

Abstract
Background and Purpose
The Kaposi sarcoma (KS)-associated herpesvirus GPCR (vGPCR) is a key molecule in the pathogenesis of KS, where it increases NF-κB gene expression and activates the NF-κB pathway. We investigated whether the less calcemic vitamin D analogue TX 527 inhibited the proliferation of endothelial cells transformed by vGPCR by modulation of the NF-κB pathway.
Experimental Approach
Endothelial cells transformed by vGPCR (SVEC-vGPCR) were treated with TX 527. Proliferation was measured by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) and cell cycle by flow cytometry. mRNA and protein levels were measured by real-time quantitative reverse transcriptase-PCR (qRT-PCR) and immunoblot analysis respectively.
Key Results
TX 527, similar to bortezomib (0.5 nM), a proteasome inhibitor that inhibits the activation of NF-κB, reduced proliferation and induced G0/G1 cell cycle arrest in SVEC-vGPCR. TX 527 like 1α,25(OH)2D3, biological active form of vitamin D, decreased the activity of NF-κB comparable with the effect of bortezomib. Time-response studies showed that TX 527 significantly decreased NF-κB and increased IκBα mRNA and protein levels. The increase of IκBα was accompanied by a reduction in p65/NF-κB translocation to the nucleus. These responses were abolished when vitamin D receptor (VDR) expression was suppressed by stable transfection of shRNA against VDR. In parallel with NF-κB inhibition, there was a down-regulation of inflammatory genes such as IL-6, CCL2/MCP and CCL20/MIP3α.
Conclusions and Implications
These results suggest that the anti-proliferative effects of the vitamin D analogue TX 527 in SVEC-vGPCR occur by modulation of the NF-κB pathway and are VDR dependent.
Keywords: TX 527, VDR, NF-κB pathway, Kaposi sarcoma
Introduction
1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3, calcitriol], the most biologically active form of vitamin D, maintains calcium homeostasis through its actions in intestine, bone, kidneys and the parathyroid glands (Feldman et al., 2007). In addition to its classical effects, 1α,25(OH)2D3 exerts anti-proliferative, pro-apoptotic and pro-differentiating actions on various malignant cells and exhibits anti-inflammatory effects through inhibition of pro-inflammatory cytokines and NF-κB signalling (Krishnan and Feldman, 2011). Most of the activity of 1α,25(OH)2D3 is mediated by the vitamin D receptor (VDR), a member of the nuclear receptor superfamily (Haussler et al., 1998). The VDR is present not only in cells and tissues involved in calcium regulation but also in a wide variety of other cells including neoplastic cells (Feldman et al., 2007). In recent years, it has been recognized that 1α,25(OH)2D3 exerts anti-proliferative and pro-differentiating effects in several malignant cells, and retards the development and growth of tumours in animal models raising the possibility of its use as an anticancer agent (Deeb et al., 2007; Krishnan et al., 2010). However, because of its calcemic effects, such as hypercalcemia, hypercalciuria and increased bone resorption, the use of the hormone for therapeutic purposes is limited (Eelen et al., 2007). The synthesis of analogues with less calcemic activity tries to overcome this problem (Verlinden et al., 2000; Bouillon et al., 2005; Eelen et al., 2007). One of such analogues, TX 527 [19-nor-14,20-bisepi-23-yne-1,25(OH)2D3] has been shown to possess markedly diminished in vivo calcemic effects in combination with enhanced anti-proliferative and pro-differentiating capacities on normal and malignant cell types when compared with 1α,25(OH)2D3 (Van Etten et al., 2000; Verlinden et al., 2000). In addition, TX 527 has enhanced immune regulatory capacities when compared with the parental compound (van Etten et al., 2003; Baeke et al., 2011) which makes this analogue a suitable candidate to treat hyper-proliferative and inflammatory disorders.
Kaposi's sarcoma (KS) is an angio-proliferative tumour and is the most common cancer in HIV-infected untreated individuals. KS-associated herpes virus (KSHV; also known as human herpes virus 8) is the infectious cause of this neoplasm (Martin and Gutkind, 2009; Mesri et al., 2010). The KS-associated herpes virus GPCR (KSHV-GPCR) is a key molecule in the pathogenesis of KS. Persistent expression and activity of viral GPCR (vGPCR) is required for tumour maintenance (Montaner et al., 2006). Thus, vGPCR and its regulated signalling pathways may represent suitable candidates for KS treatment (Martin et al., 2008). vGPCR contributes to KS development through its potent transforming and pro-angiogenic functions. At the molecular level, the angiogenic and paracrine transforming effect of vGPCR involves the activation of multiple MAPKs and small GTPases of the rho family which activities converge in the nucleus to control transcription factors such as hypoxia-inducible factor 1α and AP-1 and promotes the expression of pro-inflammatory molecules, IL-6, IL-8/CXCL8 and MIP-1/CCL3 (Sodhi et al., 2000; 2001; Montaner et al., 2001; 2004; Martin et al., 2008). NF-κB consists of a family of transcription factors including p65 (RelA), p105/p50, p100/p52, RelB and c-Rel. It has been determined that NF-κB gene expression is increased in KS and that vGPCR potently activates the NF-κB pathway which is required for direct induction of neoplasia by vGPCR, thus being an important therapeutic target for the treatment of KS (Martin et al., 2008; Martin and Gutkind, 2009; Gupta et al., 2010). NF-κB regulates the transcription of a wide array of genes involved in inflammation and also in growth regulation, apoptosis, cancer invasion/metastasis, tumour promotion and carcinogenesis (Aggarwal, 2004; Gupta et al., 2010).
We have previously demonstrated that 1α,25(OH)2D3 and TX 527 have anti-proliferative effects on the growth of endothelial cells transformed by the vGPCR in vitro and in vivo by a mechanism that depends on VDR expression (González Pardo et al., 2010). Furthermore, down-regulation of the NF-κB pathway by 1α,25(OH)2D3 in vGPCR cells was found to be part of the mechanism of inhibition (Gonzalez Pardo et al., 2012). In this work, we investigated whether the analogue TX 527 similar to 1α,25(OH)2D3 inhibits the NF-κB pathway and controls the expression of inflammatory genes and the proliferation of endothelial cells transformed by KS-associated herpes virus GPCR in a VDR-dependent manner.
Methods
Chemicals and reagents
The vitamin D analogue TX 527, originally synthesized by M. Vandewalle and P. De Clercq (University of Ghent, Belgium), was provided by Théramex (Monte Carlo, Monaco). Immobilon P (PVDF) membranes and the antibiotic G418 were supplied by Sigma-Aldrich (St. Louis, MO, USA). Puromycin was provided by Invivogen (San Diego, CA, USA). The antibodies used mouse monoclonal anti-VDR (Affinity Bioreagents, Golden, CO, USA), rabbit polyclonal anti-NF-κB and mouse monoclonal IκBα (Cell Signalling Technology, Danvers, MA, USA); anti-tubulin, anti-lamin B, anti-actin, anti-rabbit, anti-mouse and anti-rat horseradish peroxidase–conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). PI/RNase staining buffer and CellQuest Pro were obtained from BD Pharmigen (San Jose, CA, USA). High pure RNA isolation kit was provided by Roche Applied Science (Indianapolis, IN, USA). Superscript II reverse transcriptase was acquired from Invitrogen (Gent, Belgium). PCR primers and fluorogenic probes for mouse RelA (NF-κB), IκBα and β-actin were purchased from Eurogentec (Seraing, Belgium). MIP3-α, MCP primers were received from Dr. C. Mathieu (Laboratory of Clinical and Experimental Endocrinology, KU Leuven, Leuven, Belgium). TX 527 was used at 10 nM because this concentration consistently shows anti-proliferative effects (Verlinden et al., 2000; González Pardo et al., 2010).
Cell lines and transfections
SV-40 immortalized murine endothelial cells stably expressing vGPCR full length (SVEC- vGPCR) were used. Stable over-expression of vGPCR promotes tumour formation when injected into immune-suppressed mice and induces angiogenic lesions similar to those developed in KS (Montaner et al., 2003; González Pardo et al., 2010). Transfected cells were selected with 500 μg·mL−1 G418. Stable SVEC-vGPCR endothelial cells targeted with small hairpin RNA against mouse VDR (vGPCR-shVDR) or control shRNA (vGPCR-shctrl) were obtained by transduction of lentiviral particles (González Pardo et al., 2010). The stable cell lines were selected with 2 μg·mL−1 puromycin and the medium was freshly changed every other day. VDR knock-down was monitored by Western blot analysis.
Cell cycle analysis
Cell cycle distribution was analysed by flow cytometry. SVEC-vGPCR cells were incubated with TX 527 (10 nM) or vehicle (0.01% ethanol) and bortezomib (0.5 nM) alone or in combination with the analogue for 24 h. Then they were trypsinized, washed once with PBS, and fixed in 70% ethanol for at least 1 h at −20°C. Fixed cells were washed two times with PBS containing 0.05% Tween 20 and incubated with PI/RNase staining buffer for 15 min at room temperature in the dark. The stained cells were analysed in a FACS Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The program used for the acquisition and analysis of the samples was the CellQuest Pro.
Proliferation assays
SVEC-vGPCR cells were seeded in 96-well plates at 2000 cells per well. After overnight growth, the cells were starved for 24 h and then treated with TX 527 (10 nM) or vehicle (0.01% ethanol) in the presence or absence of bortezomib (0.5 nM) in triplicate in DMEM 2% FBS for 48 h. CellTiter 96® AQueous one solution cell proliferation assay containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) was used to determine cell proliferation according to the manufacturer's instructions. Absorbance was measured at 490 nm.
NF-κB activity assay
The transcription factor kit for NF-κB p65 from Thermo Scientific (Pierce Biotechnology, Rockford, IL, USA) was used to measure the activity of NF-κB from nuclear extracts following manufacturer's protocol as previously reported (Gonzalez Pardo et al., 2012). Chemiluminescence reaction was captured with a microplate reader Synergy HT Biotek (Biotek Instruments Inc., Highland Park, VT, USA).
SDS-PAGE and Western blot analysis
Whole-cell lysates from cultures treated with TX 527 as indicated for each experiment and their protein content determined by the Bradford procedure (Bradford, 1976) were resolved by SDS-PAGE and transferred to PVDF membranes. Western blot analyses were performed as reported before (Gonzalez Pardo et al., 2006). Antibodies used include monoclonal anti-VDR (1:1500), rabbit anti-NF-κB/p65 (1:2000), anti-IκBα (1:3000), anti-tubulin (1:1500), anti-actin (1:10 000), anti-lamin B (1:1000) combined with anti-rabbit (1:10 000), anti-mouse (1:5000), anti-rat (1:5000) or anti-goat (1:20 000) horseradish peroxidase–conjugated secondary antibodies.
Quantitative real-time PCR
Total RNA for real-time quantitative reverse transcriptase-PCR (qRT-PCR) analysis was isolated using the high pure RNA isolation kit (Roche). RNA (0.5–1 μg) was reverse transcribed using the Superscript II reverse transcriptase and qRT-PCR reactions were performed on the resulting cDNA (2 μL of cDNA; dilution 1/10) in a 7500 RT-PCR system (Applied Biosystems, Carlsbad, CA, USA). Specific primers were used to detect RelA/NF-κB, IκBα, VDR, IL-6, MCP, MIP3α levels and β-actin to normalize gene expression (Gonzalez Pardo et al., 2012). Sequences of forward/reverse primers and detection probes for inflammatory genes (IL-6, MCP, MIP3α) were designed by Dr. C. Mathieu (Laboratory of Clinical and Experimental Endocrinology, KU Leuven, Leuven, Belgium).
Subcellular fractionation
SVEC-vGPCR stabling expressing shRNA against VDR (vGPCR-shVDR) or control shRNA (vGPCR-shctrl) was cultured in p100 dishes, treated with TX 527, and scrapped in ice-cold TES buffer (50 mM Tris–HCl pH 7.4, 1 mM EDTA, 250 mM sucrose) containing 1 mM DTT and protease inhibitors (0.5 mM PMSF, 20 μg·mL−1 aprotinin and 20 μg·mL−1 leupeptin). Homogenization was carried out as previously reported to obtain nuclear and cytosol fraction (Gonzalez Pardo et al., 2012). Anti-lamin B antibody was employed for the immune detection of the nuclear protein marker lamin B in the different fractions and tubulin as cytosolic marker.
Statistical analysis
Data are shown as means ± SD. Results were analysed by the two-tailed t-test to evaluate differences between control (vehicle) and treated conditions 1α,25(OH)2D3, TX 527 or bortezomib. A P-value <0.01 (**) and <0.05 (*) was considered highly statistically significant and statistically significant respectively.
Results
Inhibition of proliferation and cell cycle arrest by TX 527 is NF-κB dependent
To investigate whether the vitamin D analogue TX 527 exerts its growth inhibitory effects through the regulation of NF-κB, proliferation and cell cycle assays were carried out in SVEC-vGPCR cells treated with TX 527 (10 nM) or vehicle (0.01% ethanol) in the presence or absence of bortezomib (0.5 nM) in DMEM with 2% FBS for 24–48 h. Bortezomib at low nanomolar concentration is sufficient to inhibit the activation of NF-κB (Blackburn et al., 2010). Proliferation was measured using the CellTiter 96 AQueous one solution cell proliferation assay as described in Methods section. The results shown in Figure 1A demonstrated that TX 527 significantly decreased the proliferation of SVEC-vGPCR cells (16% after 48 h of incubation). Bortezomib alone or in combination with TX 527 also significantly decreased (26%) the proliferation of SVEC-vGPCR cells. To investigate the effect of TX 527 on the progression of the cell cycle, the percentages of cells in the G0/G1, S and G2/M phases were determined by flow cytometry analysis of propidium iodide stained cells. As shown in Figure 1B, TX 527 increased the percentage of SVEC-vGPCR cells in G0/G1 (from 53 to 61% at 24 h), which was accompanied by a reduction in the percentage of cells in the S phase (from 27 to 22%). When the cells were incubated with the NF-κB inhibitor bortezomib, the response was similar to TX 527 and the combination of both compounds did not significantly enhance the inhibitory effect of the analogue.
Figure 1.
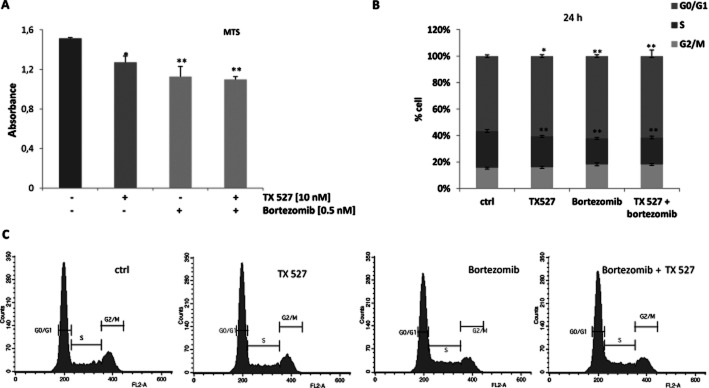
Inhibition of proliferation and cell cycle arrest is induced by TX 527 and bortezomib. SVEC-vGPCR cells were plated and incubated in serum-free DMEM for 24 h. After overnight growth, cells were treated with 10 nM TX 527 or vehicle (ctrl), bortezomib (0.5 nM) or combination of both TX 527 and bortezomib in DMEM 2% BSA for 24–48 h. (A) Proliferation assay was carried out using MTS as described in Methods section. Absorbance was measured at 490 nm. The data from three independent experiments are means ± SEM performed in triplicate. Significant differences between control and treated conditions are indicated. *P < 0.05, **P < 0.01. (B) Cells were then stained with propidium iodide and the distribution of cells in the different phases of the cell cycle was analysed by flow cytometry. The program CellQuest was used for acquisition and analysis of the FACS scans (C). A representative cell cycle analysis and percentages of each phase are shown. Data represent means ± SD of three independent experiments done in triplicate. Significant differences between cell cycle distribution of control and stimulated cells are indicated. *P < 0.05, **P < 0.01. G0/G1= cells in G0/G1 cell cycle phases; S = cells in S cell cycle phase; G2/M = cells in G2/M cell cycle phases.
TX 527 similar to 1α,25(OH)2D3 inhibits NF-κB activity in SVEC-vGPCR
The transcriptional activity of the active form of NF-κB p65 was assayed in nuclear extracts from SVEC-vGPCR. To that end, SVEC-vGPCR cells were treated with 1α,25(OH)2D3 and TX 527 (10 nM, 48 h) and bortezomib (0.5 nM) alone or in combination with TX 527. TNF-α-activated HeLa cell nuclear extracts were included in the assay as positive control. The active transcription factor (from nuclear extracts) bound to DNA consensus sequence or buffer alone (negative control) was incubated with specific primary antibody (NF-κB p65) followed by a secondary HRP-conjugated antibody, and chemiluminescence was finally detected on a luminometer. The results shown in Figure 2 demonstrated that TX 527 significantly decreased the transcriptional activity (65%) of NF-κB being more effective than 1α,25(OH)2D3 (43%). Bortezomib also decreased NF-κB transcriptional activity (68%) and in combination with TX 527 (62%) did not significantly enhance vitamin D analogue inhibitory effect.
Figure 2.
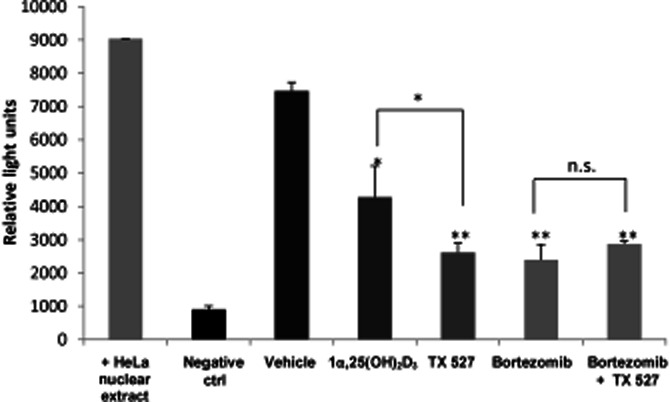
TX 527, similar to 1α,25(OH)2D3, inhibits NF-κB activity in SVEC-vGPCR. SVEC-vGPCR cells were treated with 10 nM 1α,25(OH)2D3 and TX 527 or vehicle (0.01% ethanol), bortezomib (0.5 nM), and combination of both TX 527 and bortezomib in DMEM 2% BSA for 48 h. TNFα-activated HeLa cell nuclear extracts and buffer for antibodies used alone were included in the assay as positive and negative controls respectively. NF-κB activity was measured by chemiluminescence reaction captured with a microplate reader as described in Methods section. The data from each experiment are means ± SD performed in duplicate. Significant differences between control (vehicle) and treated conditions are indicated. *P < 0.05, **P < 0.01.
TX 527 decreases NF-κB and increases IκBα mRNA and protein levels
The classic form of NF-κB is the heterodimer p50/p65 that contains the transcriptional activation domain and is sequestered in the cytoplasm as an inactive complex by the inhibitory protein IκB (Hayden and Ghosh, 2004). To investigate whether the reduced transcriptional activity of NF-κB could be related to changes in the expression levels of p65 and its inhibitory protein, SVEC-vGPCR cells were plated and incubated with TX 527 (10 nM) or vehicle (0.01% ethanol) in the presence of 2% FBS for 3–72 h. Total RNA was isolated and reverse transcribed followed by qRT-PCR using specific primers to detect NF-κB and IκBα mRNA levels and β-actin mRNA to normalize gene expression. The results shown in Figure 3 demonstrated that TX 527 significantly decreased NF-κB mRNA levels (20%) and increased IκBα mRNA levels in a time-dependent fashion (50–80%). In parallel experiments, total proteins from whole-cell lysates were prepared and subject to Western blot analysis with anti-NF-κB and IκBα antibodies. The results in Figure 3B showed that TX 527 moderately reduced the protein levels of NF-κB and significantly increased IκBα protein levels.
Figure 3.
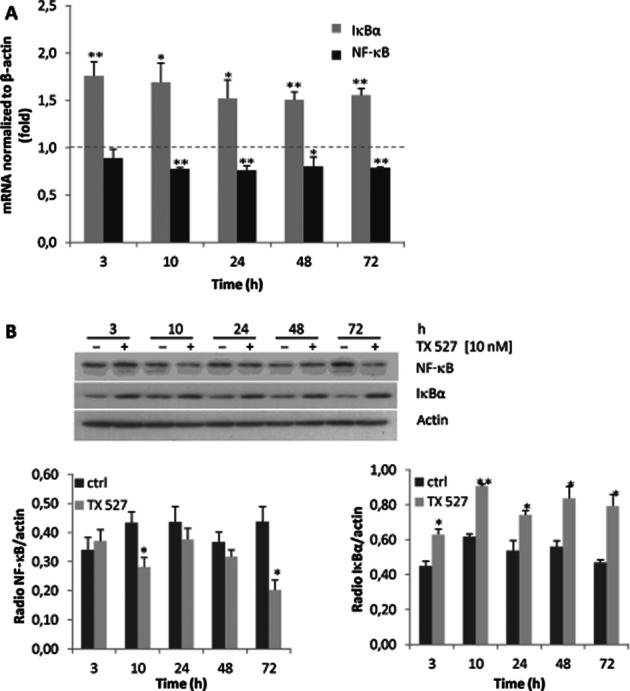
TX 527 decreases NF-κB and increases IκBα mRNA and protein levels. SVEC-vGPCR cells were cultured and treated with 10 nM TX 527 or vehicle for 3–72 h as described in Methods section. (A) Total RNA was extracted and reverse transcribed (0.5–1 μg). At each of these time points, gene expression of RelA (p65/NF-κB) IκBα and β-actin was assessed by qRT-PCR analysis. Data are expressed as a ratio between TX 527 versus vehicle-treated samples normalized to β-actin RNA levels. The statistical significance of the data was evaluated using Student's t-test. **P < 0.01, *P < 0.05. (B) Cell lysates were prepared and subject to Western blot analysis with anti-NF-κB, IκBα and actin antibodies. The bands were quantified by densitometry and the ratio between NF-κB, IκBα versus total protein (actin) from control and treated conditions was analysed statistically by Student's t-test. **P < 0.01, *P < 0.05. A representative immunoblot of three independent experiments is shown.
Decreased nuclear levels of NF-κB protein induced by TX 527 are VDR dependent
To investigate if TX 527-dependent increase of IκBα could retain p65/NF-κB in the cytosol, thus decreasing the activity of NF-κB, the localization of NF-κB and IκBα was evaluated. SVEC-vGPCR cells were cultured and treated with TX 527 (10 nM, 3–24 h) or vehicle (0.01% ethanol). The cells were collected in TES buffer and subject to differential centrifugation to obtain enriched nuclear and cytosolic fractions, as described in Methods section. Western blot analysis with anti-NF-κB and IκBα antibodies was performed and lamin B and tubulin were used to show loading controls of nuclear and cytosolic fraction respectively. The results in Figure 4A showed that NF-κB was present in the nucleus and upon TX 527 exposure (3–24 h) its localization decreased in the nucleus and increased in cytosol, whereas IκBα increases in both compartments. To determine whether an increase in the expression of the VDR was part of the mechanism of action, stable SVEC-vGPCR endothelial cells targeted with small hairpin RNA against mouse VDR (vGPCR-shVDR) or control shRNA (vGPCR-shctrl) were cultured and treated with TX 527 (10 nM, 24 h) or vehicle (0.01% ethanol). The results shown in Figure 4B demonstrated that TX 527 increased the amount of VDR and induced its translocation to the nucleus. The translocation of NF-κB was decreased in SVEC-vGPCR cells when treated with TX 527, whereas NF-κB nuclear protein levels were restored when the VDR was knocked down in vGPCR-shVDR cells. The localization of IκBα protein was not affected.
Figure 4.
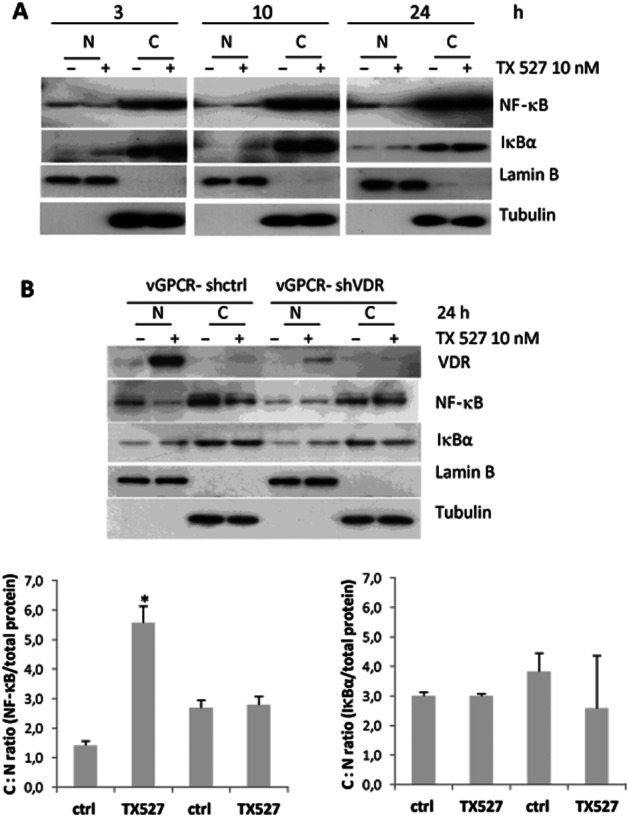
Decreased levels of NF-κB protein induced by TX 527 are VDR dependent. Stable SVEC-vGPCR cells were cultured and treated with TX 527 (10 nM, 3–24 h) or vehicle (0.01% ethanol). Cells were collected in TES buffer and subject to differential centrifugation to obtain enriched nuclear (N) and cytosolic fractions (C) as described in Methods section. (A) Western blot analyses with anti-NF-κB and IκBα antibodies were performed and lamin B and tubulin were used to show loading controls of nuclear and cytosolic fraction respectively. (B) SVEC-vGPCR cells targeted with small hairpin RNA against mouse VDR (vGPCR-shVDR) or control shRNA (vGPCR-shctrl) were treated with TX 527 (10 nM, 24 h) or vehicle (0.01% ethanol), enriched nuclear and cytosolic fractions were obtained as in (A) and Western blots were also performed with VDR antibody. Representative Western blots and its quantification by Image J (bar graphs) shown are representative of three independent experiments. The statistical significance of the data was evaluated using Student's t–test. *P < 0.05.
Dependence of VDR on NF-κB and IκBα mRNA expression
To further investigate the participation of VDR on the mRNA expression of NF-κB and its inhibitory protein IκBα, the stable VDR knock-down cell line vGPCR-shVDR was used. To that end, vGPCR-shctrl and vGPCR-shVDR cells were plated and incubated with TX 527 (10 nM) or vehicle (0.01% ethanol) in the presence of 2% FBS for 24 h. Total RNA was isolated and reverse transcribed followed by qRT-PCR using specific primers as described in Figure 3A. First, VDR expression was evaluated to determine the VDR knock-down (Figure 5A). In control cells (vGPCR-shctrl), VDR and IκBα mRNA significantly increased whereas NF-κB decreased upon TX 527 treatment. The induction of IκBα and inhibition of NF-κB expression by TX 527 were blunted when VDR was knocked down in vGPCR-shVDR cells (Figure 5A). To further investigate whether the changes in mRNA levels were reflected in changes in protein expression, Western blots from parallel experiments were performed (Figure 5B). Cell lysates were prepared and subject to immunoblot analysis with anti-VDR, NF-κB and IκBα antibodies. The results in Figure 5B showed that although the VDR induced by TX 527 was not completely blocked, reduced NF-κB and increased IκBα protein levels were still observed. As bortezomib has shown to inhibit NF-κB activity and did not enhance the effect of TX 527 when they were incubated together, this raises the possibility that both agents could act by the same mechanism. To evaluate this possibility, SVEC-vGPCR cells were cultured and treated with TX 527 (10 nM, 24 h) or vehicle (0.01% ethanol) in the presence or absence of bortezomib (0.5 nM) in 2% FBS. As shown in Figure 5C, bortezomib alone reduced NF-κB and increased IκBα mRNA expression similarly to TX 527. No additive effects were found when both bortezomib and TX 527 were added together.
Figure 5.

Dependence of VDR on NF-κB and IκBα mRNA expression modulated by TX 527. Stable SVEC-vGPCR cells targeted with small hairpin RNA against mouse VDR (vGPCR-shVDR) or control shRNA (vGPCR-shctrl) were cultured and treated with TX 527 (10 nM, 24 h) or vehicle (0.01% ethanol) for 24 h (A and B) or in the presence/absence of bortezomib (0.5 nM) (C). (A and C) Total RNA was extracted and reverse transcribed (0.5–1 μg). Gene expression of RelA (NF-κB), IκBα and VDR was assessed by qRT-PCR analysis, normalized to β-actin RNA levels. The statistical significance of the data was evaluated using Student's t-test. **P < 0.01, *P < 0.05. The data shown are representative of three independent experiments done in triplicate. (C) In parallel, total cell lysates were prepared and subject to Western blot analysis with anti-NF-κB, -IκBα and -tubulin antibodies. Representative immunoblots from three independent experiments are shown.
Down-regulation of NF-κB by TX 527 controls inflammatory gene expression
Expression and secretion of pro-inflammatory mediators are important for autocrine and paracrine neoplasia induced by vGPCR in endothelial cells (Martin and Gutkind, 2009; Mesri et al., 2010). We investigated whether TX 527 modulates cytokine IL-6 and chemokines CCL2/MCP and CCL20/MIP3α gene expression and whether the effects are VDR dependent. To this end, vGPCR-shctrl and vGPCR-shVDR cells were plated and incubated with TX 527 (10 nM) or vehicle (0.01% ethanol) in the presence of 2% FBS for 24 h. RNA was isolated and reverse transcribed followed by qRT-PCR using specific primers as described in Methods section. As shown in Figure 6A, TX 527 significantly decreased the expression of IL-6 (60%, P < 0.01), MCP (57%, P < 0.01) and MIP3α (39%, P < 0.01) in vGPCR endothelial cells (vGPCR-shctrl) compared with vehicle-treated cells. When the VDR was knocked down, the effect of TX 527 on the expression of IL-6 was partially reversed (21%, P = 0.03) whereas MCP and MIP3α gene expression was not statistically affected. To investigate the involvement of NF-κB on the effects of TX 527, SVEC-vGPCR cells incubated with NF-κB inhibitor bortezomib (0.5 nM) alone or in combination with TX 527 (10 nM) were cultured for 24 h (Figure 6B). Bortezomib by itself significantly reduced the expression of IL-6 (33%, P < 0.01), MCP (24%, P < 0.01) and MIP3α (80%, P < 0.01) compared with vehicle-treated cells but did not significantly potentiate the reduction in the expression of IL-6 (17%, P = 0.22) and MCP (10%, P = 0.56) when combined with TX 527; however, down-regulation of MIP3α by TX 527 was greatly enhanced by bortezomib (73%, P < 0.01).
Figure 6.

Down-regulation of inflammatory genes induced by TX 527 and bortezomib. Stable SVEC-vGPCR cells targeted with small hairpin RNA against mouse VDR (vGPCR-shVDR) or control shRNA (vGPCR-shctrl) were cultured and treated with TX 527 (10 nM, 24 h) or vehicle (0.01% ethanol) for 24 h (A and B) or in the presence/absence of bortezomib (0.5 nM) (B). Total RNA was extracted and reverse transcribed (0.5–1 μg). Gene expression of IL-6, MCP and MIP3α was assessed by qRT-PCR analysis, normalized to β-actin RNA levels. The statistical significance of treated versus control (vehicle) conditions was evaluated using Student's t-test and is explained in detail in Results section. The data shown are representative of three independent experiments done in triplicate. Results were analysed by the two-tailed t-test. P-value and statistical significance are given in the Results section.
Discussion and conclusions
1α,25(OH)2D3 and its synthetic analogues have anti-proliferative effects in many types of cancer and also possess potent immune modulatory activities; however, the molecular mechanism of 1α,25(OH)2D3-mediated inflammatory gene alteration and its participation in tumour development is not yet fully understood. In addition to its principal function in physiological immune reactions, NF-κB plays a pivotal role in the generation and maintenance of malignancies (Nishikori, 2005). The classic form of NF-κB is the p65/p50 heterodimer that contains the transcriptional activation domain and is sequestered in the cytoplasm as an inactive complex by IκBα (Baldwin, 1996). Acute stimuli such as TNF-α, LPS or phorbol myristate acetate lead to the activation of IκB kinases which in turn phosphorylate Ser32 and Ser36 within the N-terminal response domain of IκB (Karin and Ben-Neriah, 2000). Phosphorylated IκB undergoes ubiquitination-dependent proteolysis and the release of IκB unmasks the nuclear localization signal and results in the translocation of NF-κB to the nucleus, followed by the activation of specific target genes (Karin and Ben-Neriah, 2000). It has been described that IκB proteins play an important role in the termination of NF-κB activation. Newly synthesized IκBα enters the nucleus and binds NF-κB, thereby enhancing its dissociation from the DNA (the affinity of NF-κB to IκB appears to be higher than its affinity to κB sites on DNA) and causing its re-exportation to the cytoplasm by means of a nuclear export sequence present on IκBα. (Arenzana-Seisdedos et al., 1997). In this work, we have demonstrated that the NF-κB pathway is regulated by vitamin D analogue TX 527 at various levels. Moreover, data show that TX 527 through reduction of NF-κB activity inhibits the proliferation of SVEC-vGPCR cells through the induction of cell cycle arrest (Figures 1 and 2). Reduced NF-κB transcriptional activity was also observed in response to 1α,25(OH)2D3 in VDR-positive MCF-7 breast cancer cells (Tse et al., 2010) and our previous work in KS cellular model (Gonzalez Pardo et al., 2012). Moreover, NF-κB and IκBα mRNA and protein levels (Figure 3) were also modulated by the analogue similar to 1α,25(OH)2D3 (Tse et al., 2010). Of interest, in connection to the mode of action of TX 527, it has been reported that 20-hydroxy-vitamin D3, a product of vitamin D3 hydroxylation by P450scc, decreases NF-κB activity by increasing IκBα levels in human keratinocytes (Janjetovic et al., 2009). In our research, TX 527 decreases nuclear translocation of NF-κB by a mechanism that depends on VDR expression (Figure 4). Supporting our data, TX 527 has been found to reduce nuclear translocation of NF-κB in peripheral blood mononuclear cells from patients with Chron's disease compared with healthy subjects treated with TX 527 possibly through a decrease in TNF-α production (Stio et al., 2007). Furthermore, in keratinocytes lacking VDR, 20-hydroxy-vitamin D3 did not affect IkBα mRNA levels, which indicates that VDR is required for its action on NF-κB activity (Janjetovic et al., 2009). More recently, it has been suggested that 1α,25(OH)2D3 suppressed inflammation (induced by expression of plasminogen activator inhibitor-1) by blocking NF-κB activity and its nuclear translocation through the induction of IκBα in mouse embryonic fibroblasts (MEFs) (Chen et al., 2011). In addition, the Vitamin D analogue paricalcitol has been shown to inhibit renal inflammation by promoting VDR-mediated sequestration of NF-κB and inhibiting the ability of p65 to trans-activate gene transcription (Tan et al., 2008). In preliminary observations, we found that phosphorylated IκBα was present in vehicle-treated vGPCR cells, and after TX 527 stimulation total IκBα increased and the ratio p-IκBα/total IκBα decreased and thus NF-κB activation. A possible explanation is that p-IκBα undergoes proteosomal degradation and the bulk of IκBα increases rapidly rather than its phosphorylation. Nevertheless, further experiments are needed to confirm this hypothesis. We have also demonstrated that TX 527 induced a VDR-dependent up-regulation of inhibitory protein IκBα (Figure 5A). Our results are consistent with a study that shows a marked decrease of basal levels of IκBα in VDR-deficient MEFs compared with VDR-positive MEFs (Sun et al., 2006). A previous study showed that IκBα may be a direct target gene of activated VDR because a DR3-type vitamin D-responsive element was found in IκBα (NFκB1a) gene promoter (Wang et al., 2005). On the contrary, other reports showed that nuclear hormone receptors, such as PPAR (Delerive et al., 2002) and GR (Deroo and Archer, 2001), may induce IκBα transcription by a DNA-binding-independent manner. The expression of pro-inflammatory mediators is important for autocrine and paracrine neoplasia induced by vGPCR in endothelial cells (Martin and Gutkind, 2009), suggesting that inhibition of pro-inflammatory mediators could be important as therapeutic strategy. We demonstrated that TX 527 reduced cytokine IL-6 and chemokines CCL2/MCP and CCL20/MIP3α gene expression by a mechanism that did not require direct participation of the VDR but involves the inhibition of activated NF-κB in SVEC-vGPCR cells (Figure 6). Several studies have reported that 1α,25(OH)2D3 and analogues exhibit anti-inflammatory and immune modulatory effects in various cell types (Yee et al., 2005; Equils et al., 2006; Suzuki et al., 2009; Baeke et al., 2010). In our cell system, we observed that bortezomib inhibits NF-κB activity and modulates inflammatory gene expression and that its action did not enhance TX 527 effects (Figures 5B and 6), therefore suggesting that both agents may act through the same mechanism. Taken together, our findings strongly suggest that the vitamin D analogue TX 527 regulates the NF-κB pathway and indicates that TX 527 through reduction of NF-κB activity inhibited the proliferation of SVEC-vGPCR cells inducing cell cycle arrest. The analogue strongly increased IκBα mRNA and protein levels, and decreased the nuclear translocation of NF-κB by a mechanism that depends on VDR expression. The inhibition of nuclear NF-κB activity is implicated in TX 527-reduced cytokine IL-6 and chemokines CCL2/MCP and CCL20/MIP3α gene expression in SVEC-vGPCR cells.
In conclusion, vitamin D analogue TX 527 exerts anti-proliferative and anti-inflammatory effects in KS (SVEC-vGPCR) cells by down-regulation of the NF-κB pathway. Due to its hypocalcemic activity, TX 527 may be a suitable candidate to treat hyper-proliferative and inflammatory disorders, although it is necessary to test its potential therapeutic properties in animal models bearing vGPCR tumours.
Acknowledgments
This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT), Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET) and Universidad Nacional del Sur. We also wish to thank Dr. Silvio Gutkind for his support and cell donation. Fund for Scientific Research (FWO G.0587.09 and G.0859.11) and the Catholic University of Leuven (GOA 2009/10, EF05/007) are also acknowledged.
Glossary
- CCL2/MCP
chemokine (C-C motif) ligand 2/monocyte chemotactic protein-1
- CCL20/MIP3α
chemokine (C-C motif) ligand 20/macrophage inflammatory protein-3
- IL-8/CXCL8
interleukin 8
- IκBα
nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, α
- KS
Kaposi sarcoma; MEFs, mouse embryonic fibroblasts
- MIP-1/CCL3
macrophage inflammatory protein 1-α/chemokine (C-C motif) ligand 3
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- qRT-PCR
real-time quantitative reverse transcriptase-PCR
- RelA
factor de transcripción p65
- RelB
v-rel reticuloendotheliosis viral oncogene homologue B
- SVEC-vGPCR
endothelial cells transformed by vGPCR
- TX 527
[19-nor-14,20-bisepi-23-yne-1,25(OH)2D3]
- VDR
vitamin D receptor
- 1α,25(OH)2D3
1α,25-dihydroxyvitamin D3
- vGPCR
viral GPCR
- vGPCR-shVDR or -(shctrl)
endothelial cells targeted with small hairpin RNA against mouse VDR or control
Conflict of interest
None.
References
- Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, et al. Nuclear localization of IkBα promotes active transport of NF-kB from the nucleus to the cytoplasm. J Cell Sci. 1997;110:369–378. doi: 10.1242/jcs.110.3.369. [DOI] [PubMed] [Google Scholar]
- Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol. 2010;10:482–496. doi: 10.1016/j.coph.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Baeke F, Korf H, Overbergh L, Verstuyf A, Thorrez L, Van Lommel L, et al. The vitamin D analog, TX 527, promotes a human CD4+ CD25 high CD127 low regulatory T cell profile and induces a migratory signature specific for homing to sites of inflammation. J Immunol. 2011;186:132–142. doi: 10.4049/jimmunol.1000695. [DOI] [PubMed] [Google Scholar]
- Baldwin AS. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese F, et al. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S β5-subunit. Biochem J. 2010;430:461–476. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillon R, Verlinden L, Eelen G, De Clercq P, Vandewalle M, Mathieu C, et al. Mechanisms for the selective actions of vitamin D analogs. J Steroid Biochem Mol Biol. 2005;97:21–30. doi: 10.1016/j.jsbmb.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for quantification of micrograms quantities of proteins utilizing the principle of protein biding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Chen Y, Kong J, Sun T, Li G, Szeto FL, Liu W, et al. 1,25-Dihydroxyvitamin D3 suppresses inflammation-induced expression of plasminogen activator inhibitor-1 by blocking nuclear factor-κB activation. Arch Biochem Biophys. 2011;507:241–247. doi: 10.1016/j.abb.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684–700. doi: 10.1038/nrc2196. [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Vanden Berghe W, Fruchart JC, Haegeman G, Staels B. DNA binding-independent induction of Ikappa B alpha gene transcription by PPAR alpha. Mol Endocrinol. 2002;16:1029–1039. doi: 10.1210/mend.16.5.0826. [DOI] [PubMed] [Google Scholar]
- Deroo BJ, Archer TK. Glucocorticoid receptor activation of the I kappa B alpha promoter within chromatin. Mol Biol Cell. 2001;12:3365–3374. doi: 10.1091/mbc.12.11.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eelen G, Gysemans C, Verlinden L, Vanoirbeek De Clercq EP, Van Haver D, Mathieu C, et al. Mechanism and potential of the growth-inhibitory actions of vitamin D and analogs. Curr Med Chem. 2007;14:1893–1910. doi: 10.2174/092986707781058823. [DOI] [PubMed] [Google Scholar]
- Equils O, Naiki Y, Shapiro AM, Michelsen K, Lu D, Adams J, et al. 1,25-Dihydroxyvitamin D inhibits lipopolysaccharide-induced immune activation inhuman endothelial cells. Clin Exp Immunol. 2006;143:58–64. doi: 10.1111/j.1365-2249.2005.02961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D, Malloy PJ, Krishnan AV, Balint E. Vitamin D: biology, action and clinical implications. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd edn. Vol. 1. San Diego: Academic Press; 2007. pp. 317–382. [Google Scholar]
- Gonzalez Pardo V, Boland R, de Boland AR. 1α,25(OH)2-vitamin D3 stimulates intestinal cell p38 MAPK activity and increases c-fos expression. Int J Biochem Cell Biol. 2006;38:1181–1190. doi: 10.1016/j.biocel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Gonzalez Pardo V, D'Elia N, Verstuyf AM, Boland R, Boland AR. NFκB pathway is down-regulated by 1 α,25(OH)2- vitamin D3 in endothelial cells transformed by Kaposi sarcoma-associated herpesvirus G protein coupled receptor. Steroids. 2012;77:1025–1032. doi: 10.1016/j.steroids.2012.05.006. [DOI] [PubMed] [Google Scholar]
- González Pardo V, Martin D, Gutkind JS, Verstuyf A, Bouillon R, Russo de Boland A, et al. 1alpha,25-dihydroxyvitamin D3 and its TX527 analog inhibit the growth of endothelial cells transformed by Kaposi sarcoma-associated herpes virus G protein-coupled receptor in vitro and in vivo. Endocrinology. 2010;151:23–31. doi: 10.1210/en.2009-0650. [DOI] [PubMed] [Google Scholar]
- Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–787. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Janjetovic Z, Zmijewski MA, Tuckey RC, DeLeon DA, Nguyen MN, Pfeffer LM, et al. 20-Hydroxycholecalciferol, product of vitamin D3 hydroxylation by P450scc, decreases NF-kappaB activity by increasing IkappaB alpha levels in human keratinocytes. PLoS ONE. 2009;19:e5988. doi: 10.1371/journal.pone.0005988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-kB activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Krishnan AV, Feldman D. Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annu Rev Pharmacol Toxicol. 2011;51:311–336. doi: 10.1146/annurev-pharmtox-010510-100611. [DOI] [PubMed] [Google Scholar]
- Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin North Am. 2010;39:401–418. doi: 10.1016/j.ecl.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Gutkind JS. Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene. 2009;27:S31–S42. doi: 10.1038/onc.2009.351. [DOI] [PubMed] [Google Scholar]
- Martin D, Galisteo R, Ji Y, Montaner S, Gutkind JS. An NFκB gene expression signature contributes to Kaposi's sarcoma virus vGPCR-induced direct and paracrine neoplasia. Oncogene. 2008;27:1844–1852. doi: 10.1038/sj.onc.1210817. [DOI] [PubMed] [Google Scholar]
- Mesri EA, Cesarman E, Boshoff C. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/ protein kinase B. Cancer Res. 2001;61:2641–2648. [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, He Y, et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23–26. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Servitja JM, Ramsdell AK, Barac A, Sawai ET, et al. The small GTPase Rac1 links the Kaposi sarcoma associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia. Blood. 2004;104:2903–2911. doi: 10.1182/blood-2003-12-4436. [DOI] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Ramsdell AK, Martin D, Hu J, Sawai ET, et al. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic target for the treatment of Kaposi's sarcoma. Cancer Res. 2006;66:168–174. doi: 10.1158/0008-5472.CAN-05-1026. [DOI] [PubMed] [Google Scholar]
- Nishikori M. Classical and alternative NF-kB activation pathways and their roles in lymphoid malignancies. J Clin Exp Hematopathol. 2005;45:15–24. [Google Scholar]
- Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, et al. The Kaposi's sarcoma-associated herpes virus G protein coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60:4873–4880. [PubMed] [Google Scholar]
- Sodhi A, Montaner S, Miyazaki H, Gutkind JS. MAPK and Akt act cooperatively but independently on hypoxia inducible factor-1alpha in rasV12 upregulation of VEGF. Biochem Biophys Res Commun. 2001;287:292–300. doi: 10.1006/bbrc.2001.5532. [DOI] [PubMed] [Google Scholar]
- Stio M, Martinesi M, Bruni S, Treves C, Mathieu C, Verstuyf A, et al. The vitamin D analogue TX 527 blocks NF-Bα activation in peripheral blood mononuclear cells of patients with Crohn's disease. J Steroid Biochem Mol Biol. 2007;103:51–60. doi: 10.1016/j.jsbmb.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Sun J, Kong J, Duan Y, Szeto FL, Liao A, Madara JL, et al. Increased NF-kB activity in fibroblasts lacking the vitamin D receptor. Am J Physiol Endocrinol Metab. 2006;291:315–322. doi: 10.1152/ajpendo.00590.2005. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Ichiyama T, Ohsaki A, Hasegawa S, Shiraishi M, Furukawa S. Anti-inflammatory effect of 1alpha,25-dihydroxyvitamin D3 in human coronary arterial endothelial cells: implication for the treatment of Kawasaki disease. J Steroid Biochem Mol Biol. 2009;113:134–138. doi: 10.1016/j.jsbmb.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Tan X, Wen X, Liu Y. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling. J Am Soc Nephrol. 2008;19:1741–1752. doi: 10.1681/ASN.2007060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse AK, Zhu GY, Wan CK, Shen XL, Yu ZL, Fong WF. 1alpha,25-Dihydroxyvitamin D3 inhibits transcriptional potential of nuclear factor kappa B in breast cancer cells. Mol Immunol. 2010;47:1728–1738. doi: 10.1016/j.molimm.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Van Etten E, Branisteanu DD, Verstuyf A, Waer M, Bouillon R, Mathieu C. Analogs of 1,25-dihydroxyvitamin D3 as dose-reducing agents for classical immunosuppressants. Transplantation. 2000;69:1932–1942. doi: 10.1097/00007890-200005150-00032. [DOI] [PubMed] [Google Scholar]
- Van Etten E, Decallonne B, Verlinden L, Verstuyf A, Bouillon R, Mathieu C. Analogs of 1alpha,25-dihydroxyvitamin D3 as pluripotent immunomodulators. J Cell Biochem. 2003;88:223–226. doi: 10.1002/jcb.10329. [DOI] [PubMed] [Google Scholar]
- Verlinden L, Verstuyf A, Van Camp M, Marcelis S, Sabbe K, Zhao XY, et al. Two novel 14-epi-analogues of 1,25-dihydroxyvitamin D3 inhibit the growth of human breast cancer cells in vitro and in vivo. Cancer Res. 2000;60:2673–2679. [PubMed] [Google Scholar]
- Wang TT, Tavera-Mendoza LE, Laperriere D, Libby E, MacLeod NB, Nagai Y, et al. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol Endocrinol. 2005;19:2685–2695. doi: 10.1210/me.2005-0106. [DOI] [PubMed] [Google Scholar]
- Yee YK, Chintalacharuvu SR, Lu J, Nagpal S. Vitamin D receptor modulators for inflammation and cancer. Mini Rev Med Chem. 2005;5:761–778. doi: 10.2174/1389557054553785. [DOI] [PubMed] [Google Scholar]