

Abstract
Background and Purpose
B cell lymphoma 2 (Bcl-2) is a central regulator of cell survival that is overexpressed in the majority of small-cell lung cancers (SCLC) and contributes to both malignant transformation and therapeutic resistance. The purpose of this work was to study the key factors that determine the sensitivity of SCLC cells to Bcl-2 homology domain-3 (BH3) mimetic S1 and the mechanism underlying the resistance of BH3 mimetics.
Experimental Approaches
Western blot was used to evaluate the contribution of Bcl-2 family members to the cellular response of SCLC cell lines to S1. Acquired resistant cells were derived from initially sensitive H1688 cells. Quantitative PCR and gene silencing were performed to investigate Bcl-2 up-regulation.
Key Results
A progressive increase in the relative levels of Bcl-2 and phosphorylated Bcl-2 (pBcl-2) characterized the increased de novo and acquired resistance of SCLC cell lines. Furthermore, acute treatment of S1 induced Bcl-2 expression and phosphorylation. We showed that BH3 mimetics, including S1 and ABT-737, induced endoplasmic reticulum (ER) stress and then activated MAPK/ERK pathway. The dual function of MAPK/ERK pathway in defining BH3 mimetics was illustrated; ERK1/2 activation leaded to Bcl-2 transcriptional up-regulation and sustained phosphorylation in naïve and acquired resistant SCLC cells. pBcl-2 played a key role in creating resistance of S1 and ABT-737 not only by sequestrating pro-apoptotic proteins, but also sequestrating a positive feedback to promote ERK1/2 activation.
Conclusions and Implications
These results provide significant novel insights into the molecular mechanisms for crosstalk between ER stress and endogenously apoptotic pathways in SCLC following BH3 mimetics treatment.
Keywords: ERK1/2, Bcl-2 phosphorylation, BH3 mimetics, SCLC
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide, and there are more than one million new cases reported worldwide each year (Kazma et al., 2012). Small-cell lung cancer (SCLC) makes up 15–20% of all lung cancers, with a 5-year survival rate of 5–10% (Johnson et al., 2006; Hansen et al., 2010). Despite advances in treatments such as surgery, chemotherapy and radiotherapy, the clinical outcome for patients with SCLC still remains poor because of relapse (Kurup and Hanna, 2004; van Meerbeeck et al., 2011). Tumours become resistant to further chemotherapy due to the overexpression of the anti-apoptotic protein B cell lymphoma 2 (Bcl-2) (Ohmori et al., 1993; Breton et al., 1998; Sartorius and Krammer, 2001; Fennell, 2003).
Bcl-2 was an oncogene that promoted cancer cell accumulation by opposing cell death (Chao and Korsmeyer, 1998; Burlacu, 2007). Bcl-2 family proteins are key regulators of the intrinsic apoptotic pathway. A balance between anti-apoptotic Bcl-2 members (Bcl-2, Bcl-XL, Mcl-1, Bcl-w and A1) and two pro-apoptotic groups (the multidomain proteins: Bax, Bak and Bok, and the Bcl-2 homology domain-3 (BH3) only proteins: Bim, Puma, Noxa, Bad, Bid, Bmf, Bik and Hrk) through the BH3 groove they shared dictates the outcome of many death initiating signalling pathways (Youle and Strasser, 2008). The overexpression of Bcl-2 has been reported to occur in 55–90% of all SCLC cases; and Bcl-2 has been suggested to be an unfavourable prognostic factor involved in both the genesis and maintenance of SCLC (Ohmori et al., 1993; Tahir et al., 2007).
The emergence of BH3 mimetics that modulate Bcl-2 pathway by occupying the BH3 groove represents a rational approach for the treatment of this neoplasm (Zhang et al., 2007). ABT-263 (the orally analogue of ABT-737) and Obatoclax, for example, have been developed in clinical trials for SCLC treatment (Paik et al., 2010; Gandhi et al., 2011). ABT-737, which blocks the function of Bcl-2 and Bcl-XL, but not Mcl-1, has shown promising activities in preclinical models of SCLC(Hann et al., 2008). However, sensitivities of individual cell lines to ABT-737 differ, with IC50s ranging from <0.1 to >10 μmol·L−1 in SCLC cell lines (Oltersdorf et al., 2005; Tse et al., 2008). Obatoclax is a pan-Bcl-2 inhibitor, which binds to Bcl-2, Bcl-XL, Bcl-w and Mcl-1. However, it shows the limited single-agent activity against several cancer types in clinical trials and has to combine with chemotherapy in SCLC patients (Paik et al., 2010; Dean et al., 2011). The mechanism underlying SCLC resistance to BH3 mimetics is not clear.
Additionally, some studies have linked phosphorylation of the anti-apoptotic Bcl-2 family members with their ability to antagonize BH3 mimetics-induced apoptosis. High levels of phosphorylated Bcl-2 (pBcl-2) may directly counteract or reduce the activity of both ABT-737 and Obatoclax in leukaemia (Konopleva et al., 2006; Perez-Galan et al., 2008).
Recently, it was found that ABT-737 may induce activation of the MAPK/ERK pathway and up-regulation of Mcl-1 in acute myeloid leukemia cells by an as yet undetermined mechanism (Konopleva et al., 2012). The MAPK/ERK inhibitor was then testified to sensitize cells to ABT-737, perhaps due to the transcriptional suppression of Mcl-1. These inhibitors also had synergistic effect with Obatoclax through inhibition of Bcl-2 phosphorylation in chronic lymphocytic leukaemia (Perez-Galan et al., 2008).
Thus, understanding the mechanism underlying SCLC cell resistant to BH3 mimetics becomes important in the backdrop of development of these agents. It is necessary to understand the cause of resistance for planning strategies to overcome it. The investigation of the signal pathway induced by BH3 mimetics in SCLC following acute and prolonged exposure could reveal the mechanism whereby SCLC response to the disruption of mitochondrial apoptosis.
S1 is a BH3 mimetic as specific as ABT-737 that it depends on Bax/Bak completely, and it is as pan as Obatoclax that it inhibits Bcl-2, Bcl-XL and Mcl-1 with a Kd in the range of 0.5 μM. In vitro and in vivo, S1 exhibits concentration- and time-dependent cytotoxicity against multiple solid and haematology tumour cell lines (Zhang et al., 2010; 2011; Song et al., 2012). The purpose of this work was to study the key factors that determine the sensitivity of SCLC cells to S1 and the mechanism underlying the resistance of BH3 mimetics. We showed novel mechanistic data demonstrating that BH3 mimetics including S1 and ABT-737 induced endoplasmic reticulum (ER) stress and then activated MAPK/ERK pathway, which resulted in Bcl-2 transcriptional up-regulation and phosphorylation. Here, we provided the first evidence that ERK1/2 mediated the crosstalk between mitochondrial apoptosis and ER stress to define BH3 mimetics in SCLC cells.
Materials and methods
Cell culture and reagents
The 11 SCLC cell lines (NCI-H82, NCI-H1417, NCI-H1688, NCI-H209, NCI-H69, NCI-H187, DMS53, NCI-H446, SHP77, NCI-H1048 and NCI-H740) were purchased from American Type Culture Collection (Manassas, VA, USA) and used within 6 months from resuscitation. Cells were cultured in RPMI 1640 (HyClone, Beijing, China) supplemented with 10% FBS (Invitrogen, Grand Island, NY, USA) and 1% penicillin/streptomycin (HyClone). All cell lines were maintained in a humidified chamber at 37°C containing 5% CO2.
The S1-acquired resistant H1688 cells were derived from H1688 cells by initially adding 200 nM S1 to their culture medium and thereafter doubling the concentration over a period of several months. Different acquired resistant clones were obtained. Several drug-resistant variants that adapted to and maintained in the presence of 0.5, 2.0, 8.0, 20.0 and 40.0 μmol·L−1 S1 were generated in this manner and were designated as H1688-SR2, H1688-SR4, H1688-SR6, H1688-SR8 and H1688-SR10.
Human Bcl-2 cDNA was cloned in pUC19 plasmid. Nucleotides corresponding to 70, 87 serine (S) or 69 threonine (T) residue were substituted to create a conservative alteration to alanine (A) or glutamic acid (E) with a site-directed mutagenesis kit (Clontech, Beijing, China) and then modified by addition of the HA-tag sequence at its NH2 terminus. Each single mutant was cloned into pET28b (+) and pCIneo mammalian expression vector (Promega Corp., Madison, WI, USA). S1 was synthesized, as previously described (Zhang et al., 2011), and dissolved in dimethyl sulfoxide (DMSO) (10 mM). Actinomycin D, okadaic acid, Staurosporine, SB202190, LY294002 and PD98059 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lipofectamine™ 2000 Transfection Reagent and G418 were from Invitrogen. Recombinant human His-TRAIL was purchased from Alexis (Lausen, Switzerland).
Cell viability assays
Cells were treated for 24 or 48 h in 96-well culture plates in a total volume of 200 μL culture medium. Each concentration was tested in duplicate at least thrice separately. Viable cells were determined using the CellTiter 96 AQueous non-radioactive cell proliferation 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay (Promega).
Western blot and co-immunoprecipitation
Western blot and co-immunoprecipitation (co-IP) were performed as described in a previous paper (Zhang et al., 2010). Signals were detected using an enhanced chemiluminescence kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). The following antibodies were used: Bcl-2, pBcl-2 (Ser70) and Mcl-1 from Abcam (Cambridge, MA, USA), ERK1/2, phospho (p)-ERK1/2 (Thr202/Tyr204), phospho (p)-ribosomal S6 kinase 1 [RSK1; phosphorylated RSK1 (pRSK1)] (Thr359/Ser363), cAMP responsive element binding protein (CREB), phospho (p)-CREB (Ser133), PKCα, AKT, phospho (p)-AKT (Ser473), Bad, Bim, Noxa, Bcl-XL, Bax, Bak and PARP were from Cell Signaling Technology (Danvers, MA, USA). Ubiquitin, glucose-regulated protein 78 (GRP78), RSK1, β-actin and cytochrome C were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Quantitative reverse transcription (RT)-PCR
Total RNA was isolated using the Trizol method (Invitrogen). A total of 5 μg of RNA were reverse-transcribed and amplified using One Step SYBR® PrimeScript® PLUS RT-PCR Kit (TaKaRa, Dalian, China) and the Thermal Cycler Dice instrument (TaKaRa), according to the manufacturer's instructions. RT-PCR primers for GADPH and Bcl-2 are listed: GADPH (451 bp) sense: 5′-accacagtccatgccatcac-3′; antisense: 5′-tccaccaccctgttgctgta-3′; Bcl-2 (349 bp) sense: 5′-cacccctggcatcttctcct-3′; and antisense: 5′-gttgacgctccccacacaca-3′. Results were normalized to GAPDH.
Gene silencing and stably transfection
Gene silencing was achieved by either transfecting siRNA using Lipofectamine 2000 Transfection Reagent following the manufacturer's instructions. ERK1/2 siRNA (#6560) and Bcl-2 siRNA (#1441) were purchased from Cell Signaling Technology, Inc. Control siRNA (SC-37007) was from Santa Cruz Biotechnology.
Transfection of H1688 cell line was performed with Lipofectamine according to the manufacturer's instructions. Under our condition, 20–30% of cells are routinely transfected. Then the stably transfected cells were selected by addition of Geneticin (G418), purchased from Invitrogen, in the medium at a concentration of 800 μg·mL−1. After 3 weeks, stably transfected selected cells were further cultured with G418 at a concentration of 400 μg·mL−1. Different expression clones (WT-Bcl-2, AAA-Bcl-2 and EEE-Bcl-2) were selected for other experiments.
Statistical analysis
Synergism, additive effects and antagonism were assessed using Calcusyn software v2.0 (Biosoft, Ferguson, MO, USA). The interaction among drugs was found synergistic when combination index (CI) values were <1. Relative protein quantification was done with Kodak Carestream Molecular Imaging software (New Haven, CT, USA). Results are expressed as mean ± SD of three independent experiments.
Results
Expression levels of Bcl-2 and Bcl-2 phosphorylation associate with cellular response to S1 in a subset of SCLC cells
We assessed the ability of S1 to inhibit cell proliferation as a single agent against a panel of 11 SCLC cell lines with ABT-737 as a control (Figure 1A). The cellular response based on the cell proliferation EC50 values ranged from as low as 360 nM for NCI-H82 cells to >100 μM for NCI-H740 cells following S1 treatment for 48 h. Improved killing ability was found for ABT-737. Co-IP experiments revealed that 500 nM S1 brings about Bax, Bak and Bim releasing from Bcl-2 and Mcl-1 in H1688 cells (Supporting Information Fig. S1A). Cytochrome C releasing, caspase-3/9 activation and the cleavage of PARP coincided with a dose-dependent decrease in cell viability following S1 treatment, suggesting that S1 inhibits cell proliferation through the induction of apoptosis (Supporting Information Fig. S1B and C).
Figure 1.
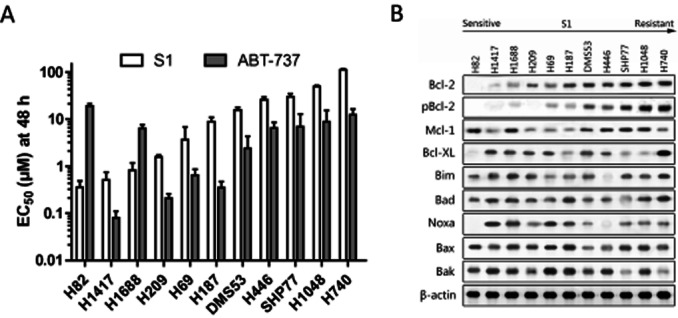
Cytotoxic activity of S1 against a panel of SCLC cell lines associated with Bcl-2 expression and phosphorylation. (A) Cell viability EC50 values of various SCLC cell lines following treatment for 48 h with S1 or ABT-737 were determined by MTS assay. Columns, average (n = 3) of triplicate experiments; bars, SD. (B) Western blot analysis of the Bcl-2 family proteins indicated in the 11 cell lines. Cell lines are arranged according to their resistance to S1.
Expressions of Bcl-2 family members were evaluated to determine if their expression patterns associated with response to S1. pBcl-2 (Ser70) was also measured because it could antagonize other BH3 mimetics such as ABT-737 and Obatoclax (Konopleva et al., 2006; Perez-Galan et al., 2008). In good agreement with previous studies, either Mcl-1 or pBcl-2 protein levels were higher in cells that were resistant to ABT-737 than those in sensitive cells (Konopleva et al., 2006; Tahir et al., 2007). Bcl-2 and pBcl-2 protein levels, however, associated directly with the cellular response to S1 (Figure 1B) such that Bcl-2 protein levels and phosphorylation were higher in cells that were more resistant to S1. As for the other Bcl-2 family proteins, Mcl-1, Bcl-XL, Bax, Bak, Noxa and Bad, there was no clear correlation between protein expression and cellular response to S1.
Bcl-2 transcriptionally up-regulates and sustains phosphorylation in H1688 cells following long-term and acute exposure to S1
To further study the mechanism contributing to resistance of S1, the acquired resistant variants were derived from the sensitive H1688 cells following continuous exposure of S1 for several months. Verapamil was used to inhibit resistance based on increased expression of drug efflux pumps in cells treated with S1 and untreated controls. The dose and time were increased after cell viability was maintained and a series of drug-resistant variants were adapted. Cells were capable of maintaining viability with continuous exposure to S1 at 500 nM for the H1688-SR2 cell line and 40 μM for the H1688-SR10 cell line (Figure 2A). The cellular sensitivity for these variants ranged from an EC50 of 0.8 μM for the parental cells up to >50 μM for H1688-SR10. These variants also exhibited more resistance to ABT-737 and apoptotic cytokines such as TRAIL than the parental cells (Figure 2A and Supporting Information Fig. S1D).
Figure 2.
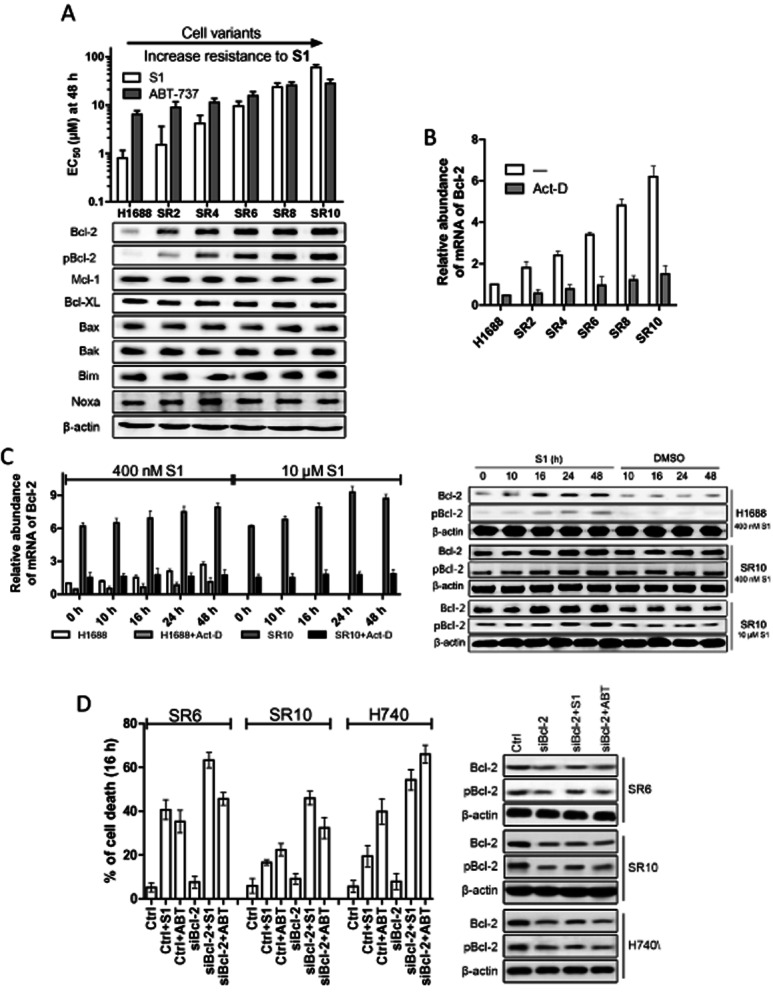
Bcl-2 is transcriptionally up-regulated in H1688-acquired resistant cells. (A) EC50 values (S1 and ABT-737 48 h) and expressions of Bcl-2 family proteins of parental and S1-acquired resistant H1688 cells. Columns, average (n = 3) of triplicate experiments; bars, SD. (B) Bcl-2 transcript level was analysed by quantitative PCR in H1688-derived cells. H1688-derived resistant cells were cultured in the absence of S1 for 3 weeks and cells were treated or not with 3 μg·mL−1 actinomycin D (Act-D) for 1 h before RNA was isolated. (C) Bcl-2 is up-regulated in both transcriptional, protein and phosphorylation levels in parental H1688 and resistant H1688-SR10 cells after transient treatment with S1. H1688 and H1688-SR10 cells were treated with S1 (400 nM for H1688 and H1688-SR10, 10 μM for H1688-SR10) for indicated time and Bcl-2 transcript level was analysed by quantitative PCR. H1688-SR10 cells were removed from S1-containing media for 3 weeks. H1688 and H1688-SR10 cells were treated with S1 (400 nM for H1688, 10 μM for H1688-SR10) or DMSO for indicated time. H1688 and H1688-SR10 cells were treated actinomycin D (3 μg·mL−1) for 1 h before the addition of S1. Whole-cell lysates were made after treatment and analysed by immunoblot. Columns, average (n = 3) of triplicate experiments; bars, SD. (D) The indicated cell lines were transfected with control (Ctrl) or Bcl-2 siRNA for 48 h and then treated with and without 10 μM S1 or ABT-737 for an additional 16 h. Protein levels were examined by Western blot. Cell viability was assessed by MTS assay.
We investigated whether the acquired resistance was mediated by changes in the expression pattern of Bcl-2 family proteins. Increases in Bcl-2 expression and phosphorylation status were observed by Western blot upon progressive adaptation to S1 in H1688 cells (Figure 2A). Expressions of other Bcl-2 family members remained constant across all H1688-derived cell lines. Together with the similar trends observed in the naïve SCLC panel of cells, it is suggestive that the up-regulation of Bcl-2 in protein and phosphorylation levels may contribute mechanistically to the cellular response to S1.
Next, we investigated whether Bcl-2 up-regulation is due to increased transcript abundance. mRNA from sensitive and resistant H1688 cells (all cultured in the absence of S1) was isolated and RT-PCR followed by quantitative real-time PCR (qPCR) were performed (Figure 2B). A more than sixfold increased Bcl-2 mRNA was found in H1688-SR10 cells than that in H1688 cells, and it could be inhibited by pretreatment with actinomycin D, suggesting a transcriptional increase rather than a change in the mRNA stability.
Furthermore, an additional dynamic increase in Bcl-2 transcript abundance was found upon treatment with S1 within hours both in parental and resistant cells, while actinomycin D could inhibit this effect (Figure 2C left). Consistently, Bcl-2 protein and phosphorylation levels were inducible upon acute treatment with S1 (Figure 2C right). Okadaic acid (an inhibitor of protein phosphatases) was applied to exclude the possibility that Bcl-2 expression level was up-regulated by inhibiting proteasome-mediated degradation (Supporting Information Fig. S2). To further demonstrate the key role of Bcl-2 and pBcl-2 in the acquired resistance, Bcl-2 siRNA was used. The levels of Bcl-2 and pBcl-2 were reduced by transfection of Bcl-2 siRNA. Correspondingly, Bcl-2 siRNA-transfected cells increased sensitivity to S1 and ABT-737 (Figure 2D).
In summary, these results indicate that Bcl-2 prevent S1-induced SCLC apoptosis through not only expression level, but also post-translational modification. In addition to the increased Bcl-2 expression and phosphorylation in resistant cells, Bcl-2 expression and phosphorylation could be inducible by acute S1 treatment.
ERK1/2 activation through ER stress regulates Bcl-2 expression in SCLC cell lines
Bcl-2 and pBcl-2 are up-regulated in resistant cells and are inducible upon S1 treatment, suggesting a disruption of the intracellular pathways but not selecting of the pre-existing resistant cells. We speculated that certain signal pathways were activated by S1 and up-regulated downstream effectors leading to both Bcl-2 phosphorylation and activation of transcriptional factors. Among the kinase pathways, MAPK/ERK, PI3K/AKT and p38 pathways appear to be prominently involved in cancer drug resistance and regulate Bcl-2 through both expression level and phosphorylation (Pugazhenthi et al., 2000; Wang et al., 2005; De Chiara et al., 2006; Subramanian and Shaha, 2007; Creson et al., 2009; Galante et al., 2009). We then examined whether S1 treatment leads to p38, PI3K/AKT or MAPK/ERK activation in SCLC cells. Treatment of H1688-derived cells with S1 for 16 h did not increase activated phosphor-p38 or phosphor-AKT levels. The results of qPCR showed the p38 inhibitor SB202190 and PI3K inhibitor LY294002 could not inhibit Bcl-2 transcription up-regulation upon (Supporting Information Fig. S3). Similar results were found in H209 and H740 cell lines upon S1 or ABT-737 treatment (Supporting Information Fig. S3). These results suggested that p38 or AKT is not involved in S1-induced Bcl-2 up-regulation pathway.
Then, we checked the effect of S1 on MAPK/ERK pathway. The Western blot analysis revealed an increase in the phosphorylation of ERK1/2 accompanied with a progressive resistance to S1 in H1688-acquired resistant cells. The MAPK inhibitor PD98059 was able to suppress ERK1/2 phosphorylation while having reduced Bcl-2 protein levels in acquired resistant H1688 cells (Figure 3A). These data suggest that the long-term S1 exposure-induced Bcl-2 up-regulation is mediated by MAPK/ERK pathway activation in acquired resistant cells. Additionally, analysis of ERK1/2 activation in the 11 naïve SCLC panel of cells showed ERK1/2 was activated in S1-resistant cells, which were consistent with the high expression of Bcl-2 (Figures 1B and 3B). These data indicates that S1 resistance in both naïve SCLC cell lines and acquired resistant cells is associated with ERK1/2 activation.
Figure 3.

S1 induces up-regulation of Bcl-2 by an ER stress-induced ERK1/2 activation-dependent mechanism. (A) H1688-derived cells were incubated with 5 μM PD98059 for 16 h. DMSO treatment (left) was used as a control. Whole cell lysates were prepared and Western blots were probed using specific antibodies against phosphorylated ERK1/2, whole ERK1/2 and Bcl-2. Blot shows ERK phosphorylation correlated with up-regulation of Bcl-2 (left). (B) Whole-cell lysates from 11 SCLC cell lines were analysed with Western blot for the levels of phosphorylated ERK1/2 and ERK1/2. Cell lines are arranged according to their resistance to S1. (C) H1688 and H1688-SR10 cells were treated with S1 (400 nM for H1688 and 10 μM for H1688-SR10) alone or together with 5 μM PD98059 for 16 h. Protein levels were examined by Western blot. H1688-SR10 cells were cultured in the absence of S1 for 3 weeks before used. (D) H209 and H740 cells were treated with 400 nM S1 alone or together with 5 μM PD98059 for 16 h. ABT-737 (100 nM) was tested in parallel. DMSO was used as a control. Whole cell lysates were prepared and Western blots were probed using indicated antibodies. (E) The indicated cell lines were transfected with control (Ctrl) or ERK1/2 siRNA for 48 h and then treated with and without 10 μM S1 or ABT-737 for an additional 16 h. Protein levels were examined by Western blot. H1688-SR10 and H740 cells viability were assessed by MTS assay.
In addition, the H1688-derived cell lines were exposed to S1 and dynamical ERK1/2 activation was found (Figure 3 panel 2 and 6).While having reduced Bcl-2 protein (Figure 3C panels 3, 4, 7 and 8) and transcriptional levels, 5 μM PD98059 blocked the activation of ERK1/2 regardless of S1 treatment (Supporting Information Fig. S4A). As we found previously (Zhong et al., 2012), S1 could up-regulate GRP78 [a commonly used indicator of activation of the ER stress (Jiang et al., 2007)], which indicates that ERK1/2 is activated through S1-induced ER stress (Figure 3C panels 2 4, 6 and 8). Additionally, GRP78 was inducible upon S1 treatment regardless of whether ERK1/2 was inhibited by PD98059, indicating that ER stress induced by S1 acts upstream of MAPK/ERK pathway. Then, we tested the effect of ER stress on MAPK/ERK pathway in S1-sensitive H209 and -resistance H740 cells with ABT-737 in parallel. While up-regulating Bcl-2 expression, 100 nM ABT-737 or 400 nM S1, alone, up-regulated GRP78, induced ER stress and promoted phosphorylation of ERK1/2 (Figure 3D). To investigate S1-triggered ER stress pathway in SCLC cells, we examined the signalling pathway(s) of the ER stress. It is notable that the protein kinase RNA-activated-like endoplasmic reticulum kinase (PERK) and inositol-requiring kinase 1 (IRE1) were activated upon S1 treatment while ABT-737 activated PERK. In contrast, exposure to S1 and ABT-737 did not have any notable effect on the activation levels of ATF6 and CCAAT/enhancer-binding protein homologous protein (Supporting Information Fig. S4B). These observations indicate that both PERK and IRE1 signalling pathways of the ER stress play roles in S1-induced ER stress.
ERK1/2 siRNA was used to robustly demonstrate the involvement of ERK1/2 signalling in S1-induced Bcl-2 expression. Transfection of ERK1/2 siRNA reduced the basal levels of ERK1/2 and inhibited S1-induced increase in pERK1/2 in H1688-SR10 and H740 cells. ERK1/2 siRNA transfected cells restored sensitivity to S1 and ABT-737 (Figure 3E). Correspondingly, Bcl-2 up-regulation by S1 was observed only in cells transfected with control siRNA.
Taken together, these results suggest that the activation of MAPK/ERK pathway by ER stress could be a common mechanism by which SCLC cells antagonize BH3 mimetics.
CREB and STAT3 activation through ERK1/2 contributes to S1 induces Bcl-2 expression
It was reported that the increased transcript abundance of Bcl-2 could be mediated by the transcription factor CREB and STAT3 (Bhattacharya et al., 2005; Meller et al., 2005). S1 treatment increased activated pCREB and STAT3 levels, but not total CREB and STAT3 levels (Figure 4A), suggesting that CREB and STAT3 may be downstream of ERK1/2 and mediate Bcl-2 transcriptional up-regulation upon S1 treatment in SCLC cell lines. Downstream of ERK1/2 phosphorylation induced by S1, pRSK1 levels increased while no effect was found on total RSK1 levels (Figure 4A). The activation of RSK1, CREB and STAT3 were inhibited by using PD98059 and ERK1/2 siRNA in the sensitive and resistant cells with and without S1 treatment (Figures 4A and S5A), indicating that RSK1 is a pivotal junction point linking together MAPK/ERK pathway and CREB/STAT3. Similar results were obtained in H209 and H740 cells with S1 and ABT-737 treatment (Figure 4B). An RNAi strategy was used to specifically block RSK1 to further evaluate the role of RSK1 as a junction point for the ERK1/2 and CREB/STAT3 pathways on Bcl-2 up-regulation in H1688-SR10 cells (Supporting Information Fig. S5B and C). RSK1 siRNA reduced CREB and STAT3 activation and also attenuated S1-induced Bcl-2 up-regulation while pERK1/2 was not altered.
Figure 4.
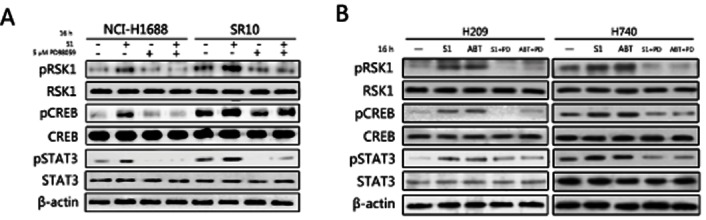
CREB and STAT3 were activated upon S1 treatment in SCLC cell lines. (A) H1688 and H1688-SR10 cells were treated with S1 (400 nM for H1688 and 10 μM for H1688-SR10) alone or together with 5 μM PD98059 for 16 h. The activations of RSK1, CREB and STAT3 were examined by Western blot. (B) H209 and H740 cells were treated with 400 nM S1 alone or together with 5 μM PD98059 for 16 h. ABT-737 (100 nM) was tested in parallel. DMSO was used as a control. Whole cell lysates were prepared and Western blots were probed using indicated antibodies.
These results indicated that Bcl-2 level was regulated by activation of the transcription factor CREB and STAT3. Therefore, a link between S1 stimulation, ER stress, phosphorylation of ERK1/2, activation of CREB and STAT3, and Bcl-2 transcriptional up-regulation could be established in both acquired resistant and naïve SCLC cell lines.
Bcl-2 undergoes phosphorylation by ERK1/2 and sequestrates more pro-apoptotic proteins in acquired resistant cells
As a Bcl-2 kinase, ERK1/2 could contribute to S1-induced Bcl-2 phosphorylation (Konopleva et al., 2006; Deng et al., 2009). As expected, PD98059 was able to block Bcl-2 phosphorylation and reduce the pBcl-2/Bcl-2 ratios in H1688-SR6, H1688-SR10, H1048 and H740 cells (Figure 5A).
Figure 5.

Bcl-2 is phosphorylated by ERK1/2 in H1688-derived cells and contributes to the resistance to S1. (A) PD98059 was able to block Bcl-2 phosphorylation and reduce the pBcl-2/Bcl-2 ratios. H1688-derived H1688-SR6, H1688-SR10 cell, H1048 and H740 cells were incubated with 5 μM PD98059 for 16 h. DMSO was used as a control. Total protein extracts (50 μg) from these cells were analysed by Western blot for pBcl-2 and Bcl-2 expression. The mean of pBcl-2/Bcl-2 ratio is shown for each cell line. (B) pBcl-2 sequestrated more pro-apoptotic proteins in acquired resistant cells. Immunoprecipitation of H1688-derived cell lysates. Mcl-1, Bcl-2 and pBcl-2 immunoprecipitations were performed, and immunoprecipitated fractions were analysed by Western blotting for the indicated proteins. (C) H1688-SR10 cells were treated with 20 μM S1 for 16 h. Mcl-1, Bcl-2 and pBcl-2 immunoprecipitations were performed, and immunoprecipitated fractions were analysed by Western blotting for the indicated proteins. (D) Phosphorylated Bcl-2 activates ERK1/2. Parental H1688 cells and H1688-expressing WT-Bcl-2, AAA-Bcl-2 or EEE-Bcl-2 were treated with and without 10 μM S1 or ABT-737 for 16 h and then lysed. Protein levels were examined by immunoblotting. Cell viability was assessed by MTS assay.
pBcl-2 is another pivotal role in antagonizing BH3 mimetics. Previous reports showed that Bcl-2 phosphorylation may regulate its interaction with mutidomain (Bax) and BH3-only pro-apoptotic (Bim and Bid) members (Bassik et al., 2004). To test whether there was a change in pro-apoptotic Bcl-2 family proteins associated with Bcl-2 and Mcl-1 in the resistant derivative, where more Bcl-2 was phosphorylated, we performed co-IP in parental and acquired resistant cells. We found that resistant cell lines appeared significantly less Bim and Bak complexed with Mcl-1 than those in H1688 cells, while more pro-apoptotic proteins were associated with Bcl-2 and pBcl-2 (Figure 5B). Non-pro-apoptotic members were substituted by 20 μM S1 from pBcl-2 in H1688-SR10 cells using a pBcl-2 (Ser70) antibody except for a slight disruption of Mcl-1/Bak complex (Figure 5C). Since S1 could disrupt Bcl-2 dimmers, as shown in Supporting Information Fig. S6, Bcl-2 phosphorylation prevent S1 interaction with Bcl-2. Similar results were found in H740 cells upon S1 and ABT-737 treatment (Supporting Information Fig. S7). These results suggest that Bcl-2 phosphorylation is meaningful in determining the sensitivity of SCLC cells to S1.
As expected, a synergy between PD98059 and S1 was found in H1688-SR10, H1048 and H740 cells (CI = 0.48, 0.52 and 0.63, respectively, Supporting Information Fig. S8A and B). The combination of PD98059 with ABT-737 was also synergistic in H1688 and SR10 cells (CI = 0.65 and 0.41, respectively, Supporting Information Fig. S8C). Co-IP experiment data showed that when Bcl-2 expression level and phosphorylation were inhibited by PD98059 S1 treatment led to massive release of pro-apoptotic members from Bcl-2 and Mcl-1 (Supporting Information Fig. S8D). These data suggested that the activation of MAPK/ERK pathway results in enhanced resistance due to the protection of pBcl-2 against S1.
ERK1/2 is activated by pBcl-2 but not non-pBcl-2
It was reported that Bcl-2 could activate ERK signalling pathway (Feng et al., 2004). Together with pBcl-2's key role in resistance to S1, we speculated that Bcl-2 phosphorylation could affect the activation of MAPK/ERK pathway. Different Bcl-2 expression vectors (HA-wt-Bcl-2, non-phosphomimetic mutant HA-AAA-Bcl-2 and a phosphomimetic mutant HA-EEE-Bcl-2) were used to perform stable transfection. As shown in panels 1, 2 and 3 of Figure 5D, although the exogenous Bcl-2 levels of HA-wt-Bcl-2, HA-AAA-Bcl-2 and HA-EEE-Bcl-2 stably transfected H1688, cells were similar. Expression of EEE-Bcl-2 induced highest increase in the activition of ERK1/2. However, AAA-Bcl-2 did not affect the activation of ERK1/2. Figure 5D also revealed that phosphomimetic, but not non-phosphomimetic, Bcl-2 could promote CREB phosphorylation through activating ERK1/2. These results demonstrate that pBcl-2, but not non-pBcl-2, would feedback activate ERK1/2 in SCLC cell lines. Our experiments also demonstrated that EEE-Bcl-2 H1688 cells are more resistant to S1 and ABT-737 than cells expressing wt-Bcl-2 and AAA-Bcl-2 (Figure 5D), suggesting that Bcl-2 phosphorylation opposes the pro-apoptotic action of S1 and ABT-737.
Discussion
As a new and promising treatment for SCLC, BH3 mimetics, ABT-263 and Obatoclax, have been developed in phase I and II clinical trials respectively (Reed, 1997). To understand the mechanisms contributing to the response, especially the spontaneous select for resistance to these ‘drugs’, has direct implications for the development of BH3 mimetics in SCLC treatment.
S1 is a Bcl-2/Mcl-1/Bcl-XL triple inhibitor and a specific BH3 mimetic, which kills through Bax/Bak (Zhang et al., 2010; Song et al., 2012). Compared with ABT-737 and Obataclax, S1 may provide full and exact understanding of model of cell death induced by BH3 mimetics. In this study, we used S1 together with ABT-737 to reveal a novel mechanism whereby ERK1/2 mediated crosstalk between mitochondrial apoptosis and ER stress, and found that this mechanism determines BH3 mimetics' killing in SCLC cells.
Although the cause of inherent resistance always differs from the biological basis for acquired resistance in a certain cancer following the same therapy, we still identified that MAPK/ERK pathway regulated BH3 mimetics' effects on SCLC cells, both in a panel of 11 SCLC cell lines and H1688-derived S1-acquired resistant cells.
Firstly, we found that the increased expression and phosphorylation of Bcl-2 play a key role in defining S1. In our SCLC model of acquired resistance, S1 induced progressive increases in Bcl-2 transcript and Bcl-2 phosphorylation following a prolonged exposure; additionally, Bcl-2 expression was dynamically increased upon acute treatment with S1, and pBcl-2 was also inducible by S1 treatment with hours. Together with the association between high levels of Bcl-2 and pBcl-2 in naïve S1 resistant SCLC cell lines, it seems that the selection for regulation of Bcl-2 on both transcriptional level and post-modification level is the determination of S1 resistance.
We then revealed there is a crosstalk between ER stress and mitochondrial apoptosis by which SCLC cells define S1 and ABT-737 through regulation of Bcl-2 family members. We have previously reported that S1 triggered IRE1 and GRP78 in human glioma U251 cells (Zhong et al., 2012). The findings in this study showed that GRP78 was activated by S1 and ABT-737 in SCLC cell lines and acquired resistance model. Together with these results, we conclude that S1 and ABT-737 induced ER stress and then activated MAPK/ERK pathway. The ERK1/2 activation mediated Bcl-2 regulation on transcriptional level; and post modification level was identified by siRNA transfection and MAPK inhibitor PD98059 treatment. Western blot analysis identified that it was ERK1/2, not p38 or AKT, that is involved into the BH3 mimetics-induced apoptosis. Consequently, the downstream factor RSK1 stimulated phosphorylation of CREB and STAT3, which are major promoter of Bcl-2. Even if the increased Bcl-2 transcript cannot fully define S1, since it's the target of S1, the increased Bcl-2 phosphorylation resulted from ERK1/2 activation antagonized S1, most likely by impede of S1 to access its BH3 pocket. Although there is a discrepancy between S1 and ABT-737 in inducing anti-apoptotic Bcl-2 members' alternation, we still found the same crosstalk by which SCLC defined ABT-737. Bcl-2 was up-regulated by S1 while Mcl-1 showed relative constancy accompanied with the activation of ERK1/2. By contrast, Mcl-1 was up-regulated by ABT-737 in the paralleled experiment in this study. In the previous investigations, it was also found that the up-regulation of Mcl-1 determined ABT-737 resistance in lymphoma and other tumour cells following both chronic and acute exposure (Tahir et al., 2007; Yecies et al., 2010). We believe it is due to the different selectivity of the two molecules. When these molecules activated ER stress, the downstream transcriptional factors of both Mcl-1 and Bcl-2 were activated. Both the two proteins have the potential to be up-regulated. Because S1 efficiently hit Mcl-1, which is a short-life protein (Marriott et al., 2005), no significant reduce in Mcl-1 level was found in S1 treatment. ABT-737, however, hits Bcl-2 only. As such, Mcl-1 was up-regulated freely and contributed to ABT-737 resistance. Further bolstering our confidence of the crosstalk between ER stress and mitochondrial apoptosis is the progressive activation of ERK1/2 as H1688 cells adapted to S1, which resulted from the long-term ER stress upon S1 treatment.
Numerous investigations had illustrated that ER stress can potentially activate apoptosis signalling pathways while some studies found that ER stress may result in Mcl-1 protein up-regulation, which encountered apoptosis (Puthalakath et al., 2007; Jiang et al., 2008; Tabas and Ron, 2011). In our study, we identified that the crosstalk between ER stress and mitochondrial apoptosis resulted in the activation of ERK1/2 and up-regulation of Bcl-2 members, in inherent sensitive, inherent resistant and acquired resistant SCLC cells. This regulation exhibits anti-apoptotic effects against BH3 mimetics.
pBcl-2 is another key feature in creating resistance to S1 and, in other reports, it is also against ABT-737 and Obatoclax's killing ability (Konopleva et al., 2006; Perez-Galan et al., 2008). Here, we identified that pBcl-2 rendered resistance against S1 and ABT-737 in SCLC cell lines because the two molecules can only disturb Bcl-2 dimers, but not pBcl-2 dimers. The co-IP results showed pBcl-2 sequestrated more Bim and Bax in resistant H1688-SR6 and H1688-SR10 cells that S1 could not disturb. In naïve resistant H740 cells, neither S1 nor ABT-737 could displace Bax or Bak from pBcl-2. Additionally, it is reported that in ABT-737-acquired resistant cells, the Bcl-2 complexes with Bim detected by co-IP using Bcl-2 antibody are different from those in parental ones (Tahir et al., 2007). Because of the cross reaction of Bcl-2 antibody, it may also recognize pBcl-2. As such, we speculate that in ABT-737 resistant cell, it is pBcl-2 resulted from ERK activation that regulate its interaction with mutidomain and BH3-only pro-apoptotic members. Probably, the divergence in BH3 grooves of Bcl-2 and pBcl-2 accounts for the regulation of the interaction with both Bcl-2 family members and BH3 mimetics. Most likely, the altered BH3 groove of pBcl-2 cannot accommodate S1 and ABT-737. As such, Bcl-2 phosphorylation is a common mechanism that cells creating resistance against BH3 mimetics, which result from the ER stress-activated MAPK/ERK pathway.
Moreover, for the first time, we identified that pBcl-2 feeds back to promote ERK1/2 activation. This means ERK1/2 is not only a hub to convey ER stress to mitochondria, but may amplify the signal by a positive feedback loop to enhance its downstream Bcl-2 phosphorylation.
Till now, the dual function of MAPK/ERK pathway in resistance of BH3 mimetics is illustrated. It increases Bcl-2 and Mcl-1 transcript and phosphorylates Bcl-2 protein (maybe Mcl-1 can also be phosphorylated that we did not investigated here). Bcl-2-like protein could protect cells through either increased poorly targeted level or poorly targeted modified status by the BH3 mimetics, or both.
Recent reports indicate that MAPK inhibitor reduced Mcl-1 expression and showed synergism with ABT-737 in acute lymphoblastic leukemia as well as in S1 in this study; however, the underlying mechanism was not clearly identified (Konopleva et al., 2012). Here, we provided the biological basis for the resistance of S1 and other BH3 mimetics, which revealed the mechanism by which the combination of BH3 mimetics, with strategies that inhibit MAPK/ERK, is favourable for SCLC treatment.
In summary, we have provided evidence that activation of MAPK/ERK by ER stress governs cell survival during BH3 mimetics treatment (Figure 6). We propose that this is an important physiological response when cancer cells encounter stimulation of BH3 mimetics. These results provide significant novel insights into the molecular mechanisms for crosstalk between ER stress and endogenous apoptotic pathways.
Figure 6.
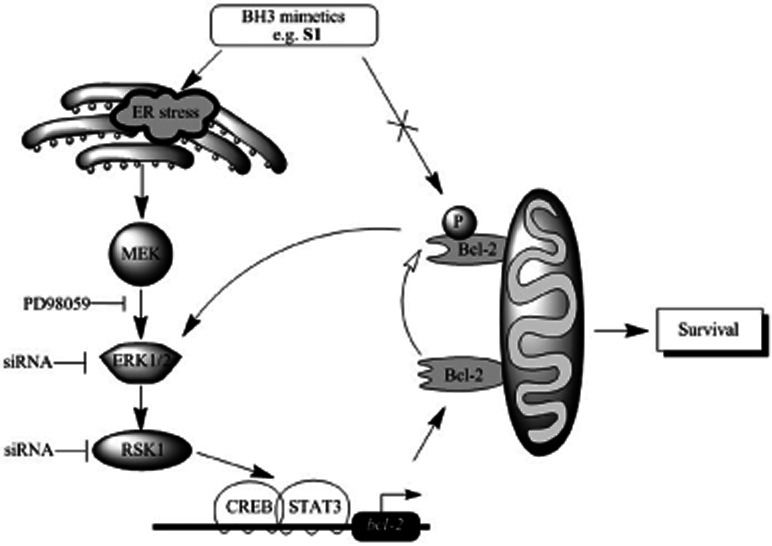
Schematic working model for ER stress/ERK1/2-mediated signalling of BH3 mimetics (e.g. S1)-induced Bcl-2 expression and phosphorylation. BH3 mimetics, such as S1 and ABT-737, stimulate ER stress then induce MAPK/ERK cascades with respect to Bcl-2 transcriptional up-regulation (solid arrowhead) and phosphorylation (hollow arrowhead). Excess pBcl-2 leads to cell survival by promoting ERK1/2 activation and sequestrating more pro-apoptotic members, which BH3 mimetics cannot release.
Acknowledgments
The authors would like to gratefully acknowledge support from National Natural Science Foundation of China (81272876 and 81273436).
Conflict of interest
No potential conflicts of interest were disclosed.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 S1 induces apoptosis by intrinsic apoptotic pathway in lung cancer cell lines. (A) H1688 cells were treated with 400 nM S1 for 16 h. Bcl-2 and Mcl-1 immunoprecipitations were performed and analyzed by western blot for Bak, Bax and Bim proteins. (B) Caspase-3/9 activation and dose response of cell viability after 24 h of treatment of S1 in H1688 cells. Points, mean; bars, SD. (C) H1688 cells were exposed to S1 (indicated doses) and at the indicated times cytosol and mitochondrial fractions were analyzed by western blotting for cytochrome c (cyt c) and PARP cleavage. (D) The S1 acquire resistant H1688 cells were incubated with 20 ng/mL His-TRAIL, and cell proliferation was determined in MTT and BrdU assays 72 h thereafter. Absorbance values obtained with untreated cells maintained under identical conditions were taken as 100%. Columns, average (n = 3) of triplicate experiments; bars, SD.
Figure S2 Bcl-2 expression level was not up-regulate by inhibiting proteasome-mediated degradation. Ubiquitinated Bcl-2 was found similar with/without S1 or OA treatment in H1688 and H1688-SR10 cells respectively. S1 and OA induced Bcl-2 phosphorylation without affect Bcl-2 ubiquitination. More Bcl-2 was ubiquitinated in H1688-SR10 cells suggested that ubiquitination was enhanced when Bcl-2 was accumulated. These data indicated that Bcl-2 expression level was not up-regulate by inhibiting proteasome-mediated degradation through phosphorylation. H1688 and H1688-SR10 cells were pretreated with S1 (400 nM for H1688, 10 μM for H1688-SR10) or OA (1 μM, an inhibitor of serine/threonine protein phosphatases which can induce modification of Bcl-2 phosphorylation) for 16 h. Cell lysates were immunoprecipitated with anti-Bcl-2 antibody, and the immune complexes were analyzed for ubiquitin by Western blotting.
Figure S3 S1 and ABT-737 regulation Bcl-2 expression level does not through p38 or PI3K/AKT pathway in lung cancer cell lines. Treatment with S1 or ABT-737 did not increased activated phosphor-p38 (p-p38) or phosphor-AKT (pAKT) levels in H1688-derived cells, H209 and H740 cells. p38 inhibitor SB202190 and PI3K inhibitor LY294002 did not regulate Bcl-2 expression levels. These results suggest that S1 or ABT-737 promote Bcl-2 expression by a mechanism does not involving p38 or PI3K/AKT pathway. (A) H1688, H1688-SR6 and H1688-SR10 cells were treated with DMSO or S1 (400 nM for H1688, 5 μM for H1688-SR6, 20 μM for H1688-SR10) for 16 h and activated p-p38 and pAKT levels were tested by immunoblot. (B) H1688-derived cells were incubated with DMSO, 10 μM SB202190 or 25 μM LY294002 with/without S1 treatment for 16 h and Bcl-2 transcript level was analyzed by quantitative PCR. Columns, average (n = 3) of triplicate experiments; bars, SD. (C) H209 and H740 cells were treated with DMSO, S1 (200 nM for H209, 10 μM for H740) or ABT-737 (100 nM for H209, 10 μM for H740) for 16 h and activated p-p38 and pAKT levels were tested by immunoblot. (D) H209 and H740 cells were incubated with DMSO, 10 μM SB202190 or 25 μM LY294002 with/without S1 treatment for 16 h and Bcl-2 transcript level was analyzed by quantitative PCR. Columns, average (n = 3) of triplicate experiments; bars, SD.
Figure S4 (A) PD98059 reduced Bcl-2 transcriptional levels in H1688-derived cells. H1688 and H1688-SR10 cells were treated with S1 (400 nM for H1688 and 10 μM for H1688-SR10) alone or together with 5 μM PD98059 for 16 h. Bcl-2 transcript level was analyzed by quantitative PCR. Columns, average (n = 3) of triplicate experiments; bars, SD. (B) PERK and IRE1 signaling pathways of the ER stress play roles in S1 induced ER stress. H1688 and H1688-SR10 cells were treated with S1 (400 nM for H1688 and 10 μM for H1688-SR10) alone or together with 5 μM PD98059 for 16 h. Protein levels were examined by western blot. H1688-SR10 cells were cultured in the absence of S1 for 3 weeks before used. H209 and H740 cells were treated with 400 nM S1 alone or together with 5 μM PD98059 for 16 h. ABT-737 (100 nM) was tested in parallel. DMSO was used as a control. Whole cell lysates were prepared and Western blots were probed using indicated antibodies.
Figure S5 RSK1 is a pivotal junction point linking together MAPK/ERK pathway and CREB/STAT3. (A) The indicated cell lines were transfected with control (Ctrl) or ERK1/2 siRNA for 48 h and then treated with and without 10 μM S1 or ABT-737 for an additional 16 h. Protein levels were examined by western blot. (B) RSK1 knockdown inhibits CREB, STAT3 activation and attenuates Bcl-2 expression level in H1688-SR10 cells. Two RSK1-specific siRNAs were tested for RSK1 knockdown efficiency. H1688-SR10 cells were treated or not with 10 μM S1 and transfected with the siRNAs for 2 days before cells were collected and processed for RSK1, pCREB, pSTAT3, Bcl-2 specific immunoblotting and Bcl-2 qPCR. Columns, average (n = 3) of triplicate experiments; bars, SD. (C) RSK1 knockdown inhibits CREB, STAT3 activation and attenuates Bcl-2 expression and phosphorylation level in H209 and H740 cells. RSK1-specific siRNA1 were tested for RSK1 knockdown efficiency. H209 and H740 cells were treated or not with 10 μM S1 and transfected with the siRNA for 2 days before cells were collected and processed for RSK1, pCREB, pSTAT3, pBcl-2, Bcl-2 specific immunoblotting.
Figure S6 S1 could disrupt npBcl-2/Bak, npBcl-2/Bax and npBcl-2/Bim. H1688-SR10 cells were treated with 20 μM S1 for 16 h. Supernatants of pBcl-2 immunoprecipitation were used for Bcl-2 immunoprecipitation. Immunoblotting was performed as described. No pBcl-2 was found in supernatants of pBcl-2 immunoprecipitation, indicating that immunoprecipitates of Bcl-2 contained npBcl-2 but not pBcl-2. Our data showed that S1 could disrupt npBcl-2/Bak, npBcl-2/Bax and npBcl-2/Bim heterodimers.
Figure S7 Bcl-2 is phosphorylated contributes to the resistance to S1 and ABT-737. S1 and ABT-737 cannot disrupt the complexes contain pBcl-2. H740 cells were treated with 20 μM S1 or 5 μM ABT-737 for 16 h. Bcl-2 and pBcl-2 immunoprecipitations were performed, and immunoprecipitated fractions were analysed by western blotting for the indicated proteins.
Figure S8 PD98059 significantly enhances the killing of S1 in resistant cells. (A) H1688-SR10 cells were treated with the indicated doses of S1 alone for 24 h, together with 5 μM PD98059. Viability was assessed by MTS assay. H1688 was tested in parallel. CIs were calculated. Columns, average (n = 3) of triplicate experiments; bars, SD. (B) H1048 and H740 cells were treated with the indicated doses of S1 alone for 24 h, together with 5 μM PD98059. Viability was assessed by MTS assay. CIs were calculated. Columns, average (n = 3) of triplicate experiments; bars, SD. (C) H1688 and SR10 cells were treated with the indicated doses of ABT-737 alone for 24 h, together with 5 μM PD98059. Viability was assessed by MTS assay. CIs were calculated. Columns, average (n = 3) of triplicate experiments; bars, SD. (D) H1688-SR10 cells (500 μg) treated or not with 5 μM S1 and 5 μM PD98059 for 12 h. The immunoprecipitates were analyzed for the presence for the indicated proteins by immunoblotting.
References
- Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. 2005;392(Pt 2):335–344. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton C, Story MD, Meyn RE. Bcl-2 expression correlates with apoptosis induction but not loss of clonogenic survival in small cell lung cancer cell lines treated with etoposide. Anticancer Drugs. 1998;9:751–757. doi: 10.1097/00001813-199810000-00002. [DOI] [PubMed] [Google Scholar]
- Burlacu A. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 2007;7:249–257. doi: 10.1111/j.1582-4934.2003.tb00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Ann Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Creson TK, Yuan P, Manji HK, Chen G. Evidence for involvement of ERK, PI3K, and RSK in induction of Bcl-2 by valproate. J Mol Neurosci. 2009;37:123–134. doi: 10.1007/s12031-008-9122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, et al. BCL-2 phosphorylation by p38 MAPK. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- Dean EJ, Cummings J, Roulston A, Berger M, Ranson M, Blackhall F, et al. Optimization of circulating biomarkers of obatoclax-induced cell death in patients with small cell lung cancer. Neoplasia. 2011;13:339. doi: 10.1593/neo.101524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Gao F, May WS. Protein phosphatase 2A inactivates Bcl2's antiapoptotic function by dephosphorylation and up-regulation of Bcl2-p53 binding. Blood. 2009;113:422–428. doi: 10.1182/blood-2008-06-165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Xiang H, Mao YW, Wang J, Liu JP, Huang XQ, et al. Human Bcl-2 activates ERK signaling pathway to regulate activating protein-1, lens epithelium-derived growth factor and downstream genes. Oncogene. 2004;23:7310–7321. doi: 10.1038/sj.onc.1208041. [DOI] [PubMed] [Google Scholar]
- Fennell DA. Bcl-2 as a target for overcoming chemoresistance in small-cell lung cancer. Clin Lung Cancer. 2003;4:307–313. doi: 10.3816/clc.2003.n.012. [DOI] [PubMed] [Google Scholar]
- Galante JM, Mortenson MM, Bowles TL, Virudachalam S, Bold RJ. ERK/BCL-2 pathway in the resistance of pancreatic cancer to anoikis. J Surg Res. 2009;152:18–25. doi: 10.1016/j.jss.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Camidge DR, de Oliveira MR, Bonomi P, Gandara D, Khaira D, et al. Phase I study of navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L, et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res. 2008;68:2321–2328. doi: 10.1158/0008-5472.CAN-07-5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen O, Sørensen P, Hansen KH. The occurrence of hyponatremia in SCLC and the influence on prognosis: a retrospective study of 453 patients treated in a single institution in a 10-year period. Lung Cancer. 2010;68:111–114. doi: 10.1016/j.lungcan.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Jiang CC, Chen LH, Gillespie S, Wang YF, Kiejda KA, Zhang XD, et al. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 2007;67:9750–9761. doi: 10.1158/0008-5472.CAN-07-2047. [DOI] [PubMed] [Google Scholar]
- Jiang CC, Lucas K, Avery-Kiejda KA, Wade M, deBock CE, Thorne RF, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 2008;68:6708–6717. doi: 10.1158/0008-5472.CAN-08-0349. [DOI] [PubMed] [Google Scholar]
- Johnson B, Crawford J, Downey R, Ettinger D, Fossella F, Grecula J, et al. Small cell lung cancer clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2006;4:602–622. doi: 10.6004/jnccn.2006.0050. [DOI] [PubMed] [Google Scholar]
- Kazma R, Babron MC, Gaborieau V, Génin E, Brennan P, Hung RJ, et al. Lung cancer and DNA repair genes: multilevel association analysis from the International Lung Cancer Consortium. Carcinogenesis. 2012;33:1059–1064. doi: 10.1093/carcin/bgs116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778–787. doi: 10.1038/leu.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kurup A, Hanna NH. Treatment of small cell lung cancer. Crit Rev Oncol Hematol. 2004;52:117–126. doi: 10.1016/j.critrevonc.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Marriott HM, Bingle CD, Read RC, Braley KE, Kroemer G, Hellewell PG, et al. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest. 2005;115:359–368. doi: 10.1172/JCI21766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meerbeeck JP, Fennell DA, De Ruysscher DKM. Small-cell lung cancer. Lancet. 2011;378:1741–1755. doi: 10.1016/S0140-6736(11)60165-7. [DOI] [PubMed] [Google Scholar]
- Meller R, Minami M, Cameron JA, Impey S, Chen D, Lan JQ, et al. CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:234–246. doi: 10.1038/sj.jcbfm.9600024. [DOI] [PubMed] [Google Scholar]
- Ohmori T, Podack E, Nishio K, Takahashi M, Miyahara Y, Takeda Y, et al. Apoptosis of lung cancer cells caused by some anti-cancer agents (MMC, CPT-11, ADM) is inhibited by BCL-2. Biochem Biophys Res Commun. 1993;192:30–36. doi: 10.1006/bbrc.1993.1377. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Paik PK, Rudin CM, Brown A, Rizvi NA, Takebe N, Travis W, et al. A phase I study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol. 2010;66:1079–1085. doi: 10.1007/s00280-010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Galan P, Roue G, Lopez-Guerra M, Nguyen M, Villamor N, Montserrat E, et al. BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15-070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukemia cells. Leukemia. 2008;22:1712–1720. doi: 10.1038/leu.2008.175. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Reed JC. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Adv Pharmacol. 1997;41:501–532. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- Sartorius UA, Krammer PH. Upregulation of bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int J Cancer. 2001;97:584–592. doi: 10.1002/ijc.10096. [DOI] [PubMed] [Google Scholar]
- Song T, Chang X, Zhang Z, Liu Y, Shen X. S1, a novel Pan-BH3 mimetic, induces apoptosis in Mcl-1-overexpressing cells through Bak. J Pharmacol Sci. 2012;119:330–340. doi: 10.1254/jphs.12103fp. [DOI] [PubMed] [Google Scholar]
- Subramanian M, Shaha C. Up-regulation of Bcl-2 through ERK phosphorylation is associated with human macrophage survival in an estrogen microenvironment. J Immunol. 2007;179:2330–2338. doi: 10.4049/jimmunol.179.4.2330. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Wang C, Song J, Song D, Yong V, Shuaib A, Hao C. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differ. 2005;13:1203–1212. doi: 10.1038/sj.cdd.4401804. [DOI] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ming L, Yu J. BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug Resist Updat. 2007;10:207–217. doi: 10.1016/j.drup.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song T, Zhang T, Gao J, Wu G, An L, et al. A novel BH3 mimetic S1 potently induces Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int J Cancer. 2010;128:1724–1735. doi: 10.1002/ijc.25484. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wu G, Xie F, Song T, Chang X. 3-Thiomorpholin-8-oxo-8 H-acenaphtho [1, 2-b] pyrrole-9-carbonitrile (S1) based molecules as potent, dual inhibitors of B-cell lymphoma 2 (Bcl-2) and myeloid cell leukemia sequence 1 (Mcl-1): structure-based design and structure − activity relationship studies. J Med Chem. 2011;54:1101–1105. doi: 10.1021/jm101181u. [DOI] [PubMed] [Google Scholar]
- Zhong JT, Xu Y, Yi HW, Su J, Yu HM, Xiang XY, et al. The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett. 2012;323:180–187. doi: 10.1016/j.canlet.2012.04.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.