2OCH2), 7.44–7.59 (4H, m, 4 × ArH), 7.67–7.72 (1H, m, 1 × ArH), 8.03–8.07 (1H, m, 1 × ArH), 8.67 (1H, s, 1 × ArH), 8.96–9.00 (1H, m, 1 × ArH), 12.31 (1H, br s, NH) |
HRMS-EI, high-resolution electron impact mass spectra; Mp, melting point.
Scheme 2.
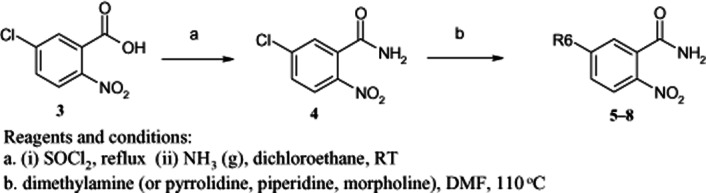
Synthesis of 2-nitro-5-substituted benzamides
Cytotoxicity of 2-aryl-6-substituted quinazolinones
The synthesized 2-aryl-6-substituted quinazolinones (MJ65–80) were assayed for cytotoxicity in vitro against five human tumour cell lines, including M21 (malignant melanoma), CH27 (lung squamous carcinoma), H460 (non-small cell lung cancer), Hep3B (hepatoma) and HSC-3 (oral cancers). As shown in Table 3, MJ65–70, 72 and 78 showed strong cytotoxic effects against the five tested cancer cell lines, with IC50 values ranging from 0.033 to 8.74 μM, while the remaining compounds MJ73–77, 79 and 80 were inactive (IC50 > 10 μM). MJ 66–70 displayed specifically anti-proliferative effects to CH27, H460 and M21 cells, with IC50 values < 1 μM, except for MJ69 with IC50 of 4.37 ± 0.33 μM in H460 cells. MJ66, 67, 69 and 70 displayed specifically anti-proliferative effects in HSC-3 cells, with IC50 values < 1 μM. As to the selectivity of these compounds for cancer cells, MJ78 showed specific selectivity to HSC-3, and MJ72 showed selective cytotoxicity to M21, H460 and Hep3B. On the whole, human hepatoma Hep3B cells were not very sensitive to the new compounds, compared with other cell lines.
Table 3.
In vitro cytotoxicity of 2-aryl-6-substituted quinazolinones
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| Compounds | M21 | CH27 | H460 | Hep3B | HSC-3 |
| 17 (MJ70) | 0.09 ± 0.006 | 0.09 ± 0.004 | 0.07 ± 0.02 | 1.94 ± 0.46 | 0.15 ± 0.03 |
| 18 (MJ74) | >10 | >10 | >10 | >10 | >10 |
| 19 (MJ79) | >10 | >10 | >10 | >10 | >10 |
| 20 (MJ69) | 0.63 ± 0.042 | 0.43 ± 0.03 | 4.37 ± 0.33 | 8.74 ± 0.63 | 0.20 ± 0.03 |
| 21 (MJ75) | >10 | >10 | >10 | >10 | >10 |
| 22 (MJ80) | >10 | >10 | >10 | >10 | >10 |
| 23 (MJ66) | 0.03 ± 0.005 | 0.05 ± 0.01 | 0.08 ± 0.01 | 1.35 ± 0.12 | 0.04 ± 0.003 |
| 24 (MJ65) | 7.94 ± 0.66 | 8.14 ± 0.53 | 8.89 ± 0.45 | 7.50 ± 0.55 | 2.01 ± 0.37 |
| 25 (MJ78) | >10 | >10 | >10 | >10 | 4.97 ± 0.82 |
| 26 (MJ68) | 0.44 ± 0.02 | 0.38 ± 0.01 | 0.38 ± 0.04 | 7.46 ± 0.90 | 1.65 ± 0.45 |
| 27 (MJ72) | 8.1 ± 0.57 | >10 | 6.86 ± 0.67 | 6.86 ± 0.81 | >10 |
| 28 (MJ73) | >10 | >10 | >10 | >10 | >10 |
| 29 (MJ67) | 0.49 ± 0.05 | 0.78 ± 0.07 | 0.44 ± 0.04 | 1.11 ± 0.14 | 0.63 ± 0.06 |
| 30 (MJ76) | >10 | >10 | >10 | >10 | >10 |
| 31 (MJ77) | >10 | >10 | >10 | >10 | >10 |
| HMJ38 | 3.44 ± 0.39 | >10 | >10 | >10 | 2.69 ± 0.38 |
Cell viability was determined by Trypan blue exclusion assay. The IC50 values of 2-aryl-6-substituted quinazolinones were determined from dose–response curves. All results are expressed as the mean percentage of control ± SD of triplicate determinations from three independent experiments.
CH27, human lung squamous carcinoma; H460, human lung non-small carcinoma cells; Hep3B, human hepatoma; HSC-3, human oral cancer cells; M21, human malignant melanoma.
Effects of MJ65–70 on microtubule polymerization of CH27 and HSC-3 cells
We examined the effect of MJ65–70 on tubulin distribution to determine if tubulins were targets in the MJ65–70-induced cancer cell death. As shown in Figure 2, mitotic spindles of cells blocked in anaphase by MJ65–70 were positioned in the vicinity of the spindle poles. Treatment of HSC-3 or CH27 cells with MJ65–70 resulted in significant mitotic arrest accompanied by increasing multiple spindle poles of microtubules (Figure 2). The observed changes were not the result of interaction of a Tubulin Tracker Green reagent with polymerized tubulin as the control cells did not show the inhibition of cell division. In this study, MJ72 had no significant effect on the formation of multiple asters of microtubules (Figure 2).
Figure 2.
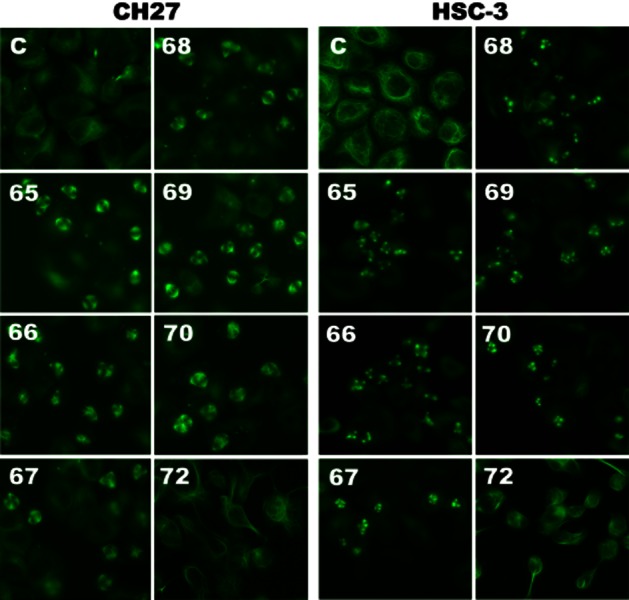
MJ65–70 induced changes in microtubule formation of CH27 and HSC-3 cells. Cells were incubated with 0.1% DMSO or IC50 concentrations of MJ65–70 for 12 h. The concentration of MJ72 used in CH27 and HSC-3 is 10 μM. After treatment, cells were incubated for 30 min with 250 nM Tubulin Tracker Green reagent. The specimens were observed by fluorescence microscopy. All results are representative of three independent experiments.
Effects of MJ65–70 on cell cycle distribution and the protein expression of cell cycle regulators, cyclin B1 and Cdk1, in CH27 and M21 cells
As microtubules are essential for mitosis, we therefore examined whether treatment with compounds MJ65–70 induced G2/M phase arrest of cell cycle in CH27 and M21 cells. Cell cycle analysis revealed S- and G2/M-phase arrest after treatment of CH27 and M21 cells, with IC50 concentrations of MJ65–70 for 12 h. Significant S- and G2/M-phase arrest was indicated by a decreasing proportion of cells in the G0/G1-phase (Figure 3). It is interesting to note that MJ72 had a slight effect on S- and G2/M-phase arrest after treatment of CH27 or M21 cells with 10 or 8.1 μM, respectively, for 12 h (Figure 3). These data indicated that MJ65–70 induced cancer cell death through the deranged spindle formation and followed by mitotic arrest. To further evaluate the involvement of specific regulators of cell cycle such as cyclin B1 and Cdk1 in the S- and G2/M-phase arrest induced by MJ65–70, the protein levels of cyclin B1 and Cdk1 were analysed by Western blot analysis in CH27 and M21 cells. As shown in Figure 4, the levels of cyclin B1 protein were consistently increased in MJ65–70-treated CH27 and M21 cells. However, there was no consistent Cdk1 protein expression in CH27 and M21 cells after MJ65–70 treatment. MJ65–70 induced no significant changes in the protein amounts of Cdk1 in CH27 cells (Figure 4). Treatment of M21 cells with MJ65–67 caused a slight increase in Cdk1 protein levels (Figure 4). However, expression of Cdk1 was decreased in MJ72-treated CH27 and M21 cells (Figure 4).
Figure 3.

MJ65–70 induced cell cycle arrest of CH27 and M21 cells. Cells were treated with 0.1% DMSO or IC50 doses of MJ65–70 for 12 h. The concentrations of MJ72 used in CH27 and M21 are 10.0 and 8.1 μM respectively. After treatment, cells were stained with propidium iodide and analysed by flow cytometry. Results are representative of three independent experiments.
Figure 4.
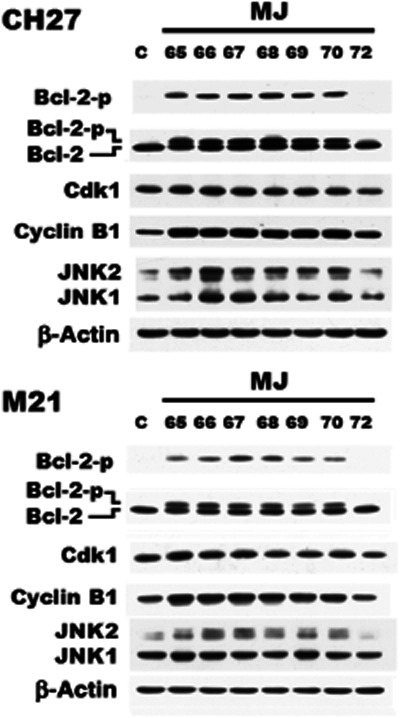
Effects of MJ65–70 on the expression of cyclin B1, Cdk1 and JNK protein in CH27 and M21 cells. The effect of MJ65–70 on the protein levels of cyclin B1, Cdk1 and JNK were assessed by Western blot analysis. Cells were incubated with 0.1% DMSO or IC50 concentrations of MJ65–70 for 12 h. The concentrations of MJ72 used in CH27 and M21 are 10 and 8.1 μM respectively. Cell lysates were analysed by SDS-PAGE (10% for cyclin B1; 12% for JNK and β-actin; 13% for Bcl-2, pS70-Bcl-2 and Cdk1) and then probed with primary antibodies as described. All results are representative of three independent experiments.
Effects of MJ65–70 on JNK protein levels and Bcl-2 phosphorylation of CH27 and M21 cells
The result in Figure 4 shows significant increase in the protein levels of JNK2 after treatment with MJ65–70. Western blot analysis of the protein expression of Bcl-2 exhibits two bands in this study. The upper and lower bands correspond to phosphorylated and non-phosphorylated Bcl-2 respectively. In this study, exposure of CH27 and M21 cells to MJ65–70 resulted in a significant increase in phosphorylated Bcl-2 protein level (Figure 4). We also demonstrated that MJ65–70 induced Bcl-2 phosphorylation at Ser70 in CH27 and M21 cells (Figure 4). Based on the above data, this study demonstrated that Bcl-2 phosphorylation, a marker of M-phase events, was induced after treatment with MJ65–70 for 12 h.
Docking simulation of MJ65–70 with JNK protein
Because JNK was overexpressed by MJ65–70 (see Western blots; Figure 4), we performed a docking simulation of these compounds with JNK2 protein and found them to bind adequately with JNK2. The virtual comparison of MJ66 with lead compound HMJ38 in JNK2 was shown in Figure 1D. The scoring function values are listed in Table 1-2. MJ66 was taken as an example to illustrate the interactions between ligand and binding site. As shown in Figure 1E, the interaction between MJ66 and 3e7o involved the van der Waals interactions of the quinazoline ring with Val40, Ala53, Met108 and Leu168;the 6-(piperidin-1-yl) group with Ile32; and the 2-(naphthalene-1-yl) group with Lys55, Ile86, Met108, Leu168 and Asp169. These interactions caused MJ66 to bind readily to JNK2. The 35F, co-crystal in the 3e7o, was re-docked into the JNK-35F domain to generate an RMSD value of 0.8931 Å (Figure 1F).
Table 1-2.
The docking results of MJ65-80 and HMJ38 with JNK2 (3e7o)
| Name | LigScore1 | LigScore2 | (-)PLP1 | (-)PLP2 | Jain | (-)PMF | Dock_Score |
|---|---|---|---|---|---|---|---|
| MJ65 | 2.52 | 4.96 | 73.83 | 66.95 | 2.75 | 80.56 | 48.35 |
| MJ66 | 3.36 | 5.21 | 69.95 | 64.42 | 2.24 | 61.93 | 46.09 |
| MJ67 | 2.62 | 4.74 | 72.03 | 66.16 | 4.38 | 63.35 | 48.03 |
| MJ68 | 1.89 | 4.98 | 81.62 | 78.49 | 4.70 | 88.18 | 49.30 |
| MJ69 | 3.26 | 5.12 | 68.63 | 62.94 | 1.98 | 59.88 | 44.09 |
| MJ70 | 3.22 | 5.09 | 68.89 | 63.57 | 2.42 | 57.78 | 43.16 |
| MJ72 | 2.71 | 5.12 | 78.35 | 70.38 | 2.47 | 81.48 | 49.65 |
| MJ73 | 1.88 | 5.17 | 79.09 | 74.58 | 3.73 | 74.92 | 50.86 |
| MJ74 | 2.35 | 4.71 | 70.57 | 63.66 | 2.58 | 72.69 | 44.52 |
| MJ75 | 2.58 | 4.94 | 75.59 | 68.10 | 2.06 | 74.48 | 44.90 |
| MJ76 | 2.77 | 4.73 | 68.08 | 60.69 | 3.69 | 60.60 | 48.02 |
| MJ77 | 2.84 | 4.91 | 68.95 | 61.98 | 3.87 | 58.70 | 49.62 |
| MJ78 | 2.65 | 5.07 | 78.26 | 71.41 | 3.09 | 71.34 | 48.96 |
| MJ79 | 3.04 | 5.15 | 67.72 | 61.38 | 0.87 | 54.99 | 45.47 |
| MJ80 | 2.55 | 4.96 | 74.91 | 67.94 | 1.94 | 68.32 | 46.00 |
| HMJ38 | 2.43 | 4.82 | 67.73 | 62.74 | 2.72 | 69.11 | 52.21 |
Discussion
In this study, HMJ38 was used as a lead structure to develop novel and potent anti-tubulin compounds. We optimized the structure of HMJ38 based on the results of the previous SAR study and complementary molecular docking simulations. In the process of docking simulation with tubulin, we found that there is more available space in the ligand-binding region encompassing available amino acid residues. Therefore, we extended the structural skeleton through introduction of substituents at the C-2 and C-6 positions. The results of the docking simulations indicated that our designed MJ series bind to tubulin mainly at the vinblastine site, but not the colchicine or paclitaxel site. Therefore, the optimized 2-aryl-6-substituted quinazolinones, like vinblastine, were supposed to be tubulin-binding agents via inhibiting tubulin polymerization. These findings are consistent with the pharmacological results in our previous study, in which the interference in the dynamics of tubulin and cell division of MJ69 is similar to that in vinblastine-treated cells, but not paclitaxel-treated cells (Hour et al., 2013).
The novel optimized 2-aryl-6-substituted quinazolinones were then synthesized and demonstrated as anti-proliferative agents towards M21, CH27, H460, Hep3B and HSC-3 cells. SAR studies indicated that compounds bearing 2-(naphthalen-1-yl) groups (MJ66–70) exhibited strong cytotoxic activity regardless of which one of the five functional groups substituted at their position 6. As for compounds bearing 2-(1H-indol-3-yl) groups, only those substituted with 6-(pyrrolidin-1-yl) or 6-(piperidin-1-yl) (MJ65 and 72) were active; however, their activity decreased significantly. All the compounds bearing 2-(benzo[b]thiophen-3-yl) groups (MJ73, 77–80) were inactive in spite of which functional groups substituted at their 6-positions, except for MJ78, which was moderately active against HSC-3 cells. The selectivity of MJ78 against HSC-3 cells will be investigated further in our laboratory.
Overall, we concluded that 2-(naphthalen-1-yl) group is the most important substituent contributing to their cytotoxicity in this class of compounds, and compounds simultaneously substituted with 6-(pyrrolidin-1-yl) group led to the most effective anti-cancer activity. MJ66 bearing both 2-(naphthalen-1-yl) and 6-(pyrrolidin-1-yl) moieties showed the highest activity, with IC50 values of 0.03, 0.05, 0.08, 1.35 and 0.04 μM against M21, CH27, H460, Hep3B and HSC-3 cells respectively. MJ70 bearing both 2-(naphthalen-1-yl) and 6-methoxyl moieties showed slightly decreased activity, with IC50 values of 0.09, 0.09, 0.07, 1.94 and 0.15 μM against M21, CH27, H460, Hep3B and HSC-3 cells respectively. The cytotoxic activity of HMJ38 was also assayed in this study and exhibited IC50 values of 3.44 and 2.69 μM against M21 and HSC-3 cells respectively (Table 3). From the above findings, MJ66–70 appear to be more potent anti-proliferative agents than the lead compound HMJ38.
The aberrant expression of microtubules was associated with cancer cell death induced by compounds MJ65-70. Therefore, the anti-cancer mechanisms of these compounds involved interactions with the formation of microtubules, leading to the failure to the progression of cell cycle in CH27 and M21 cells. These results are consistent with previous observations in which interference with the dynamics of tubulin, by compounds such as taxanes and Vinca alkaloids, and cell division have been proven to arrest cells at the G2/M phase of the cell cycle, ultimately leading to apoptosis (Roninson et al., 2001; Castedo et al., 2004). As our findings suggest that MJ65–70 are anti-microtubule agents, the mitotic arrest of cell cycle may result from their anti-microtubule effects in cancer cells. Exposure of CH27 or M21 cells to MJ65–70 resulted in a significant increase in Bcl-2 phosphorylation in this study. This result appears to be consistent with earlier findings that drugs affecting the integrity of microtubules can induce Bc1-2 phosphorylation, which is only a marker of M-phase events (Haldar et al., 1997; Ling et al., 1998).
Because MJ65–70 induced the mitotic arrest of cells that preceded cell death, the cell cycle regulators, in particular the expression of cyclin B1 and Cdk1, were examined. In this study, MJ65–70 induced a marked increase in the protein level of cyclin B1 in M21 and CH27 cells. There is evidence that cyclin B1 must be destroyed by the anaphase-promoting complex to allow mitosis to proceed at the end of metaphase (Peters, 2002). Based on the above data, mitotic arrest induced by MJ65–70 may be associated with the accumulation of cyclin B1, which is not destroyed at the end of metaphase. In this study, we also demonstrated that treatment of cells with MJ65–70 resulted in many mitotic cells with multipolar spindles and cell cycle arrest at G2/M phase.
In addition, several reports suggested that activation of JNK may be a common mechanism for tubulin-binding agents (Sánchez-Perez et al., 1998; Ma et al., 2009). In this study, we also demonstrated that MJ65–70 significantly increased the protein levels of JNK2. Therefore, JNK2 was selected as another docking target in the hope of discovering a potent dual-targeted anti-cancer agent. From the results of our ligand docking experiments, MJ65–70 were well-matched with the JNK molecule and should thus be promising JNK-binding agents. Although the crystal structure of JNK1 was used as the docking target in the docking experiments, we found that MJ compounds bound more readily to JNK2 than to JNK1 (data not shown). The docking results were compatible with those from the Western blot analysis. In our previous study, MJ69-induced cell death was attenuated by the JNK inhibitor SP600125 (3 or 10 μM), not by the inhibitors of ERK or p38. Inhibition of JNK also abolished the MJ69-induced increase in the phosphorylation of Bcl-2 (Hour et al., 2013). Therefore, we suggested that the MJ compounds may be targetting JNK. In the present study, we unexpectedly showed that MJ65–70 bound to both tubulin and JNK, and induced microtubule disruption and cell cycle arrest at G2/M phase, leading to cancer cell death with the phosphorylation of Bcl-2.1
In summary, CADD techniques were used to develop new compounds and the LigScore2 was the better scoring function, which was in good agreement with our experimental results in this study (higher scores indicate better ligand-binding affinities). The MJ65–70 compounds were dual-targeted, tubulin- and JNK-binding, anti-cancer agents and induced cancer cell death through interfering in the dynamics of tubulin and JNK regulation. Treatment of CH27 or HSC-3 cells with MJ65–70 resulted in significant mitotic arrest accompanied by increasing multiple asters of microtubules. In accordance with the cell cycle arrest at G2/M phase induced by the MJ65–70 compounds, we also found a marked increase in Bcl-2 phosphorylation. Our work provides a new strategy and mechanism for developing dual-targeted anti-cancer drugs and may contribute to clinical anti-cancer drug discovery and application.
Acknowledgments
This work was supported by research grant from the National Science Council of the Republic of China (NSC 99–2320-B-039-013-MY3; NSC97-2320-B-039-024).
Glossary
- CADD
computer-aided drug design
- PQZ
2-phenyl-4-quinazolinones
- RMSD
root mean square deviation
- SAR
structure–activity relationships
Conflict of interest
The authors state no conflict of interest.
References
- Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002;1:882–894. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- Booth B, Zemmel R. Prospects for productivity. Nat Rev Drug Discov. 2004;3:451–456. doi: 10.1038/nrd1384. [DOI] [PubMed] [Google Scholar]
- Burns CJ, Fantino E, Phillips ID, Su S, Harte MF, Bukczynska PE, et al. CYT997: a novel orally active tubulin polymerization inhibitor with potent cytotoxic and vascular disrupting activity in vitro and in vivo. Mol Cancer Ther. 2009;8:3036–3045. doi: 10.1158/1535-7163.MCT-09-0076. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J Chem Inf Model. 2006;46:401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Fukui M, Zhu BT. Role of cyclin B1/Cdc2 up-regulation in the development of mitotic prometaphase arrest in human breast cancer cells treated with nocodazole. PLoS ONE. 2011;6:e24312. doi: 10.1371/journal.pone.0024312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Iwase S, Kikuchi J, Terui Y, Nakamura M, Yamada H, et al. Phosphorylation of Bcl-2 protein by CDC2 kinase during G2/M phases and its role in cell cycle regulation. J Biol Chem. 2000;275:21661–21667. doi: 10.1074/jbc.M906893199. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Hadfield JA, Ducki S, Hirst N, McGown AT. Tubulin and microtubules as targets for anticancer drugs. Prog Cell Cycle Res. 2003;5:309–325. [PubMed] [Google Scholar]
- Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997;57:229–233. [PubMed] [Google Scholar]
- Hour MJ, Huang LJ, Kuo SC, Xia Y, Bastow K, Nakanishi Y, et al. 6-Alkylamino and 2,3-dihydro-3′-methoxy-2-phenyl-4-quinazolinones and related compounds: their synthesis, cytotoxicity and inhibition of tubulin polymerization. J Med Chem. 2000;43:4479–4487. doi: 10.1021/jm000151c. [DOI] [PubMed] [Google Scholar]
- Hour MJ, Lee KT, Wu YC, Wu CY, You BJ, Chen TL, et al. A novel antitubulin agent, DPQZ, induces cell apoptosis in human oral cancer cells through Ras/Raf inhibition and MAP kinases activation. Arch Toxicol. 2013;87:835–846. doi: 10.1007/s00204-012-0991-3. [DOI] [PubMed] [Google Scholar]
- Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- Lee HZ, Yang WH, Hour MJ, Wu CY, Peng WH, Bao BY, et al. Photodynamic activity of aloe-emodin induces resensitization of lung cancer cells to anoikis. Eur J Pharmacol. 2010;648:50–58. doi: 10.1016/j.ejphar.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Ling YH, Tornos C, Perez-Soler R. Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem. 1998;273:18984–18991. doi: 10.1074/jbc.273.30.18984. [DOI] [PubMed] [Google Scholar]
- Ma D, Warabi E, Yanagawa T, Kimura S, Harada H, Yamagata K, et al. Peroxiredoxin I plays a protective role against cisplatin cytotoxicity through mitogen activated kinase signals. Oral Oncol. 2009;45:1037–1043. doi: 10.1016/j.oraloncology.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Manfredi JJ, Parness J, Horwitz SB. Taxol binds to cellular microtubules. J Cell Biol. 1982;94:688–696. doi: 10.1083/jcb.94.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell. 2002;9:931–943. doi: 10.1016/s1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4:303–313. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- Sánchez-Perez I, Murguía JR, Perona R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene. 1998;16:533–540. doi: 10.1038/sj.onc.1201578. [DOI] [PubMed] [Google Scholar]
- Wu YC, Hour MJ, Leung WC, Wu CY, Liu WZ, Chang YH, et al. 2-(Naphthalene-1-yl)-6-pyrrolidinyl-4-quinazolinone inhibits skin cancer M21 cell proliferation through aberrant expression of microtubules and the cell cycle. Pharmacol Exp Ther. 2011;338:942–951. doi: 10.1124/jpet.110.176115. [DOI] [PubMed] [Google Scholar]
- Yang JS, Hour MJ, Kuo SC, Lee MR. Selective induction of G2/M arrest and apoptosis in HL-60 by a potent anticancer agent, HMJ38. Anticancer Res. 2004;24:1769–1778. [PubMed] [Google Scholar]