

Abstract
Background and Purpose
Pre-synaptic nicotinic ACh receptors (nAChRs) and adenosine A2A receptors (A2ARs) are involved in the control of dopamine release and are putative therapeutic targets in Parkinson's disease and addiction. Since A2ARs have been reported to interact with nAChRs, here we aimed at mapping the possible functional interaction between A2ARs and nAChRs in rat striatal dopaminergic terminals.
Experimental Approach
We pharmacologically characterized the release of dopamine and defined the localization of nAChR subunits in rat striatal nerve terminals in vitro and carried out locomotor behavioural sensitization in rats in vivo.
Key Results
In striatal nerve terminals, the selective A2AR agonist CGS21680 inhibited, while the A2AR antagonist ZM241385 potentiated the nicotine-stimulated [3H]dopamine ([3H]DA) release. Upon blockade of the α6 subunit-containing nAChRs, the remaining nicotine-stimulated [3H]DA release was no longer modulated by A2AR ligands. In the locomotor sensitization experiments, nicotine enhanced the locomotor activity on day 7 of repeated nicotine injection, an effect that no longer persisted after 1 week of drug withdrawal. Notably, ZM241385-injected rats developed locomotor sensitization to nicotine already on day 2, which remained persistent upon nicotine withdrawal.
Conclusions and Implications
These results provide the first evidence for a functional interaction between nicotinic and adenosine A2AR in striatal dopaminergic terminals, with likely therapeutic consequences for smoking, Parkinson's disease and other dopaminergic disorders.
Keywords: nicotine, dopamine, striatum, rat, nAChRs, adenosine A2A receptor, adenosine A2B receptor, locomotor sensitization, adenosine
Introduction
The striatum is a major relay nucleus between the neocortex and the basal ganglia. It receives extensive cortical and thalamic inputs, which – after being integrated at the striatal level – are processed by the basal ganglia output nuclei and subsequently sent back to thalamic and cortical areas (Graybiel, 1991). This processing occurs through an orchestrated interaction among several neuromodulators at the pre- and post-synaptic levels (Girault, 2012), where the dopaminergic inputs from the substantia nigra play a prominent role (Gerfen and Surmeier, 2011). Accordingly, the dopaminergic system is crucial in different functions processed through striatal circuits, such as locomotor activity, habit formation or associative and mnemonic functions (Wickens et al., 2007; Dagher and Robbins, 2009; Lovinger, 2010; Cools, 2011). Also, the manipulation of different neuromodulators, including ACh and adenosine, can affect the striatal dopaminergic system and, hence, is a potential strategy to manage striatal-related brain diseases associated with dopaminergic dysfunction such as addiction and Parkinson's disease (Schiffmann et al., 2007; Quik et al., 2011).
In fact, the activation of pre-synaptic ionotropic nicotinic ACh receptors (nAChRs – the nomenclature of receptors follows Alexander et al., 2011) by ACh or by the widely abused alkaloid nicotine can trigger dopamine (DA) release in the striatum, thereby modulating locomotion, drug addiction or memory processes (Calabresi and Di Filippo, 2008; Livingstone and Wonnacott, 2009; Drenan et al., 2010; Gotti et al., 2010; Quik et al., 2011; Threlfell et al., 2012). In parallel, the chronic consumption of caffeine, an adenosine receptor antagonist (Fredholm et al., 1999), or the administration of adenosine A2A receptor (A2AR) ligands can counteract different neuropsychiatric conditions involving the dopaminergic system in the basal ganglia, such as motor disorders, psychoses or addiction (Fredholm et al., 2005; Ferré et al., 2007; Cunha et al., 2008), and functional A2ARs are located in striatal dopaminergic terminals controlling the release of DA (Chowdhury and Fillenz, 1991; Gomes et al., 2006; 2009). Notably, nicotine use is often accompanied with an increased caffeine intake, and the two psychoactive substances may reinforce each other's action (Swanson et al., 1994). Furthermore, both caffeine and nicotine are pointed out as neuroprotectant in Parkinson's disease (Ross and Petrovitch, 2001). Since chronic caffeine consumption mainly acts through A2ARs (Ferré, 2008; Cunha and Agostinho, 2010), we now tested if there is a functional interaction between pre-synaptic A2ARs and nAChRs controlling the function of striatal dopaminergic nerve terminals. Here, we show that A2ARs activation diminishes the nicotine-stimulated release of DA in isolated nerve terminals, which translates into an ability of the A2ARs to control nicotine-induced locomotor sensitization in vivo.
Methods
Subjects
All studies were conducted in accordance with the principles and procedures outlined as ‘Replacement, Refinement and Reduction of Animals in Research’ (3Rs) in the guidelines of the European Union (86/609/EEC), Federation for Laboratory Animal Science Associations and the National Centre for the 3Rs [the Animals in Research: Reporting In Vivo Experiments (ARRIVE); Kilkenny et al., (2010)] and were approved by the Animal Care Committee of the Center for Neuroscience and Cell Biology of Coimbra. We also applied the principles of the ARRIVE guideline for the design and the execution of the in vitro pharmacological experiments (see below) as well as for data management and interpretation, according to McGrath et al., (2010).
The 75 animals used in this work were male Wistar rats (10–14 weeks old) obtained from Charles River (Barcelona, Spain). Importantly, different tissues from these animals were used in other on-going projects at our research centres. The animals were housed under controlled temperature (23 ± 2°C), subject to a fixed 12 h light/dark cycle, with free access to food and water. All efforts were made to reduce the number of animals used and to minimize their stress and discomfort. The animals used to perform the in vitro studies were deeply anaesthetized with 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane; no reaction to handling or tail pinch, while still breathing) before decapitation with a guillotine.
Preparation of synaptosomes
Purified nerve terminals, termed synaptosomes (Whittaker et al., 1964), represent an excellent tool to study pre-synaptic processes free of polysynaptic and glial influences (Raiteri and Raiteri, 2000).
Partially purified synaptosomes (P2 fraction) for release experiments were obtained as previously described (Ferreira et al., 2009). Briefly, the caudate-putamen region without the nucleus accumbens (hereafter simply: striatum) were quickly dissected out into 2 mL ice-cold sucrose solution (0.32 M, containing 5 mM HEPES, pH 7.4). After homogenization with a Teflon homogenizer, and centrifugation at 5000× g for 5 min, the supernatant was collected and centrifuged at 13 000× g for 10 min to obtain the P2 synaptosomal fraction.
Synaptosomes purified by a 45% Percoll gradient for Western blotting were obtained as previously described (Rebola et al., 2005). Briefly, the two striata from one animal were homogenized in an ice-cold sucrose-HEPES medium containing 0.32 M sucrose, 1 mM EDTA, 0.1% BSA and 10 mM HEPES (pH 7.4). The homogenate was spun at 3000× g for 10 min at 4°C and the supernatant was spun again at 14 000× g for 12 min. The pellet (P2 fraction) was resuspended in 1 mL of Percoll 45% (v/v) made up in Krebs–HEPES–Ringer (KHR) medium (in mM: NaCl 140, EDTA 1, KCl 5, glucose 5 and HEPES 10, pH 7.4) and spun again at 14 000× g for 2 min. The synaptosomes (top layer) were then removed and washed once with KHR medium at 14 000× g for 2 min. The synaptosomal pellet obtained was solubilized in 5% SDS supplemented with 100 μM PMSF, 2 mM DTT and a protease inhibitor cocktail. The protein concentration was then determined using the bicinchoninic acid (BCA) protein assay reagent and the samples added to a 1/6 volume of 6 × SDS-PAGE sample buffer [30% (v/v) glycerol, 0.6 M dithiothreitol (DTT), 10% (w/v) SDS and 375 mM Tris–HCl, and 0.012% bromophenol blue, pH 6.8] and the volume adjusted with milliQ water to normalize for a maximum of 2 μg·μL−1.
Western blot
The samples were denaturated by boiling at 95°C for 5 min and separated by SDS-PAGE electrophoresis using 10% polyacrylamide resolving gels and 4% polyacrylamide concentrating (stacking) gels under reducing conditions at 80–120 mV. Pre-stained precision protein standards (Bio-Rad, Amadora, Portugal) were run simultaneously with the samples to help identify the proteins of interest. The proteins in the gel were then electrophoretically transferred (1A current, for 1.5 h at 4°C with constant agitation) to previously activated PVDF membranes (0.45 μm). After blocking for 1 h at room temperature with 5% essential fatty acid-free BSA in Tris-buffered saline (Tris 20 mM, NaCl 140 mM, pH 7.6) containing 0.1% Tween 20 (TBS-T) to prevent nonspecific binding, the membranes were incubated overnight at 4°C with the primary antibody diluted in TBS-T with 1% BSA. After three washing periods of 15 min with TBS-T, the membranes were incubated with the appropriate alkaline phosphatase-tagged secondary antibody diluted in TBS-T containing 1% BSA for 2 h at room temperature. After three 15 min washes with TBS-T, the membranes were incubated with enhanced chemifluorescence (ECF) substrate and visualized in a VersaDoc 3000 imaging system with the assistance of Quantity One software (both from Bio-Rad). The membranes were then reprobed and tested for β-actin immunoreactivity to confirm that similar amounts of protein were applied to the gels.
Tritiated dopamine ([3H]DA) release from rat striatal synaptosomes
The experiments were carried out as previously reported (Ferreira et al., 2009; Martíre et al., 2011). The P2 synaptosomal fraction was diluted to 0.5 mL with Krebs–HEPES solution (in mM: NaCl 113, KCl 3, KH2PO4 1.2, MgSO4 1.2, CaCl2 2.5, NaHCO3 25, glucose 10, HEPES 15, pH 7.4, 37°C) containing the monoamine oxidase B inhibitor, pargyline (10 μM). Synaptosomes were then incubated for 10 min in the presence of tritiated dopamine ([3H]DA; ∼60 Ci × mmol−1, final concentration, 150 nM). A 16-microvolume chamber superfusion set-up was filled with the pre-loaded synaptosomes, which were trapped by layers of Whatman GF/C filters and superfused continuously at a rate of 0.8 mL·min−1 at 37°C until the end of the experiment. This system allows testing seven pharmacological treatments in duplicate (each averaged as n = 1) to reduce the number of animals utilized, in accordance with the ARRIVE guidelines. After a 10 min washout period, nine 2 min samples were collected for liquid scintillation assay.
The radioactivity content of each sample and of the filters with the trapped synaptosomes was counted by a Tricarb β-counter (PerkinElmer, Waltham, MA, USA). Disintegrations per minute values were expressed as fractional release (FR%), that is, the percent of actual content in the effluent as a function of the total synaptosomal content of radioactivity.
After collecting four 2 min samples as a baseline, nicotine and adenosine receptor ligands, alone or in combination, were applied through the superfusion solution. nAChR antagonists were present since the beginning of the 10 min washout period, until the end of the experiment. For the calculation of treatment effects, please consult the simulated examples presented in the Supporting Information Figure S1.
Adenosine release from rat striatal synaptosomes
Adenosine release was assayed both in batch-like conditions as well as upon superfusion of synaptosomes. In batch-like conditions, striatal synaptosomes (∼1.2 mg protein × mL−1) were incubated at 37°C for 15 min in the presence of the adenosine deaminase inhibitor, erythro-9-(2-hydroxy-3-nonyl)adenine hydrochloride (EHNA; 20 μM). Half of the synaptosomal samples were challenged with nicotine (1 μM) for 8 min at 37°C and the other half served as control. The mixtures were then centrifuged, at 14000× g for 10 min at 4°C and the supernatant was used for HPLC analysis, in duplicate. In superfusion conditions, the synaptosomes were superfused for 15 min in a manner similar to the [3H]DA release assay. The synaptosomes were then exposed for 5 min to EHNA (20 μM) or EHNA combined with nicotine (1 μM) and the effluents were collected for HPLC analysis.
The separation and quantification of adenosine and its metabolites was carried out by HPLC, as previously described (Cunha and Sebastião, 1993), employing a LiChroCart-RT 125-4 C-18 reverse-phase column (particle size, 5 μm) combined with a UV detector set to 254 nm. The mobile phase consisted of KH2PO4 (100 mM) and methanol (85/15 v/v%) at pH 6.50 with the flow rate of 1 mL × min−1 and a loop volume of 50 μL. The identification and quantification of adenosine and its metabolites was achieved by calculating the peak areas then converted to concentration values (expressed as μmol × mg protein−1) by calibration with known standards ranging from 0.1 to 10 μM.
Locomotor behavioural analysis and drug administration
The nicotine-induced locomotor sensitization was assessed based on previous reports (Werling et al., 2009; Wellman et al., 2011). The open-field test was performed in a sound-attenuated room with low-intensity light maintained constant during the testing period (Prut and Belzung, 2003). During the tests, the experimenter stayed in a room adjacent to the one where the test was performed. To remove the odour traces left by the previous animal, the floor and walls of the equipment were cleaned with 10% ethanol before testing the next animal. Locomotor behaviour was monitored in a square open-field arena, with 100 × 100 cm and 60 cm height, made of dark grey polyvinyl chloride plastic. The locomotor activity was evaluated by measuring the total distance travelled over a period of 30 min. Data were analysed using the Any-maze video tracking software (Stoelting, IL, USA).
The nicotine solution was prepared fresh each day by dissolving nicotine bitartrate in an isotonic saline solution (0.9% NaCl) neutralized to pH 7.2. ZM241385 (ZM) was dissolved in a saline solution with 5% dimethylsulfoxide (DMSO). Nicotine (0.5 mg·kg−1, as nicotine-tartrate salt) or saline were injected subcutaneously in a volume of 1 mL·kg−1 of body weight immediately before the test period, whereas ZM (1 mg·kg−1) or vehicle solution were administered intraperitoneally 30 min before the test period. The dose of each drug was chosen based on previous studies in rats showing the induction of a robust and long-lasting locomotor sensitization by nicotine (Schoffelmeer et al., 2002; Le Foll et al., 2003; Kayir et al., 2009) and the efficient antagonism of A2ARs by ZM (Poucher et al., 1996; Prediger and Takahashi, 2005; Montandon et al., 2008) at the current doses used.
For this assay, the rats were randomly divided into four experimental groups. On the first 2 days, all the rats were adapted to the open-field arena for 30 min each day (habituation). For the next 8 consecutive days, all rats received two daily injections before the test period. ZM or its vehicle (0.9% saline plus 5% DMSO) was injected 30 min before the test, while nicotine or its vehicle (0.9% saline) was injected 30 min later, immediately before the behavioural test. Rats were grouped based on the following injection scheme: vehicle–vehicle (n = 3), vehicle–nicotine (n = 4), ZM–vehicle (n = 4) and ZM–nicotine (n = 5).
Following the 8 day test period, the rats from all groups remained drug-free for 7 consecutive days. On drug-free day 8, all rats were injected with nicotine (0.5 mg·kg−1) and immediately placed in the open-field arena (challenge day).
Materials and chemicals
PMSF, DL-DTT, protease inhibitor cocktail (leupeptin, pepstatin A, chymostatin and antipain), halothane, pargyline, BSA, (-)-nicotine hydrogen tartrate salt and Whatman GF/C filters were obtained from Sigma (Sintra, Portugal). SDS and the Quantity one software were from Bio-Rad. PVDF membranes, pre-stained precision protein standards and ECF were purchased from Amersham Biosciences (Amadora, Portugal). BCA protein assay reagents were from Thermo Scientific (Pierce Biotechnology, Rockford, IL, USA). 3,4-[ring-2,5,6-3H]dihydroxyphenylethylamine ([3H]DA) was from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA). α-bungarotoxin (α-BTX), α-conotoxin-PIA (α-CTX), dihydro-β-erythroidine (DHβE), 2-[p-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamido adenosine [CGS21680 (CGS)], EHNA and N-(4-cyanophenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-acetamide [MRS1754 (MRS)] were from Tocris Bioscience (Bristol, UK); and 4-(2-[7-amino-2-(2-furyl)]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol (ZM) was from Abcam Biochemicals (Cambridge, UK). Any-maze was from Stoelting. Non-water soluble materials were dissolved in DMSO and further diluted in H2O, aliquoted and kept at −20°C until use.
The antibodies used were as follows: rat anti-nicotinic α7 receptor (1:3000) and rabbit anti-nicotinic α4 receptor (1:3000; Abcam Biochemicals); rabbit anti-nicotinic α6 receptor (1:3000) and rabbit anti-nicotinic β2 receptor (1:15000; Merck Millipore, Darmstadt, Germany); mouse anti-β-actin (Sigma); alkaline phosphatase-labelled (AP) goat anti-rabbit (1:20000) or anti-mouse (1:20000) antibodies (GE Healthcare, Lisbon, Portugal); and AP chicken anti-rat (1:3000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Data presentation
All data are expressed as means ± SEM of the indicated number of independent observations (n). Raw effect data from release and sensitization experiments were normalized to the respective vehicle control except when noted. Normalized data were tested for normality by the Kolmogorov–Smirnov normality tests and statistical significance was calculated by one-sample t-test against a hypothetical value of 100 or 0 (when normalized). If more than two groups were compared, one-way anova with Dunett's post hoc test was performed. Data from paired experiments were compared with the pairwise version of the above mentioned tests, and a value of P < 0.05 was accepted as a significant difference.
Results
Nicotine stimulates the release of [3H]DA from rat striatal synaptosomes in an adenosine A2A receptor-dependent manner
Nicotine (1, 30, 300 nM and 3 μM) stimulated the release of [3H]DA in a concentration-dependent manner [EC50 = 68.0 ± 17.2 nM; maximal effect (Emax) = 6.80 ± 0.67 FR%; n = 10–21 rats, in duplicate; Figure 1A and B]. Next, we evaluated the effect of A2AR ligands on the release of DA; the A2AR agonist CGS (30 nM) also stimulated the release of [3H]DA (0.63 ± 0.25 FR%, n = 10, P < 0.05, t = 2.503, d.f. = 9; Figure 2), while the A2AR antagonist ZM (100 nM) was without effect (0.16 ± 0.24 FR%, n = 18, P > 0.5, t = 0.6823, d.f. = 17; Figure 2). Since ZM can also antagonize adenosine A2B receptors (A2BRs) with lower potency than A2ARs, we also tested the A2BR antagonist MRS (200 nM), which was found without effect per se (0.53 ± 0.22 FR%, n = 7, P > 0.05, t = 0.2383, d.f. = 6; Figure 2). Interestingly, the combined application of ZM and MRS stimulated the release of [3H]DA (1.61 ± 0.51 FR%, n = 9, P < 0.05, t = 3.165, d.f. = 8; Figure 2), suggesting a cooperative interaction between A2ARs and A2BRs similar to that observed in splenocytes (Moriyama and Sitkovsky, 2010). Additionally, the non-selective adenosine receptor antagonist, caffeine (50 μM), also stimulated the release of [3H]DA (0.53 ± 0.13 FR%, n = 38, P < 0.0001, t = 3.903, d.f. = 37; Figure 2).
Figure 1.

Nicotine stimulates [3H]dopamine ([3H]DA) release from rat striatal synaptosomes. (A) Time course of the averaged release of [3H]DA. Synaptosomes were treated with various concentrations of nicotine (1, 30, 300 nM or 3 μM) for 8 min, as indicated by the horizontal bar. (B) Concentration-response curve for nicotine to trigger the release of [3H]DA. *P < 0.001 versus non-stimulated control; n = 10–21 independent observations in duplicate.
Figure 2.
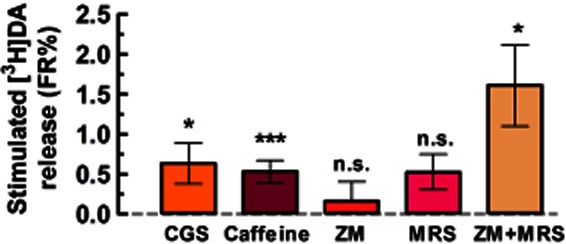
The adenosine A2AR agonist CGS (30 nM), but neither the A2AR antagonist ZM (100 nM) nor the A2BR antagonist MRS (200 nM), stimulates [3H]DA release from rat striatal synaptosomes. The non-selective adenosine receptor antagonist, caffeine (50 μM), also stimulates [3H]DA release, which may be explained by the observation that the simultaneous blockade of A2ARs and A2BRs by ZM and MRS also facilitated [3H]DA release. Data are mean ± SEM of 7–38 experiments performed in duplicate. *P < 0.05, ***P < 0.01 versus 0 FR% (i.e. no change in baseline); n.s., not significant.
In experiments combining A2AR ligands with nicotine, we observed that CGS (30 nM) inhibited the effect of nicotine on [3H]DA release at the two lowest concentrations of nicotine (1 and 30 nM), while it had no significant impact on the effect of higher nicotine concentrations (300 nM and 3 μM; Figure 3A and B). In the presence of CGS, the effect of nicotine (1 nM) was abolished (−0.54 ± 0.28 FR%, n = 8, P < 0.05 vs. CGS alone), while the effect of nicotine at 30 nM (2.53 ± 0.21 FR%, n = 18) was reduced by 53.1 ± 11.6% (P < 0.01, t = 4.457, d.f. = 6) to 1.47 ± 0.43 FR%, n = 7. The effect of CGS was concentration-dependent (Figure 3B), displaying an IC50 of 11.0 ± 6.3 nM and an Imax of 53.8 ± 7.4% for the inhibition of 30 nM nicotine-induced DA release (n = 9–11; curve not shown).
Figure 3.
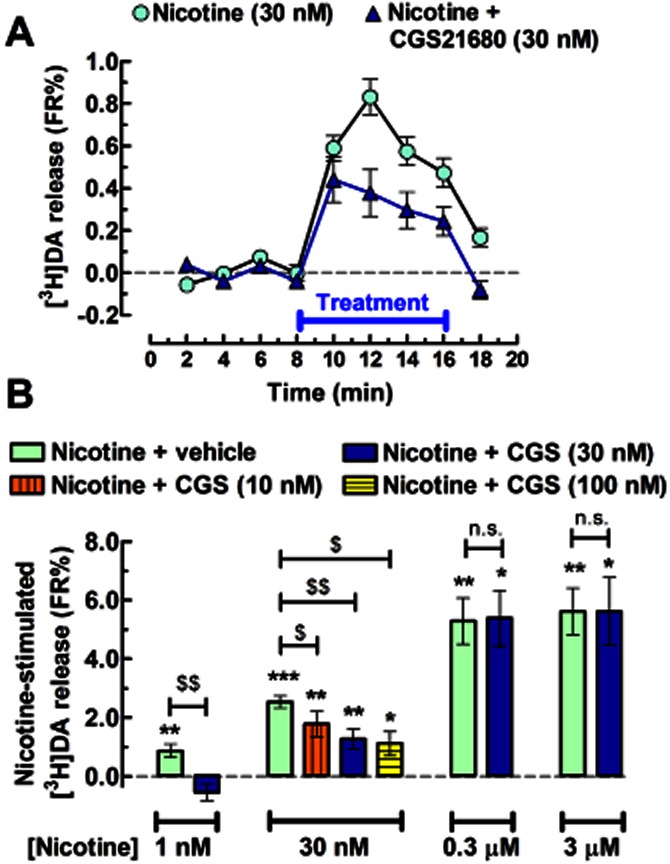
Adenosine A2AR activation inhibits the nicotine-induced [3H]DA release from rat striatal synaptosomes. (A) Time course and (B) bar graph displaying the averaged release of [3H]DA induced by various concentrations of nicotine alone or in the presence of A2AR agonist, CGS (10–100 nM). The co-administration of CGS and nicotine occurred as indicated by the horizontal bar in (A). Data are mean ± SEM of 6–18 experiments performed in duplicate. *P < 0.05, **P < 0.01 and ***P < 0.001 versus 0 FR% (i.e. no change in baseline); $P < 0.05 and $$P < 0.01 between nicotine alone (green bar) and nicotine with CGS; n.s., not significant.
In agreement with the involvement of A2ARs to inhibit the action of nicotine, the A2AR antagonist ZM (100 nM) facilitated the action of different concentrations of nicotine: at 30 nM by 63.4 ± 10.0% (to 3.66 ± 0.22 FR%, n = 7, P < 0.05, t = 3.169, d.f. = 6); at 300 nM by 26.2 ± 7.6% (to 6.33 ± 0.44 FR%, n = 8, P < 0.01, t = 3.623, d.f. = 7); and at 3 μM by 26.9 ± 5.3% (to 8.55 ± 0.58 FR%, n = 6, P < 0.01, t = 4.835, d.f. = 5; Figure 4A and B). Because of its relevance as the most widely consumed psychoactive drug worldwide (Fredholm et al., 1999), we next tested the impact of caffeine on nicotine-induced DA release. The acute administration of caffeine (10 μM, data not shown and 50 μM) failed to facilitate the action of nicotine (30 and 300 nM) and actually diminished that of 3 μM nicotine (to 4.06 ± 0.73 FR%, n = 8, P < 0.01, t = 3.795, d.f. = 7; Figure 4C). Because caffeine (Fredholm et al., 1999) as well as ZM have also been reported to antagonize the A2BRs of rat and human (Lasley et al., 2007; Li et al., 2007) albeit with a lower potency than A2ARs (Poucher et al., 1995; Ji and Jacobson, 1999), we next tested the effects of a selective A2BR antagonist, MRS in our assay. MRS (200 nM) antagonized the 30 nM nicotine-stimulated release of [3H]DA by 58.7 ± 11.3% (0.89 ± 0.24 FR%, n = 6, P < 0.05, t = 3.716, d.f. = 5; Figure 4D). When combined with ZM, the two antagonists did not significantly alter the 30 nM nicotine-induced release of DA (to 1.33 ± 0.89 FR%, n = 7, P > 0.05 vs. nicotine alone; Figure 4D). These results indicated that the facilitation of nicotine's action by ZM was not mediated by A2BRs antagonism, although it appears that A2BRs may also be involved in the control (qualitatively opposite to the role of A2ARs) of nicotinic receptor function in dopaminergic terminals of the striatum.
Figure 4.
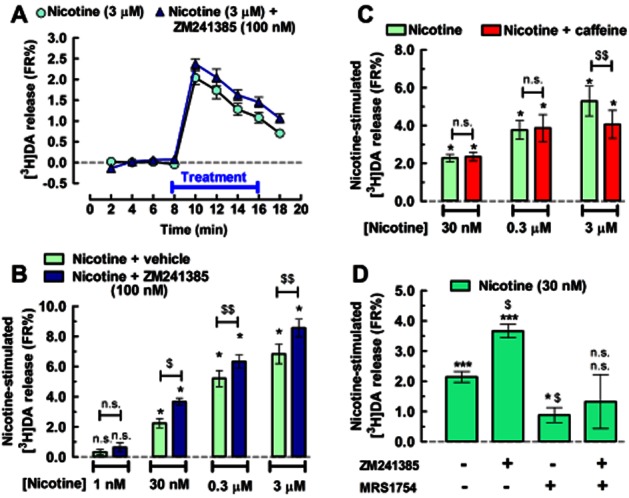
Adenosine A2AR blockade increases the nicotine-induced [3H]DA release from rat striatal synaptosomes. (A) Time course and (B) bar graph displaying the averaged release of [3H]DA induced by nicotine alone or in the presence of the A2AR antagonist, ZM (100 nM). The co-administration of ZM and nicotine occurred as indicated by the horizontal bar in (A). (C) In a similar experimental paradigm, the non-selective adenosine receptor antagonist, caffeine (50 μM), failed to mimic the action of ZM, that is, to facilitate the effect of nicotine. (D) This lack of caffeine effect may be due to the involvement of A2BRs because the selective A2BR antagonist MRS (200 nM) inhibited the effect of nicotine and prevented the facilitatory action of ZM when the two antagonists were combined. Data are mean ± SEM of 6–13 experiments performed in duplicate. *P < 0.05 and ***P < 0.001 versus 0 FR% (i.e. no change in baseline); $P < 0.05 and $$P < 0.01 between the indicated bars in (B) and (C) and when compared to control (nicotine in the absence of any adenosine receptor antagonist, displayed with the leftmost bar) in (D); n.s., not significant.
The fact that A2AR and A2BR antagonists modify DA release implies the existence of endogenous adenosine presumably released from the synaptosomes both tonically and upon nAChR activation. To evaluate this hypothesis, we directly quantified the levels of adenosine in incubated (batch conditions) or superfused synaptosomes. Figure 5 shows that adenosine and its metabolites (inosine and hypoxanthine) were present in concentrations of 6 nmol·mg−1 protein in incubated synaptosomes (n = 4), whereas their levels were below the limit of detection in the superfusate (n = 4; figure not shown). Additionally, we found that nicotine (1 μM) failed to modify the extracellular levels of adenosine or its metabolites either in incubated or in superfused synaptosomes (n = 4, P > 0.05; Figure 5).
Figure 5.
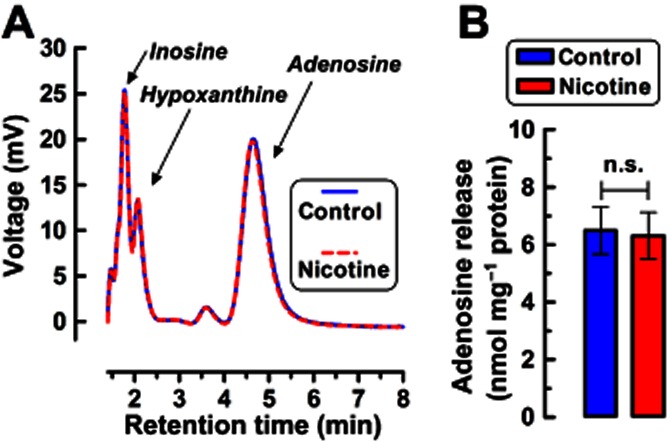
HPLC analysis reveals that striatal synaptosomes release adenosine. (A) Mean chromatograms and (B) mean ± SEM values for the quantified extracellular levels of adenosine and its metabolites in the presence of the adenosine deaminase inhibitor, EHNA (20 μM), upon incubation of synaptosomes (∼1.2 mg protein × mL−1) in the absence or in the presence of nicotine (1 μM); similar results were obtained with 30 nM nicotine (not shown). n.s., not significant.
Blockade of the α6-containing nAChRs abrogates the action of A2AR ligands on nicotine-stimulated [3H]DA release
To map the nAChRs subtypes underlying the nicotine-stimulated [3H]DA release from striatal synaptosomes, we surveyed the synaptic membranes with antibodies against different subunits of nAChRs by Western blotting. We found that the subunits α4, α6, α7 and β2 were enriched in the striatal nerve terminals (Figure 6A and B), which is in accordance with previous studies (Kaiser and Wonnacott, 2000; Grady et al., 2002; 2007; Zoli et al., 2002; Meyer et al., 2008; Livingstone and Wonnacott, 2009). The functional relevance of these subunits was next probed using nAChR antagonists tested against the concentration of nicotine (30 nM) that was found to be sensitive to both CGS and ZM.
Figure 6.

The adenosine A2AR-mediated inhibition of the nicotine-induced [3H]DA release mainly depended on α6β2-containing nicotinic ACh receptors (nAChRs). (A) Western blot analysis revealed specific bands for the different major nAChR subunits in pre-synaptic membrane preparations, with the molecular weight indicated on the right of each excerpt. (B) Quantification of the density of the different subunits obtained from three (α4, α7 and β2) and four (α6) rats under three different protein loads to ensure that the signal is not saturated (maximum = 100% = saturated signal). (C) Time course displaying the averaged [3H]DA release induced by nicotine (30 nM) in the absence or in the presence of either the α6 subunit antagonist α-CTX (30 nM), the α7 subunit antagonist α-BTX (100 nM) or the β2 subunit antagonist DHβE (100 nM). (D) Time course displaying the averaged [3H]DA release, induced by nicotine (30 nM) in the presence of α-CTX (30 nM), when combined with either the A2AR agonist CGS (30 nM) or the A2AR antagonist ZM (100 nM). (E) Bar graph summarizing the sensitivity of 30 nM nicotine-stimulated [3H]DA release under the difference conditions tested in (C) and (D). Data are mean ± SEM of nine experiments performed in duplicate. *P < 0.05, **P < 0.01 and ***P < 0.001 versus 0 FR% (i.e. no change in baseline); $$$P < 0.001 between nicotine alone (blue bar) and nicotine with antagonists of nicotinic acetylcholine receptors; n.s., not significant. Note that neither CGS nor ZM affected the non-α6 subunit-containing nAChR-induced release of [3H]DA, suggesting that it is these α6 subunit-containing nAChRs that are modulated by A2ARs to control striatal dopamine release.
As illustrated in Figure 6C and E, α-BTX (100 nM), an antagonist of α7 nAChRs, failed to significantly affect the action of nicotine (30 nM) on [3H]DA release (mean difference, 16.9 ± 15.1%, P > 0.05 by repeated measures anova with Dunett's post hoc test).
The majority of nicotine binding sites in the brain contains the β2 subunit (Grady et al., 2002; Toyohara and Hashimoto, 2010); accordingly, the β2 subunit-preferring competitive antagonist, DHβE (10 μM), prevented the nicotine-stimulated [3H]DA release by 88.1 ± 5.0% (n = 5, P < 0.001, by repeated measures anova with Dunett's post hoc test; Figure 6C and E). The α6-containing nAChR antagonist, α-CTX (30 nM), largely inhibited the action of nicotine by 69.8 ± 7.5% (n = 6, P < 0.001, by repeated measures anova with Dunett's post hoc test; Figure 6C and E). This suggests that ∼70% of the nicotine-stimulated (30 nM) [3H]DA release involves the activation of α6β2-containing receptors. Upon blockade of α6-containing nAChRs (in the presence of α-CTX, 30 nM), the nicotine-induced (30 nM) [3H]DA release was no longer affected by either CGS (30 nM, n = 9, P > 0.05 vs. α-CTX + nicotine) or ZM (100 nM, n = 9, P > 0.05 vs. α-CTX + nicotine; Figure 6D and E). Altogether, these data advocate that A2ARs are selectively coupled to the inhibition of α6β2-containing nAChRs in dopaminergic terminals of the rat striatum.
Nicotine-induced motor sensitization is facilitated by in vivo A2AR blockade
The interplay between nAChRs and A2ARs controlling DA release from striatal nerve terminals prompted us to test the in vivo relevance of this A2ARs-α6β2-containing nAChRs interaction. One simple measure of nicotine action is its ability to induce hyperlocomotion (Grottick et al., 2000), which can be rated using the open-field test (Walsh and Cummins, 1976; Prut and Belzung, 2003).
After 2 days of rats' habituation to the open-field arena, one daily injection of nicotine (0.5 mg·kg−1) significantly (P < 0.05) enhanced locomotor activity (i.e. sensitization) after 7 days compared to the control rats (i.e. vehicle–vehicle treated; Figure 7). Moreover, the nicotine-induced sensitization was blunted by the subsequent drug-free period (withdrawal; Figure 7), as indicated by similar locomotor activity between groups that are nicotine–vehicle treated and vehicle–vehicle treated (n = 4, P > 0.05). ZM (1 mg·kg−1) did not alter per se the locomotor activity of the animals (Figure 7). Remarkably, ZM-pretreated rats developed a sensitization to nicotine already at day 2, which was no longer blunted by nicotine withdrawal (n = 5, P < 0.05), that is, locomotor activity remained higher until the challenge day when compared to the ZM–vehicle group (Figure 7).
Figure 7.
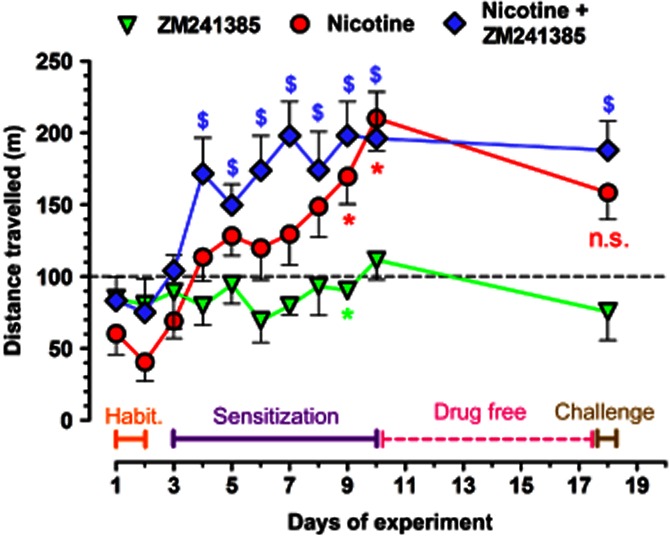
Adenosine A2AR blockade in vivo facilitates the sensitization to nicotine-stimulated hyperlocomotion. Sixteen rats were used to create factorial × groups for vehicle–vehicle, ZM–vehicle, vehicle–nicotine and ZM–nicotine injections. The A2AR antagonist ZM (1 mg·kg−1) or its vehicle was injected 30 min before the test, while nicotine (0.5 mg·kg−1) or its vehicle was injected immediately before the test. After the first 2 days of 30 min habituation to the open-field arena each day, the rats received daily injections for 8 days. Significant sensitization to nicotine developed on day 7, which was blunted by the subsequent week of abstinence, as determined by a single injection of nicotine on the day 8 of abstinence (challenge), resulting in no statistically significant difference between the vehicle–vehicle group (to which all data points were normalized, represented by the dashed line) and the nicotine–vehicle group, represented by the red circles at day 18 of the experiment. ZM had no or a minimal hypolocomotor effect throughout the duration of the experiment (green upside-down triangles). However, ZM-injected rats developed sensitization to nicotine already on day 2 (blue diamonds), which was not blunted by the drug-free period, as indicated by a statistically significant difference between the ZM–vehicle group (green upside-down triangles) and the ZM–nicotine group (blue diamonds) at day 18 of the experiment. Data are mean ± SEM of distance travelled during each 30 min of open-field observation. A *P < 0.05 representing statistical differences between the nicotine-injected (red circles) or ZM-injected (green triangles) and the vehicle-injected (dashed line) rats; and $P < 0.05 representing statistical differences between the ZM-injected and the ZM + nicotine-injected rats (blue diamonds), as determined with anova of repeated measures and Dunett's post hoc analysis.
Discussion and conclusions
The present findings provide pharmacological evidence for adenosine A2ARs exerting a negative control on the α6β2-containing nAChR-mediated stimulation of DA release from striatal dopaminergic terminals. This observation strengthens the notion that A2ARs mainly act as fine-tuners of different other neurotransmitters systems (Sebastião and Ribeiro, 2009). In fact, striatal pre-synaptic A2ARs can negatively control metabotropic receptors such as adenosine A1Rs (Ciruela et al., 2006), cannabinoid CB1Rs (Martíre et al., 2011) and glutamate group 5 receptors (Rodrigues et al., 2005), or potentiate catalytic receptors such as glial cell line-derived neurotrophic factor (GDNF) receptors (Gomes et al., 2006; 2009). In other brain areas, A2ARs also have been shown to control the function of ionotropic receptors such as NMDA (Rebola et al., 2008), AMPA (Dias et al., 2012) or GABAA receptors (Roseti et al., 2008). Additionally, A2ARs also control the rate of desensitization of different nAChRs in peripheral preparations including the myenteric plexus (Duarte-Araújo et al., 2004) and the carotid body (Fitzgerald et al., 2009) or in heterologous expression systems (Di Angelantonio et al., 2011). The present report extends this rule to the CNS, in particular to α6β2-containing nAChR in striatal dopaminergic terminals, which we showed to be controlled by A2ARs.
The present Western blot data, combined with the pharmacological characterization, indicates an important role of the α6β2-containing nAChR to mediate the DA-releasing action of nicotine. This is in agreement with previous findings that the absence of β2 subunits abrogates the ability of nicotine to trigger DA release from synaptosomes (Grady et al., 2002) and that the α6 subunit has an important role in the regulation of mesolimbic DA release (Calabresi and Di Filippo, 2008; Drenan et al., 2008; Meyer et al., 2008; Quik et al., 2011). Moreover, in striatal terminals of rats, we found nAChR subunits other than the α6 subunit, namely, the α4 and the α7 subunits. This stems from the fact that the majority of the synaptic proteins in striatal synaptic Western blot samples come from glutamatergic and GABAergic terminals, whereas dopaminergic terminals only represent about one-fifth of the total number of nerve terminals (Borycz et al., 2007; Gomes et al., 2009). Thus, whereas α7 nAChRs directly control striatal glutamate release (Kaiser and Wonnacott, 2000; Marchi et al., 2002), this subunit seems to be absent in mesolimbic dopaminergic cells (Zoli et al., 2002). This is not the case for the α4 subunit, which others have reported to be present in dopaminergic terminals in the dorsal striatum (Kaiser and Wonnacott, 2000; Zoli et al., 2002; Exley et al., 2012) and may also play a role in the control of DA release under different experimental conditions (Gotti et al., 2010; Smith et al., 2010; Exley et al., 2012).
This ability of pre-synaptic A2ARs to control the α6β2-containing nAChR-induced release of DA was extended to an in vivo setting, by showing that A2ARs also controlled the locomotor sensitization induced by nicotine. This locomotor sensitization to nicotine is known to involve the recruitment of β2-containing nAChRs (Picciotto et al., 1998) and a differential participation of α4-containing and α6-containing, but not α7, nAChRs (Kempsill and Pratt, 2000; Tapper et al., 2004; Gotti et al., 2010; Smith et al., 2010) controlling the release of DA in different regions of the basal ganglia. This differential adaptation of different α4-containing and α6-containing nAChRs upon repeated nicotinic exposure (Tapper et al., 2004; Perry et al., 2007; Perez et al., 2008; Smith et al., 2010) is a likely explanation for the potentiation of nicotinic locomotor sensitization by the tested A2AR antagonist. This is in general agreement with previous studies showing that the non-selective adenosine receptor antagonist, caffeine, bolsters the nicotine-induced increase of locomotor activity (Celik et al., 2006; Cohen et al., 1991). However, it is worth noting that the rewarding properties of nicotine, tested in a place-conditioning paradigm, were decreased in global A2AR knockout mice (Castañé et al., 2006), heralding the hypothesis that different subtypes of nAChRs might be differently controlled by A2ARs.
Apart from this ability of A2ARs to control the α6β2-containing nAChR-induced release of DA, the present results also showed that CGS per se stimulated the release of DA; this is in agreement with the functional and morphological data identifying the presence of A2ARs in dopaminergic nerve endings in the striatum (Chowdhury and Fillenz, 1991; Gomes et al., 2006; 2009) and also, with the ability of striatally micro-infused CGS to increase basal DA levels in freely moving rats (Gołembiowska and Zylewska, 1997). This contention for the involvement of A2ARs was based on the antagonism of the effect of CGS by ZM, which has a 10-fold higher potency to inhibit A2ARs compared with A2BRs (Poucher et al., 1995; Ji and Jacobson, 1999). We also probed the possible involvement of A2BRs and found that the selective A2BR antagonist MRS largely inhibited the effect of nicotine at a concentration (200 nM) threefold lower than its IC50 at A2ARs. Furthermore, the concomitant inhibition of both A2ARs and A2BRs extinguished each other's effect. This may mean two subsets of dopaminergic terminals bearing either A2ARs or A2BRs, leading to a lack of net change, or alternatively that the two receptors may reside and interact in the same nerve terminals. Intriguingly, neither ZM nor MRS affected DA release per se but when combined they synergistically stimulated DA release. This was also observed when testing the non-selective adenosine receptor antagonist, caffeine, an observation that may be pertinent to the understanding of the addictive profile of caffeine (Svikis et al., 2005). Although the underlying mechanism for how A2AR activation or the simultaneous A2AR/A2BR blockade increases basal DA outflow is unclear, these data provide the first demonstration of a functional interaction between the two A2R subtypes in the CNS, in a manner similar to that previously reported to occur in splenocytes (Moriyama and Sitkovsky, 2010).
In summary, the present results show that A2ARs curtail the function of α6β2-containing nAChRs in striatal dopaminergic nerve terminals – an effect that seems relevant for the ability of A2AR antagonists to potentiate the psychomotor effects resulting from a repeated exposure to nicotine. These observations provide a mechanistic insight to explain the frequent correlation in nicotine and caffeine abuse (Swanson et al., 1994). This A2AR–nAChR interaction also paves the way to foster novel therapeutic opportunities to manage motor diseases related with dysfunctional DA signalling, such as Parkinson's disease, where both caffeine and nicotine provide a combined prophylactic benefit (Powers et al., 2008) and where A2AR antagonists are a leading non-dopaminergic therapeutic strategy (Prediger, 2010). Notably, the mechanisms underlying the A2AR-mediated amelioration of Parkinson's disease symptoms are not fully understood and it is also possible that it may involve a rescuing of nAChRs-stimulated phasic DA release (see Threlfell et al., 2012).
Acknowledgments
This work was supported by the PTDC/SAU-NEU/81064/2006, the PEst-C/SAU/LA0001/2013-2014, and the PTDC/SAU-NEU/100729/2008 grants from the Fundação para a Ciência e a Tecnologia (FCT) of the Portuguese Government, by the FEDER and COMPETE, by the FCT-CAPES Luso-Brasilian grant and by DARPA (grant 09–68-ESR-FP-010). P. G. acknowledges his FCT fellowship SFRH/BD/28722/2006. E.C. S. is grateful for the support of ERASMUS Student Mobility for Placements and Campus Hungary TÁMOP 4.2.4. B/2–11/1–2012-0001.
Glossary
- [3H]DA
tritiated dopamine
- 3Rs
Replacement, Refinement and Reduction of Animals in Research
- A2AR
adenosine A2A receptors
- A2BR
adenosine A2B receptors
- ARRIVE
Animals in Research: Reporting In Vivo Experiments
- CGS
- DA
dopamine
- DHβE
dihydro-β-erythroidine
- DMSO
dimethylsulfoxide
- MRS
MRS1754
- nAChR
nicotinic ACh receptor
- ZM
ZM241385
- α-BTX
α-bungarotoxin
- α-CTX
α-conotoxin PIA
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 A guide for the calculations and statistics of the release experiments. In a single experiment from a rat, various treatments were performed, including a pair of vehicle controls and various pairs of treatments with ligands alone or in combination. To obtain the net effect of each tested drug, the averaged duplicates of the respective controls (not shown) were subtracted from the averaged treatments, leading to the graphs pictured in panels A-D. In this simulated case, the area under the curve (AUC) value for nicotine amounts to ‘X’ fractional release % (FR%), and the same for CGS21680 gives the value of ‘Y’ FR%. Additionally, in the same experiment, we combined CGS21680 with nicotine, yielding a total AUC value of ‘V’ FR% This ‘V’ value can be perceived as a sum of ‘Y’ + ‘Z’ where ‘Z’ is the modified effect of nicotine after discounting the effect of CGS21680 per se. These raw data were then used for the following comparisons: ‘X’, ‘Y’, ‘V’ were compared to zero FR% (i.e. no effect on dopamine release) and if statistical difference was detected it was marked with *, **, or ***. Furthermore, ‘X’ was also compared to ‘Z’ to see whether CGS21680 (or any other adenosinergics alone or in combination) altered the effect of nicotine, and if statistical difference was detected it was labeled with $, $$, or $$$. The respective ‘Y’ values are displayed in Figure 3. The ‘Z’ values are displayed in Figures 3, 4 and 6, respectively, while in Figure 7, the same set of symbols are used on locomotor activity instead of dopamine release.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borycz J, Pereira MF, Melani A, Rodrigues RJ, Köfalvi A, Panlilio L, et al. Differential glutamate-dependent and glutamate-independent adenosine A1 receptor-mediated modulation of dopamine release in different striatal compartments. J Neurochem. 2007;101:355–363. doi: 10.1111/j.1471-4159.2006.04386.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M. ACh/dopamine crosstalk in motor control and reward: a crucial role for alpha 6-containing nicotinic receptors? Neuron. 2008;60:4–7. doi: 10.1016/j.neuron.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Castañé A, Soria G, Ledent C, Maldonado R, Valverde O. Attenuation of nicotine-induced rewarding effects in A2A knockout mice. Neuropharmacology. 2006;51:631–640. doi: 10.1016/j.neuropharm.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Celik E, Uzbay IT, Karakas S. Caffeine and amphetamine produce cross-sensitization to nicotine-induced locomotor activity in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:50–55. doi: 10.1016/j.pnpbp.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Chowdhury M, Fillenz M. Presynaptic adenosine A2 and N-methyl-D-aspartate receptors regulate dopamine synthesis in rat striatal synaptosomes. J Neurochem. 1991;56:1783–1788. doi: 10.1111/j.1471-4159.1991.tb02081.x. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen C, Welzl H, Bättig K. Effects of nicotine, caffeine, and their combination on locomotor activity in rats. Pharmacol Biochem Behav. 1991;40:121–123. doi: 10.1016/0091-3057(91)90331-u. [DOI] [PubMed] [Google Scholar]
- Cools R. Dopaminergic control of the striatum for high-level cognition. Curr Opin Neurobiol. 2011;21:402–407. doi: 10.1016/j.conb.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Agostinho PM. Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J Alzheimers Dis. 2010;20(Suppl. 1):95–116. doi: 10.3233/JAD-2010-1408. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM. Adenosine and adenine nucleotides are independently released from both the nerve terminals and the muscle fibres upon electrical stimulation of the innervated skeletal muscle of the frog. Eur J Physiol. 1993;424:503–510. doi: 10.1007/BF00374914. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Ferré S, Vaugeois JM, Chen JF. Potential therapeutic interest of adenosine A2A receptors in psychiatric disorders. Curr Pharm Des. 2008;14:1512–1524. doi: 10.2174/138161208784480090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher A, Robbins TW. Personality, addiction, dopamine: insights from Parkinson's disease. Neuron. 2009;61:502–510. doi: 10.1016/j.neuron.2009.01.031. [DOI] [PubMed] [Google Scholar]
- Di Angelantonio S, Piccioni A, Moriconi C, Trettel F, Cristalli G, Grassi F, et al. Adenosine A2A receptor induces protein kinase A-dependent functional modulation of human α3β4 nicotinic receptor. J Physiol. 2011;589:2755–2766. doi: 10.1113/jphysiol.2011.207282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias RB, Ribeiro JA, Sebastião AM. Enhancement of AMPA currents and GluR1 membrane expression through PKA-coupled adenosine A2A receptors. Hippocampus. 2012;22:276–291. doi: 10.1002/hipo.20894. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron. 2008;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM, et al. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci. 2010;30:9877–9889. doi: 10.1523/JNEUROSCI.2056-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte-Araújo M, Timóteo MA, Correia-de-Sá P. Adenosine activating A2A-receptors coupled to adenylate cyclase/cyclic AMP pathway downregulates nicotinic autoreceptor function at the rat myenteric nerve terminals. Neurochem Int. 2004;45:641–651. doi: 10.1016/j.neuint.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Exley R, McIntosh JM, Marks MJ, Maskos U, Cragg SJ. Striatal α5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J Neurosci. 2012;32:2352–2356. doi: 10.1523/JNEUROSCI.4985-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S. An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem. 2008;105:1067–1079. doi: 10.1111/j.1471-4159.2007.05196.x. [DOI] [PubMed] [Google Scholar]
- Ferré S, Diamond I, Goldberg SR, Yao L, Hourani SM, Huang ZL, et al. Adenosine A2A receptors in ventral striatum, hypothalamus and nociceptive circuitry implications for drug addiction, sleep and pain. Prog Neurobiol. 2007;83:332–347. doi: 10.1016/j.pneurobio.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SG, Lomaglio T, Avelino A, Cruz F, Oliveira CR, Cunha RA, et al. N-acyldopamines control striatal input terminals via novel ligand-gated cation channels. Neuropharmacology. 2009;56:676–683. doi: 10.1016/j.neuropharm.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RS, Shirahata M, Chang I. The impact of adenosine and an A2A adenosine receptor agonist on the ACh-induced increase in intracellular calcium of the glomus cells of the cat carotid body. Brain Res. 2009;1301:20–33. doi: 10.1016/j.brainres.2009.08.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83–133. [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA. Integrating neurotransmission in striatal medium spiny neurons. Adv Exp Med Biol. 2012;970:407–429. doi: 10.1007/978-3-7091-0932-8_18. [DOI] [PubMed] [Google Scholar]
- Gołembiowska K, Zylewska A. Adenosine receptors-the role in modulation of dopamine and glutamate release in the rat striatum. Pol J Pharmacol. 1997;49:317–322. [PubMed] [Google Scholar]
- Gomes CA, Vaz SH, Ribeiro JA, Sebastião AM. Glial cell line-derived neurotrophic factor (GDNF) enhances dopamine release from striatal nerve endings in an adenosine A2A receptor-dependent manner. Brain Res. 2006;1113:129–136. doi: 10.1016/j.brainres.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Gomes CA, Simões PF, Canas PM, Quiroz C, Sebastião AM, Ferré S, et al. GDNF control of the glutamatergic cortico-striatal pathway requires tonic activation of adenosine A2A receptors. J Neurochem. 2009;108:1208–1219. doi: 10.1111/j.1471-4159.2009.05876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, Corbioli S, Zanetti L, Moretti M, et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci. 2010;30:5311–5325. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Murphy KL, Cao J, Marks MJ, McIntosh JM, Collins AC. Characterization of nicotinic agonist-induced [3H]dopamine release from synaptosomes prepared from four mouse brain regions. J Pharmacol Exp Ther. 2002;301:651–660. doi: 10.1124/jpet.301.2.651. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, et al. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74:1235–1246. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM. Basal ganglia – input, neural activity, and relation to the cortex. Curr Opin Neurobiol. 1991;1:644–651. doi: 10.1016/s0959-4388(05)80043-1. [DOI] [PubMed] [Google Scholar]
- Grottick AJ, Trube G, Corrigall WA, Huwyler J, Malherbe P, Wyler R, et al. Evidence that nicotinic α7 receptors are not involved in the hyperlocomotor and rewarding effects of nicotine. J Pharmacol Exp Ther. 2000;294:1112–1119. [PubMed] [Google Scholar]
- Ji XD, Jacobson KA. Use of the triazolotriazine [3H]ZM 241385 as a radioligand at recombinant human A2B adenosine receptors. Drug Des Discov. 1999;16:217–226. [PMC free article] [PubMed] [Google Scholar]
- Kaiser S, Wonnacott S. α-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–318. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- Kayir H, Goktalay G, Yildirim M, Uzbay TI. Clozapine inhibits development and expression of nicotine-induced locomotor sensitization in rats. Synapse. 2009;63:15–21. doi: 10.1002/syn.20576. [DOI] [PubMed] [Google Scholar]
- Kempsill FE, Pratt JA. Mecamylamine but not the alpha7 receptor antagonist alpha-bungarotoxin blocks sensitization to the locomotor stimulant effects of nicotine. Br J Pharmacol. 2000;131:997–1003. doi: 10.1038/sj.bjp.0703560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasley RD, Kristo G, Keith BJ, Mentzer RM., Jr The A2a/A2b receptor antagonist ZM-241385 blocks the cardioprotective effect of adenosine agonist pretreatment in in vivo rat myocardium. Am J Physiol Heart Circ Physiol. 2007;292:426–431. doi: 10.1152/ajpheart.00675.2006. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Diaz J, Sokoloff P. Increased dopamine D3 receptor expression accompanying behavioural sensitization to nicotine in rats. Synapse. 2003;47:176–183. doi: 10.1002/syn.10170. [DOI] [PubMed] [Google Scholar]
- Li Q, Ye K, Blad CC, den Dulk H, Brouwer J, Ijzerman AP, et al. ZM241385, DPCPX, MRS1706 are inverse agonists with different relative intrinsic efficacies on constitutively active mutants of the human adenosine A2B receptor. J Pharmacol Exp Ther. 2007;320:637–645. doi: 10.1124/jpet.106.111203. [DOI] [PubMed] [Google Scholar]
- Livingstone PD, Wonnacott S. Nicotinic acetylcholine receptors and the ascending dopamine pathways. Biochem Pharmacol. 2009;78:744–755. doi: 10.1016/j.bcp.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–1078. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- Martíre A, Tebano MT, Chiodi V, Ferreira SG, Cunha RA, Köfalvi A, et al. Pre-synaptic adenosine A2A receptors control cannabinoid CB1 receptor-mediated inhibition of striatal glutamatergic neurotransmission. J Neurochem. 2011;116:273–280. doi: 10.1111/j.1471-4159.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- Meyer EL, Yoshikami D, McIntosh JM. The neuronal nicotinic acetylcholine receptors alpha 4* and alpha 6* differentially modulate dopamine release in mouse striatal slices. J Neurochem. 2008;105:1761–1769. doi: 10.1111/j.1471-4159.2008.05266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montandon G, Bairam A, Kinkead R. Neonatal caffeine induces sex-specific developmental plasticity of the hypoxic respiratory chemoreflex in adult rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:922–934. doi: 10.1152/ajpregu.00059.2008. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Sitkovsky MV. Adenosine A2A receptor is involved in cell surface expression of A2B receptor. J Biol Chem. 2010;285:39271–39288. doi: 10.1074/jbc.M109.098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez XA, Bordia T, McIntosh JM, Grady SR, Quik M. Long-term nicotine treatment differentially regulates striatal α6α4β2* and α6(non-α4)β2* nAChR expression and function. Mol Pharmacol. 2008;74:844–853. doi: 10.1124/mol.108.048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, Kellar KJ. Chronic nicotine differentially regulates α6- and β3-containing nicotinic cholinergic receptors in rat brain. J Pharmacol Exp Ther. 2007;322:306–315. doi: 10.1124/jpet.107.121228. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PW, Jones G, et al. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. Br J Pharmacol. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poucher SM, Keddie JR, Brooks R, Shaw GR, McKillop D. Pharmacodynamics of ZM 241385, a potent A2A adenosine receptor antagonist, after enteric administration in rat, cat and dog. J Pharm Pharmacol. 1996;48:601–606. doi: 10.1111/j.2042-7158.1996.tb05981.x. [DOI] [PubMed] [Google Scholar]
- Powers KM, Kay DM, Factor SA, Zabetian CP, Higgins DS, Samii A, et al. Combined effects of smoking, coffee, and NSAIDs on Parkinson's disease risk. Mov Disord. 2008;23:88–95. doi: 10.1002/mds.21782. [DOI] [PubMed] [Google Scholar]
- Prediger RD. Effects of caffeine in Parkinson's disease: from neuroprotection to the management of motor and non-motor symptoms. J Alzheimers Dis. 2010;20:S205–S220. doi: 10.3233/JAD-2010-091459. [DOI] [PubMed] [Google Scholar]
- Prediger RD, Takahashi RN. Modulation of short-term social memory in rats by adenosine A1 and A2A receptors. Neurosci Lett. 2005;376:160–165. doi: 10.1016/j.neulet.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviours: a review. Eur J Pharmacol. 2003;463:3–33. doi: 10.1016/s0014-2999(03)01272-x. [DOI] [PubMed] [Google Scholar]
- Quik M, Perez XA, Grady SR. Role of α6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem Pharmacol. 2011;82:873–882. doi: 10.1016/j.bcp.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiteri L, Raiteri M. Synaptosomes still viable after 25 years of superfusion. Neurochem Res. 2000;25:1265–1274. doi: 10.1023/a:1007648229795. [DOI] [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience. 2005;132:893–903. doi: 10.1016/j.neuroscience.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Rodrigues RJ, Alfaro TM, Rebola N, Oliveira CR, Cunha RA. Co-localization and functional interaction between adenosine A2A and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J Neurochem. 2005;92:433–441. doi: 10.1111/j.1471-4159.2004.02887.x. [DOI] [PubMed] [Google Scholar]
- Roseti C, Martinello K, Fucile S, Piccari V, Mascia A, Di Gennaro G, et al. Adenosine receptor antagonists alter the stability of human epileptic GABAA receptors. Proc Natl Acad Sci U S A. 2008;105:15118–15123. doi: 10.1073/pnas.0807277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GW, Petrovitch H. Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson's disease. Drugs Aging. 2001;18:797–806. doi: 10.2165/00002512-200118110-00001. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferré S. Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol. 2007;83:277–292. doi: 10.1016/j.pneurobio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoffelmeer A, De Vries T, Wardeh G, van de Ven H, Vanderschuren L. Psychostimulant-induced behavioural sensitization depends on nicotinic receptor activation. J Neurosci. 2002;22:3269–3276. doi: 10.1523/JNEUROSCI.22-08-03269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Tuning and fine-tuning of synapses with adenosine. Curr Neuropharmacol. 2009;7:180–194. doi: 10.2174/157015909789152128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Pivavarchyk M, Wooters TE, Zhang Z, Zheng G, McIntosh JM, et al. Repeated nicotine administration robustly increases bPiDDB inhibitory potency at alpha6beta2-containing nicotinic receptors mediating nicotine-evoked dopamine release. Biochem Pharmacol. 2010;80:402–409. doi: 10.1016/j.bcp.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svikis DS, Berger N, Haug NA, Griffiths RR. Caffeine dependence in combination with a family history of alcoholism as a predictor of continued use of caffeine during pregnancy. Am J Psychiatry. 2005;162:2344–2351. doi: 10.1176/appi.ajp.162.12.2344. [DOI] [PubMed] [Google Scholar]
- Swanson JA, Lee JW, Hopp JW. Caffeine and nicotine: a review of their joint use and possible interactive effects in tobacco withdrawal. Addict Behav. 1994;19:229–256. doi: 10.1016/0306-4603(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Toyohara J, Hashimoto K. α7 nicotinic receptor agonists: potential therapeutic drugs for treatment of cognitive impairments in schizophrenia and Alzheimer's disease. Open Med Chem J. 2010;4:37–56. doi: 10.2174/1874104501004010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh RN, Cummins RA. The open-field test: a critical review. Psychol Bull. 1976;83:482–504. [PubMed] [Google Scholar]
- Wellman PJ, Clifford PS, Rodriguez J, Hughes S, Eitan S, Brunel L, et al. Pharmacologic antagonism of ghrelin receptors attenuates development of nicotine induced locomotor sensitization in rats. Regul Pept. 2011;172:77–80. doi: 10.1016/j.regpep.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling LL, Reed SC, Wade D, Izenwasser S. Chronic nicotine alters cannabinoid-mediated locomotor activity and receptor density in periadolescent but not adult male rats. Int J Dev Neurosci. 2009;27:263–269. doi: 10.1016/j.ijdevneu.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker VP, Michaelson IA, Kirkland RJ. The separation of synaptic vesicles from nerve-ending particles (‘synaptosomes’) Biochem J. 1964;90:293–303. doi: 10.1042/bj0900293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens JR, Horvitz JC, Costa RM, Killcross S. Dopaminergic mechanisms in actions and habits. J Neurosci. 2007;27:8181–8183. doi: 10.1523/JNEUROSCI.1671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.