

Abstract
Prostate cancer is a clinically and molecularly heterogeneous disease. Understanding the biologic underpinning of prostate cancer is necessary to best determine how biology is associated with the risk of disease progression and how this understanding might provide insight into the development of novel therapeutic approaches. The focus of this review is on the recently identified common ETS and non-ETS gene rearrangements in prostate cancer. Although multiple molecular alterations have been detected in prostate cancer, a basic understanding of gene fusion prostate cancer should help explain the clinical and biologic diversity, providing a rationale for a molecular subclassification of the disease.
Keywords: ETS, gene fusion, next generation sequencing, prostate cancer, TMPRSS2:ERG, translocation
Introduction
There is an emerging view of prostate cancer suggesting a high degree of molecular complexity involving common recurrent gene fusions,1 large genomic rearrangements2 and common recurrent mutations (Barbieri et al., in press). This heterogeneity might be best viewed as a collection of homogenous subgroups defined by somatic copy number alterations, mutations and rearrangements. This review focuses on the common recurrent gene fusions involving most commonly ETS transcription factors. The most common gene fusion in prostate cancer is the TMPRSS2:ERG fusion.1 Since the initial discovery in 2005, a great deal has been learned about this common molecular event. One of the most important concepts regarding the TMPRSS2:ERG fusion is to what extent the mutation plays a role as a biologically driving event (Figure 1).
Figure 1.
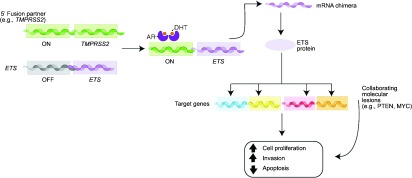
How ETS fusions function in prostate cancer. After the gene fusion even occurs between TMPRSS2 and an ETS gene, the TMPRSS2 5′prime promoter acts as an ‘on-switch' in the presence of androgens and in some settings estrogen. The chimeric transcript encodes a truncated version of the ETS transcription factor. The overexpression of these ETS genes in the setting of the prostate gland act as oncogenes that set off a series of abnormal molecular events including increased cell invasion. A number of cooperating molecular events are believed to also be critical in disease progression including PTEN loss. DHT, dihydrotestosterone; AR, androgen receptor.
Hematological malignancies, such as acute myeloid leukemia, are often subtyped based on the recurrent cytogenetic and molecular aberrations identified. Therefore, the recent discovery that at least 50% of prostate cancers harbor recurrent gene rearrangements resulting in the fusion of genes,1 may enable molecular subtyping of prostate cancers similar to what has been established for leukemias and lymphomas. Most often, these fusions juxtapose a hormone sensitive promoter and an ETS transcription factor that is believed to act in an oncogenic role conferring a distinct biology to this tumor. These fusions, unlike the bcr-abl fusion in chronic myelogenous leukemia (CML), do not create a chimeric protein but instead overexpress an ETS transcription factor in a normal, albeit truncated form. Although other molecular events play a role in prostate cancer development and progression, defining prostate cancer based on the presence or absence of the different gene fusions that drive cancer development provides novel insight into disease heterogeneity.
Discovery of gene fusion prostate cancer
Recurrent chromosomal aberrations were thought to be primarily characteristic of leukemias, lymphomas and sarcomas. Epithelial tumors (i.e., carcinomas), which are the most common human tumors contributing to a large fraction of morbidity and mortality associated with human cancer, comprised less than 1% of the known, disease-specific chromosomal rearrangements. The discovery of the ETS family transcription factor gene fusions by Tomlins et al.1 in 2005 dramatically changed this view of the solid tumor biology. Key to the discovery of TMPRSS2:ETS gene fusions was the development of a simple, statistical approach, termed ‘Cancer Outlier Profile Analysis', to identify oncogene profiles in a subset of samples within publicly available cancer profiling data sets, characteristic of genes commonly associated with known genomic rearrangements (reviewed by Rubin and Chinnaiyan3 and Hanauer et al.4). The application of Cancer Outlier Profile Analysis in prostate cancer microarray experiments revealed two consistently high-scoring and mutually exclusive candidates across 50% of prostate cancer samples that were members of the ETS family of transcription factors, ERG and ETV1. Further experiments revealed fusions of the 5′-untranslated region of TMPRSS2 (21q22.3) with the ETS transcription factor family members, either ERG (21q22.2), ETV1 (7p21.2),1 or ETV4,5 suggesting a novel mechanism for overexpression of the ETS genes in prostate cancer.
A multitude of gene fusions in prostate cancer
Since the initial discovery of ETS fusions in prostate cancer, a number of more recent studies have identified fusion events involving additional ETS family members (i.e., ELK4,6, 7 novel 5 prime (upstream) partners) and a class of non-ETS based fusions. Based on these discoveries, we have developed a classification system (Figure 2) comprised of three categories: (1) fusions involving ETS gene family members (ERG, ETV1, ETV4, ETV5 and ELK4); (2) RAF kinase family fusions and other mutually exclusive molecular events including (3) SPINK1 overexpressing cases and (4) SPOP mutant cases2, 8 (also see Barbieri et al., in press).
Figure 2.

Prostate cancer gene fusion classification. The ongoing effort to screen prostate cancer patients for gene fusions, in combination with the recent technology advances, has resulted in comprehensive gene fusion landscape. This schematic highlights gene fusions categorized into ETS rearrangements and RAF kinase gene fusions. A number of rare gene fusions that occur at less than 1% frequency have been excluded. Two other non-fusion classes that are characterized by mutual exclusivity with ETS rearrangements SPINK1 and SPOP mutations. The percentages highlight the estimated frequency of each gene fusion based on published screens.
The largest category, ETS fusions, is comprised of the highly recurrent TMPRSS2:ERG fusion, which contrasts the remaining, less common, fusion events. Interestingly, the ETS family member fusions involve a diverse set of five prime upstream partners, as exemplified by ETV1 having nine different fusion partners. In addition to TMPRSS2, three additional androgen responsive 5′ partners SLC45A3,9, 10 HERPUD111 and NDRG112 have been found to fuse with ERG. However, many of the 5′ partners appear to fuse to multiple ETS family members, such as SLC45A3 (-ERG, -ELK4, -ETV1 and -ETV5) and TMPRSS2 (-ERG, -ETV1, -ETV4 and -ETV5), both of which are androgen responsive. The majority of these androgen receptor-regulated promoters confer an organ specificity to these gene fusions. Interestingly, as these events occur as early as the precursor lesion, high-grade PIN, they suggest one of the first hormonally regulated mutations in prostate cancer development. This may have implication in how individual men respond to endogenous hormone and/or hormone manipulation as part of systemic treatment for more aggressive prostate cancer.
Recent advances in next-generation transcriptome sequencing facilitated the discovery of RAF kinase gene fusions, SLC45A3-BRAF, ESRP1-RAF1 and RAF1-ESRP1 in advanced prostate cancers.13 Although rare, detected in about 1% of prostate cancers, RAF kinase fusions represent the first ‘driver' fusion in prostate cancers that do not involve an ETS family member. We can also include two other categories, where the non-fusion molecular events are mutually exclusive to ETS rearrangements. SPINK1-positive prostate cancers is included in the classification since the outlier expression of SPINK1 occurs in ETS rearrangement-negative prostate cancers and therefore defines a specific subclass of prostate cancers.14 Another category are the SPOP mutated prostate cancers2, 8 (also see Barbieri et al., in press). SPOP is the most frequently mutated gene in localized prostate cancer seen mutated in around 13% of all studied cases (Barbieri et al., in press). SPOP encodes the substrate-binding subunit of a Cullin-based E3 ubiquitin ligase. Importantly, in over 300 cases studied to date, SPOP mutations never occur concurrently with ETS rearrangements (Barbieri et al., in press). One can argue about the immediate utility of describing cancer using a molecular classification with no proven drug targets or clear differences in clinical behavior. Yet, the emergence of these distinct molecular classes with distinct differences in hormonal regulation could prove an important even in the short term. Clinical trials should account for these key molecular events to help interpret outcomes. Eventually, small molecule targeting specific events such as the TMPRSS2:ERG rearrangement could alter the outcome of prostate cancer disease progression.</tpb -4pt?>
The TMPRSS2:ETS fusion is a common, early driver of prostate cancer
Microscopic examination of prostate cancers using an fluorescence in situ hybridization assay reveal that gene fusion occurs in neoplastic cells but not in adjacent benign nuclei or stromal cells.1, 15, 16 A larger study drawn from a wide spectrum of benign prostatic lesions and precursors of prostate cancer17 failed to detect TMPRSS2:ERG fusion in benign prostate tissue, BPH or PIA (also commonly referred to as ‘focal prostate atrophy' or ‘prostate atrophy' see de Marzo et al.18 for a review). The TMPRSS2:ERG fusion was observed in approximately 20% of high-grade PIN lesions intermingled with prostate cancer that carried the same fusion pattern. This was the same frequency previously detected by Cerveira et al. using an RT-PCR based assay.19 More recently, immunohistochemistry has been employed to evaluate the gene fusions in situ.20 Using an antibody highly specific for ERG rearrangements, one detect the earliest overexpression of the ERG oncogene in the morphologic area of high-grade PIN but not in directly adjacent benign prostate tissue. A significant clinical implication for this finding is the potential utility of assessing the TMPRSS2:ERG fusion status in problematic prostate needle core biopsies with high-grade PIN and adjacent small atypical glands.
Multiple, independent studies have corroborated the initial observation that TMPRSS2:ETS fusions are common in prostate cancer.19, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 While most studies have focused on the dominant rearrangement TMPRSS2:ERG fusion, a variety of other fusions involving TMPRSS2 and other 5′ prime partners have been described but appear to be less common.5, 32, 35, 36, 37 The prevalence of TMPRSS2:ERG prostate cancer has been reported to range from 40% to 70%, depending on the clinical cohorts investigated. The first large clinical study on a German prostatectomy cohort reported approximately 50% of cases with a TMPRSS2:ERG fusion.15 Multiple, retrospective studies from prostate-specific antigen (PSA)-screened prostatectomy cohorts have reported frequencies of the TMPRSS2:ERG fusion between 35% and 50% when fluorescence in situ assay was used to detect the rearrangement.27, 28, 31, 35, 38, 39 Other smaller studies using PCR-based methodology have reported higher frequencies.19, 21, 25, 26, 29 Only one study to date has comprehensively explored for the presence of other fusion partners and determined that an additional 5%–10% of cases may harbor other gene fusions including TMPRSS2:ETV1 and TMPRSS2:ETV4.35
In two population-based studies from Sweden and the United Kingdom, 15%–20% of men diagnosed with incidental prostate cancer had tumors that harbored TMPRSS2:ERG.27, 40, 41 It is worth highlighting that the 354 incidental cancers from the Swedish cohorts were detected in five population-based cohorts prior to PSA screening.40 All of the tumors were detected on transurethral resection of the prostate samples, which differs from the prostatectomy series. Although some have suggested that there may be a genetic component to this lower frequency in the Swedish population, we have determined that, in a PSA-screened biopsy cohort from Örebro, the percentage is approximately 45%, which is similar to all other PSA-screened hospital-based cohorts (Svensson and Rubin, unpublished. data, 2012). We have observed similar frequencies in gene fusion when examining hormone naive primary and castration resistant metastatic prostate cancers.42
As part of an Early Detection Research Network study sponsored by the National Cancer Institute, we prospectively determined that 46% of men with prostate cancer detected on 12-core needle biopsies by PSA screening harbor TMPRSS2:ERG fusion.43 This result is consistent with the surgical cohorts. Taken together, observations made over the past several years, from a number of studies since the original description of TMPRRSS2:ETS prostate cancer, suggest that the majority of prostate cancers currently detected by PSA-screening harbor either the common TMPRSS2:ERG (46%) fusion, or one of the less common fusions involving TMPRSS2 or other 5′ prime partners (5%–10%). This has important clinical implications, as the TMPRSS2:ERG transcript can be detected in urine and represents a highly specific prostate cancer biomarker.44, 45, 46
The common ETS gene fusions are widely believed to have an important oncogenic role, but that ERG or ETV1 overexpression alone is not sufficient to lead to prostate cancer. There is now mounting molecular data for an important concomitant role of TMPRSS2:ERG and the pten/PI3K/ATK pathway activation in prostate cancer oncogenesis. The study from Carver et al.47 looked at data from human samples and determined that high-grade PIN demonstrates ERG gene rearrangement in 10% of cases, whereas loss of PTEN as determined by immunohistochemistry was observed in 45% of cases. Similar to previous observations by our group on high-grade PIN with ERG rearrangement,16, 17 the associated prostate cancer also demonstrated the same gene fusion status. They interpret these data to be consistent with TMPRSS2:ERG fusion representing an early molecular event facilitating the transition from high-grade PIN to prostate cancer. They further analyzed tumor samples demonstrating that 68% (27/40) of prostate cancers had loss of PTEN expression by immunohistochemistry in contrast to 38% (15/40) of cases with ERG rearrangement.47 Interestingly, 14 of 15 ERG rearranged cases demonstrated PTEN loss. King et al.48 presented almost identical data with 14 of 17 TMPRSS2:ERG fusion prostate cancers showing PTEN loss. These human tumor data support the hypothesis that PTEN and ERG are concomitant molecular lesions.
Initial work exploring the role of the TMPRSS2:ERG fusions in cell lines demonstrates fairly consistent findings for the overexpression of ETS gene in benign epithelial cells. Studies that have overexpressed ETV1, ETV5 and ERG have demonstrated an increase in cell migration capability not an increase in proliferation or the ability to transform these cells into tumor cells.36, 37, 49 Klezovitch et al.50 confirmed that these finding demonstrate that the overexpression of ERG is associated with tumor cell migration through a proteolytic molecular program. Recently, Carver et al.47 performed a series of studies to explore the concomitant relationship between TMRPSS2:ERG and the pten/PI3K/Akt pathways. Benign prostate cells (BPH-1) demonstrate increased proliferation with constitutively overexpressed Akt (AKT-1) as compared to controls. ERG overexpression did not lead to an increase in cell proliferation. In contrast, cell migration studies demonstrated an additive effect with ERG and Akt. These results suggest that the oncogenic role of TMPRSS2:ERG fusion is in tumor cell migration that is enhanced by the proliferative effects of pten/PIK3/Akt pathway activation.
The first mouse models used a probasin promoter to overexpress ERG49, 50 and ETV1.36 These models demonstrate the ability of the trans gene (i.e., ERG or ETV1) to develop early molecular changes referred to as mouse PIN.51 These subtle changes have not reached the level of invasive cancer.
Based on observations described above in human samples, more recent work explored the in vivo significance of TMPRSS2–ERG fusion in the context of the pten/PI3K/ATK pathway activation. The Pandolfi group had previously demonstrated that a transgenic mouse models with loss of one pten allele (pten−/+) develops mouse high-grade PIN after a long latency period with incomplete penetrance.52 By developing a pten−/+ transgenic mouse that overexpresses ERG using a probasin promoter (ARR2PB), they observed a florid high-grade PIN with a shorter latency period and more complete penetrance as compared to the pten−/+ mouse. Taken together, Carver et al.47 concluded that alterations of pten/PI3K/Akt pathway play an early role in tumor cell proliferation and the TMPRSS2:ERG gene fusion contributes through cell migration. They posit that the combination leads to an aggressive phenotype as suggested by Yoshimoto et al.53 King et al. report supportive data using a different mouse model.48 They developed a model that overexpresses the most common TMPRSS2:ERG isoform under control of a probasin promoter. This TMPRSS2:ERG model does not demonstrate an apparent phenotype. However, bigenic mice with either TMPRSS2–ERG; pten−/+ or TMPRSS2–ERG and probasin-driven Akt demonstrated high-grade PIN. They also tested the potential concomitant effect of TMPRSS2–ERG with their MYC overexpressing mice (Hi-Myc) but did not see any additional effect; the Hi-Myc mice develop prostate cancer. These data are also consistent with clinical data that failed to show associations with MYC amplification and ERG rearrangement. Both of these studies suggest that pten/PI3K/Akt and TMPRSS2:ERG alterations need to be explored as important events in prostate cancer disease progression.
TMPRSS2:ERG and association with a more aggressive clinical outcome
The data generated in the search for associations with clinical outcome emerge from two types of studies: population-based Watchful Waiting cohorts and retrospective prostatectomy series. A review of the literature suggests that, in some instances, the TMPRSS2:ERG fusion is associated with a more aggressive clinical course, while conversely, others report the opposite result. We hope to clarify this confusion, but concede that large population-based studies will be required to clarify this issue in the future.
Our group initially observed an enrichment in theTMPRSS2:ERG fusion in higher stage prostate cancer.15 We then searched for associations between TMPRSS2:ERG fusion and clinical outcome in a population-based study.41 The Örebro Watchful Waiting cohort represents a treatment-naïve population drawn from a strictly defined catchment area for 190 000 inhabitants living in Örebro. The TMPRSS2:ERG gene fusion was identified in 15% (17/111) of the patients' initial transurethral resection of the prostate biopsy samples and was significantly associated with prostate cancer-specific death (cumulative incidence ratio=2.7, P<0.01, 95% confidence interval=1.3–5.8). This is a well-defined population that dramatically differs from the retrospective prostatectomy series. First, this is a population-based cohort. All men with early prostate cancer (T1a–b, Nx, M0) diagnosed by transurethral resection of the prostate for symptomatic benign prostatic hyperplasia (i.e., lower urinary track symptoms) were included. There was no PSA screening in Sweden during the collection phase of this study. Second, the patients were followed expectantly (without curative treatment) and received clinical exams, laboratory tests and bone scans every 6 months during the first 2 years following diagnosis and subsequently at 12-month intervals. Third, the endpoint of the study was lethal prostate cancer, defined as development of distant metastases or prostate cancer as the underlying cause of death (median follow-up time: 9.1 years, maximum: 28 years). Therefore, this unique study design allows one to access the biological impact of TMPRSS2:ERG prostate cancer in the absence of early intervention.
The results of this study were supported by a report from the United Kingdom, wherein associations between TMPRSS2:ERG fusion and survival of 445 men conservatively treated for prostate cancer were undertaken.27 Overall, cancers lacking TMPRSS2:ERG fusion alterations demonstrated 90% survival at 8 years of clinical follow-up. They also identified a novel association seen in TMPRSS2:ERG fusion prostate cancer where the fusion of TMPRSS2 to ERG together with interstitial deletion of sequences 5′ to ERG15 was associated with a significantly worse cause-specific survival taking into account age, Gleason score, and pre-treatment PSA. Supporting the hypothesis that overexpression of ERG is acting as an oncogene, the overall lowest cause-specific survival was associated with a duplication of the TMPRSS2:ERG fusion with an accompanying interstitial deletion (hazard ratio=6.10, 95% confidence ratio=3.33–11.15, P<0.001, 25% survival at 8 years). On multivariate analysis, the duplication of the TMPRSS2:ERG fusion with associated deletion (referred to as ‘2+Edel') was an independent predictor of clinical outcome proving additional information to Gleason score and pre-treatment PSA level. This study reported on 110 clinical T1 prostate cancer cases that had 20% TMPRSS2:ERG fusion similar to the Swedish Watchful Waiting cohort. This study supports the aggressive biological significance of the TMPRSS2:ERG fusion. Two key observations from this study were that gain of ERG and the associated interstitial deletion of the 3 Mb region between TMPRSS2 and ERG on chromosome 21 are associated with more aggressive prostate cancer. Overexpression of ERG has been associated with poor clinical outcome in acute myeloid leukemia54 and some of the genes located in the 3 Mb area of deletion (e.g., HMGN1, ETS-2) may be acting as tumor suppressor genes.15
There have been a number of retrospective studies exploring for the association between TMPRSS2:ERG and outcome following radical prostatectomy that gave mixed results.26, 30, 34 It is difficult to compare results from a surgical study that utilized PSA biochemical failure as an end point with one that used observational studies using cancer specific death as an outcome. One of the limitations of using PSA elevation following prostatectomy as a surrogate end point comes from a single institution study of men diagnosed with clinically localized prostate cancer in the pre-PSA-screening era. Porter et al.55 observed 45.5% PSA biochemical failure in a radical prostatectomy series but prostate cancer-specific death occurred in only 18.5% of the population with a follow-up time of up to 25 years. Carver et al.56 reported that, in a population of high risk men with T3 prostate cancer who underwent radical prostatectomy, 36% with PSA biochemical failure subsequently developed clinically relevant disease progression. Ward et al.57 found that, in a population of 3897 radical prostatectomy patients, only 8.3% of the men with PSA biochemical failure died of prostate cancer with a median follow-up time of 10 years. PSA elevation following surgery is associated with prostate cancer-specific death, but the majority of men with PSA biochemical failure will die of other causes. Therefore, we would argue for caution in over-interpreting the results of each of these types of clinical studies.
Based on the two large observational clinical studies with long-term follow-up, we would argue that left untreated, TMPRSS2:ERG prostate cancer will run a more aggressive clinical course than fusion negative cancer. In the setting of surgical or other interventions immediately following diagnosis, there are insufficient data to make any reasonable conclusions.
Gene fusion is a clonal event and helps understand prostate cancer heterogeneity
It is recognized that prostate cancer is multifocal. Both morphologic and molecular analysis has shown that by the time of cancer diagnosis, more than 80% of prostates harbor multiple separate cancer foci.58, 59, 60, 61, 62 These discrete lesions have both biological and clinical implications. The TMPRSS2:ERG fusion represents an excellent early clonal marker to provide insight into molecular heterogeneity.
TMPRSS2:ERG fusions, when present, are distributed evenly among all tumor nuclei within a discrete tumor lesion. We reported that 243 out of 246 prostate cancer cases demonstrated homogeneity within a discrete tumor nodule.15 This observation was extended when multiple microdissected foci of cancer from individual patients were examined by RT-PCR for gene fusions and demonstrated either all or no foci overexpressed ERG and its family members ETV1 and ETV4.63 Thus, within a discrete nodule, the fusion rearrangement must occur early as all of the tumor nuclei harbor the fusion when present. However, when we undertook studies to evaluate rearrangement between the multiple nodules within a single prostate gland from one individual, each discrete lesion may occur independently from one another. This has been observed in three independently conducted studies.64, 65, 66 For example, in the study by Barry et al.,65 32 prostatectomy samples with clear cut discrete tumors, demonstrated fusion by balanced translocation and fusion by interstitial deletion occurring as distinct events, suggesting that these are clonal mechanisms for achieving TMPRSS2:ERG fusion. Interestingly, that study found a high rate (41%) of interfocal heterogeneity for fusion status. These observations have both biological and clinical implications. Biologically, the presence of multiple clonally distinct lesions suggests that within a single gland complex, molecular events such as the gene rearrangement can occur in some but not all lesions. This makes classifying prostate cancers more challenging. From a clinical perspective, how does one determine the most aggressive nodule to target? It has been long assumed that the dominant nodule harbors the most aggressive tumor and therefore dictates the clinical course. As discussed below, if TMPRSS2:ERG prostate cancers are more biologically aggressive, strategies will be needed to detect them regardless of their size as these may be the tumors with the highest propensity for metastatic dissemination.42
Diagnostic and clinical therapy implications
PSA has a diminished role in detecting prostate cancer, thus there is the requirement for a new molecular detection test. Several studies to date have demonstrated the detection of the TMPRSS2:ERG fusion transcripts in urine.44, 45, 67 These studies, and other unpublished reports, demonstrate a high specificity. Unlike PSA, which can be elevated in benign conditions as well as cancer, the presence of TMPRSS2:ERG transcripts has only been reported in neoplastic cells. In addition to the sensitive and specific detection of TMPRSS2:ERG in urine sediment,67 recent work has demonstrated improved detection of prostate cancer using multiple biomarkers. Multiplexed detection of GOLM1, SPINK1, PCA3 and TMPRSS2:ERG was a more significant predictor of prostate cancer than serum PSA or PCA3 alone.67 These results are promising, and with some refinement, could be adopted as a clinical supplement to serum PSA for prostate cancer detection.
Given the heterogeneity demonstrated between tumor nodules, a positive TMPRSS2:ERG urine test and a biopsy negative for cancer would suggest that the cancer has been missed. If the cancer is detected but fusion-negative, the sampling would have missed the fusion cancer. The finding of interfocal heterogeneity for TMPRSS2:ERG fusion has direct relevance in the context of a urine test result that is positive for fusion and a subsequent prostate biopsy with cancer that is negative for fusion. Given the potential prognostic role of determining the mode of rearrangement (deletion through translocation versus through interstitial deletion), a biopsy fluorescence in situ hybridization test would allow for an accurate determination of the presence and type of gene fusions.
Recent trials in the setting of castration-resistant prostate cancer suggest that targeting androgen and estrogen might be an effective approach. Data suggest that low levels of intraprostatic testosterone or dihydrotestosterone still are present when men have undergone chemical castration with anti-androgens. Therefore, novel approaches have been developed to reduce these low levels of androgens and estrogens by blocking steroid synthesis. Abiraterone acetate is a selective small molecule inhibitor of cytochrome (CYP) 17, which effectively blocks the production of androgen and estrogen.68 It was recently tested in a phase I clinical trial and demonstrated a decrease in PSA following treatment in 50% of all men with castration independent prostate cancer.69, 70 In the study, 83% (5/6) with TMPRSS2:ERG fusion prostate cancer had a decrease in PSA following abiraterone treatment. Although this study was not designed to test the potential role of abiraterone with respect to TMPRSS2:ERG fusion status, future phase II and III studies will examine this hypothesis based on these initial observations.
The RAF kinase fusions, although rare, are of immediate therapeutic significance given the numerous approved and investigational agents in late-stage of development. Palanisamy et al.13 demonstrated that the RAF kinase fusions were sensitive to sorafenib, an Federal Drug Administration-approved RAF inhibitor that has also been demonstrated to target additional kinases.71 This suggests that screening patients for RAF fusions may identify a subset of the population that may benefit from existing targeted therapies similar to the current clinical application of ALK inhibitors to EML4–ALK non-small-cell lung carcinoma patients.72, 73 We envision that other targetable gene fusions and driving mutations will be discovered in the coming years.
Ateeq et al.74 recently demonstrated that SPINK1 prostate cancer can be targeted using cetuximab, an EGFR inhibitor. SPINK1 harbors a high homology with EGF. Preclinical models using recombinant SPINK1 support targeting the extracellular domain of SPINK1. This early work provides a rationale for both the development of humanized mAbs to SPINK1 and evaluation of EGFR inhibition in SPINK1+/ETS− prostate cancers.
Emerging understanding of prostate cancer genomic complexity
Genomic rearrangements appear to be nonrandom, locus-specific and depend, in part, on the proximity of chromosomal regions in the nucleus.75 Moreover, there is mounting evidence suggesting that transcription factors are associated with DNA double-strand breaks, thus predisposing transcribed regions to genomic rearrangements. For example, both androgen and estrogen signaling recruits the enzyme topoisomerase 2B (TOP2B) to target gene promoters, which creates DNA double-strand breaks and facilitates transcription.76, 77 The androgen receptor and TOP2B are coexpressed in human prostate cancer precursor lesions in which TMPRSS2:ERG rearrangements are known to occur, suggesting a critical role of TOP2B in the recurrent ETS rearrangements. Three recent studies have also shown that androgen signaling promotes TMPRSS2:ERG fusion formation,78, 79, 80 in part, by recruiting DNA break-inducing enzymes (e.g., activation of induced cytidine deaminase (AID)) to translocation breakpoint sites.79 More recently, we demonstrated that rearrangement breakpoints were enriched near open chromatin, androgen receptor and tERG DNA-binding sites in the setting of the ETS gene fusion TMPRSS2:ERG, but inversely correlated with these regions in tumors lacking ETS fusions.2 Hence, transcription factors can contribute to the formation of genomic rearrangements by facilitating the juxtaposition of chromosomal loci and recruiting enzymatic machinery involved in DNA breaks to these target loci. This work also suggests that inhibitors of repair enzymes such as PARP1 and DNA-PK decrease the susceptibility to gene fusions. It also raises concerns that TOP2B inhibitors such as etoposide or doxorubicine might facilitate gene fusions and rearrangements by enhancing double stranded DNA breaks. Ongoing research is exploring the clinical implications of these observation.
Conclusions
ETS fusions are the most common genetic mutation identified in prostate cancer. Although a number of ETS and non-ETS family members have been observed to be fused with TMPRSS2 or other 5′ partners, the vast majority of fusions involve TMPRSS2:ERG. This fusion can be studied in large numbers, as it was identified in approximately 45% of all PSA-screened prostate cancers. Associations with disease specific death have been made in clinical observational studies. The amplification of the TMPRSS2:ERG fusion and the interstitial deletion associated with the translocation add additional statistical power to predicting lethal prostate cancer. Morphologic features, functional in vitro and in vivo studies and a specific gene signature support the view that the TMPRSS2:ERG fusion cancers represent a distinct molecular subclass. The more recent discovery of the RAF fusions also demonstrates that some of the gene fusions will be targets for clinical intervention.
Acknowledgments
This review is taken from original work by several important collaborators: Scott Tomlins, Francesca Demichelis, Sven Perner, John Presner and Arul M Chinnaiyan.
A patent for the diagnostic and therapeutic uses of gene fusion prostate cancer has been licensed to Gen-Probe for commercial development. Dr Rubin is listed as a co-inventor on this patent application. Dr Rubin serves as a consultant to Ventana/Roche.
References
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin MA, Chinnaiyan AM. Bioinformatics approach leads to the discovery of the TMPRSS2:ETS gene fusion in prostate cancer. Lab Invest. 2006;86:1099–102. doi: 10.1038/labinvest.3700477. [DOI] [PubMed] [Google Scholar]
- Hanauer DA, Rhodes DR, Sinha-Kumar C, Chinnaiyan AM. Bioinformatics approaches in the study of cancer. Curr Mol Med. 2007;7:133–41. doi: 10.2174/156652407779940431. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, et al. SLC45A3–ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–8. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- Esgueva R, Perner S, J LaFargue C, Scheble V, Stephan C, et al. Prevalence of TMPRSS2–ERG and SLC45A3–ERG gene fusions in a large prostatectomy cohort. Mod Pathol. 2010;23:539–46. doi: 10.1038/modpathol.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Mehra R, Dhanasekaran SM, Yu J, Menon A, et al. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res. 2008;68:7629–37. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci USA. 2009;106:12353–8. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflueger D, Rickman DS, Sboner A, Perner S, LaFargue CJ, et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia. 2009;11:804–11. doi: 10.1593/neo.09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16:793–8. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Yu J, Varambally S, Mehra R, et al. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell. 2008;13:519–28. doi: 10.1016/j.ccr.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–41. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- Mosquera JM, Perner S, Genega EM, Sanda M, Hofer MD, et al. Characterization of TMPRSS2–ERG fusion high-grade prostatic intraepithelial neoplasia and potential clinical implications. Clin Cancer Res. 2008;14:3380–5. doi: 10.1158/1078-0432.CCR-07-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner S, Mosquera JM, Demichelis F, Hofer MD, Paris PL, et al. TMPRSS2–ERG fusion prostate cancer: an early molecular event associated with invasion. Am J Surg Pathol. 2007;31:882–8. doi: 10.1097/01.pas.0000213424.38503.aa. [DOI] [PubMed] [Google Scholar]
- de Marzo AM, Platz EA, Epstein JI, Ali T, Billis A, et al. A working group classification of focal prostate atrophy lesions. Am J Surg Pathol. 2006;30:1281–91. doi: 10.1097/01.pas.0000213289.50660.be. [DOI] [PubMed] [Google Scholar]
- Cerveira N, Ribeiro FR, Peixoto A, Costa V, Henrique R, et al. TMPRSS2–ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia. 2006;8:826–32. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Tomlins SA, Mudaliar KM, Chiu YL, Esgueva R, et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia. 2010;12:590–8. doi: 10.1593/neo.10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlers CM, Figg WD. ETS–TMPRSS2 fusion gene products in prostate cancer. Cancer Biol Ther. 2006;5:254–5. doi: 10.4161/cbt.5.3.2603. [DOI] [PubMed] [Google Scholar]
- Hermans KG, van Marion R, van Dekken H, Jenster G, van Weerden WM, et al. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006;66:10658–63. doi: 10.1158/0008-5472.CAN-06-1871. [DOI] [PubMed] [Google Scholar]
- Iljin K, Wolf M, Edgren H, Gupta S, Kilpinen S, et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006;66:10242–6. doi: 10.1158/0008-5472.CAN-06-1986. [DOI] [PubMed] [Google Scholar]
- Liu W, Chang B, Sauvageot J, Dimitrov L, Gielzak M, et al. Comprehensive assessment of DNA copy number alterations in human prostate cancers using Affymetrix 100K SNP mapping array. Genes Chromosomes Cancer. 2006;45:1018–32. doi: 10.1002/gcc.20369. [DOI] [PubMed] [Google Scholar]
- Soller MJ, Isaksson M, Elfving P, Soller W, Lundgren R, et al. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes Chromosomes Cancer. 2006;45:717–9. doi: 10.1002/gcc.20329. [DOI] [PubMed] [Google Scholar]
- Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–51. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–63. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Merson S, Jhavar S, Flohr P, Edwards S, et al. Diversity of TMPRSS2–ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–73. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- Lapointe J, Kim YH, Miller MA, Li C, Kaygusuz G, et al. A variant TMPRSS2 isoform and ERG fusion product in prostate cancer with implications for molecular diagnosis. Mod Pathol. 2007;20:467–73. doi: 10.1038/modpathol.3800759. [DOI] [PubMed] [Google Scholar]
- Nam RK, Sugar L, Wang Z, Yang W, Kitching R, et al. Expression of TMPRSS2 ERG gene fusion in prostate cancer cells is an important prognostic factor for cancer progression. Cancer Biol Ther. 2007;6:40–5. doi: 10.4161/cbt.6.1.3489. [DOI] [PubMed] [Google Scholar]
- Rajput AB, Miller MA, de Luca A, Boyd N, Leung S, et al. Frequency of the TMPRSS2:ERG gene fusion is increased in moderate to poorly differentiated prostate cancers. J Clin Pathol. 2007;60:1238–43. doi: 10.1136/jcp.2006.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnes M, Lissbrant E, Damber JE, Stenman G. Molecular genetic analyses of the TMPRSS2–ERG and TMPRSS2–ETV1 gene fusions in 50 cases of prostate cancer. Oncol Rep. 2007;17:1033–6. [PubMed] [Google Scholar]
- Yoshimoto M, Ludkovski O, Bayani J, Graham C, Zielenska M, et al. Microdeletion and concurrent translocation associated with a complex TMPRSS2:ERG prostate cancer gene fusion. Genes Chromosomes Cancer. 2007;46:861–3. doi: 10.1002/gcc.20470. [DOI] [PubMed] [Google Scholar]
- Saramaki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, et al. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–44. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia. 2006;8:465–9. doi: 10.1593/neo.06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JJ, Rohan S, Kao J, Kitabayashi N, Mathew S, et al. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod Pathol. 2007;20:921–8. doi: 10.1038/modpathol.3800903. [DOI] [PubMed] [Google Scholar]
- Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst. 2008;100:815–25. doi: 10.1093/jnci/djn150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichelis F, Fall K, Perner S, Andren O, Schmidt F, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–9. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- Mehra R, Tomlins SA, Yu J, Cao X, Wang L, et al. Characterization of TMPRSS2–ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–90. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, et al. Prevalence of TMPRSS2–ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res. 2009;15:4706–11. doi: 10.1158/1078-0432.CCR-08-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessels D, Smit FP, Verhaegh GW, Witjes JA, Cornel EB, et al. Detection of TMPRSS2–ERG fusion transcripts and prostate cancer antigen 3 in urinary sediments may improve diagnosis of prostate cancer. Clin Cancer Res. 2007;13:5103–8. doi: 10.1158/1078-0432.CCR-07-0700. [DOI] [PubMed] [Google Scholar]
- Laxman B, Tomlins SA, Mehra R, Morris DS, Wang L, et al. Noninvasive detection of TMPRSS2:ERG fusion transcripts in the urine of men with prostate cancer. Neoplasia. 2006;8:885–8. doi: 10.1593/neo.06625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Aubin SM, Siddiqui J, Lonigro RJ, Sefton-Miller L, et al. Urine TMPRSS2:ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Sci Transl Med. 2011;3:94ra72. doi: 10.1126/scitranslmed.3001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, et al. Cooperativity of TMPRSS2–ERG with PI3–kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, et al. Role of the TMPRSS2–ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci USA. 2008;105:2105–10. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the bar harbor meeting of the mouse models of human cancer consortium prostate pathology committee. Cancer Res. 2004;64:2270–305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008;21:1451–60. doi: 10.1038/modpathol.2008.96. [DOI] [PubMed] [Google Scholar]
- Marcucci G, Baldus CD, Ruppert AS, Radmacher MD, Mrozek K, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–42. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
- Porter CR, Kodama K, Gibbons RP, Correa R, Jr, Chun FK, et al. 25-year prostate cancer control and survival outcomes: a 40-year radical prostatectomy single institution series. J Urol. 2006;176:569–74. doi: 10.1016/j.juro.2006.03.094. [DOI] [PubMed] [Google Scholar]
- Carver BS, Bianco FJ, Jr, Scardino PT, Eastham JA. Long-term outcome following radical prostatectomy in men with clinical stage T3 prostate cancer. J Urol. 2006;176:564–8. doi: 10.1016/j.juro.2006.03.093. [DOI] [PubMed] [Google Scholar]
- Ward JF, Blute ML, Slezak J, Bergstralh EJ, Zincke H. The long-term clinical impact of biochemical recurrence of prostate cancer 5 or more years after radical prostatectomy. J Urol. 2003;170:1872–6. doi: 10.1097/01.ju.0000091876.13656.2e. [DOI] [PubMed] [Google Scholar]
- Arora R, Koch MO, Eble JN, Ulbright TM, Li L, et al. Heterogeneity of Gleason grade in multifocal adenocarcinoma of the prostate. Cancer. 2004;100:2362–6. doi: 10.1002/cncr.20243. [DOI] [PubMed] [Google Scholar]
- Cheng L, Song SY, Pretlow TG, Abdul-Karim FW, Kung HJ, et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J Natl Cancer Inst. 1998;90:233–7. doi: 10.1093/jnci/90.3.233. [DOI] [PubMed] [Google Scholar]
- Greene DR, Wheeler TM, Egawa S, Dunn JK, Scardino PT. A comparison of the morphological features of cancer arising in the transition zone and in the peripheral zone of the prostate. J Urol. 1991;146:1069–76. doi: 10.1016/s0022-5347(17)38003-5. [DOI] [PubMed] [Google Scholar]
- Qian J, Bostwick DG, Takahashi S, Borell TJ, Herath JF, et al. Chromosomal anomalies in prostatic intraepithelial neoplasia and carcinoma detected by fluorescence in situ hybridization. Cancer Res. 1995;55:5408–14. [PubMed] [Google Scholar]
- Sakr WA, Macoska JA, Benson P, Grignon DJ, Wolman SR, et al. Allelic loss in locally metastatic, multisampled prostate cancer. Cancer Res. 1994;54:3273–7. [PubMed] [Google Scholar]
- Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- Clark J, Attard G, Jhavar S, Flohr P, Reid A, et al. Complex patterns of ETS gene alteration arise during cancer development in the human prostate. Oncogene. 2008;27:1993–2003. doi: 10.1038/sj.onc.1210843. [DOI] [PubMed] [Google Scholar]
- Barry M, Perner S, Demichelis F, Rubin MA. Interfocal heterogeneity for TMPRSS2–ERG fusion in multifocal prostate cancer. Urology. 2007;70:630–3. doi: 10.1016/j.urology.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra R, Han B, Tomlins SA, Wang L, Menon A, et al. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer Res. 2007;67:7991–5. doi: 10.1158/0008-5472.CAN-07-2043. [DOI] [PubMed] [Google Scholar]
- Laxman B, Morris DS, Yu J, Siddiqui J, Cao J, et al. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res. 2008;68:645–9. doi: 10.1158/0008-5472.CAN-07-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, et al. Pharmacology of novel steroidal inhibitors of cytochrome P45017α (17α-hydroxylase/C17–20 lyase) J Steroid Biochem Mol Biol. 1994;50:267–73. doi: 10.1016/0960-0760(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–8. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, et al. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008 Oct;7:3129–40. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, et al. EML4–ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Ateeq B, Tomlins SA, Laxman B, Asangani IA, Cao Q, et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci Transl Med. 2011. p. 72ra17. [DOI] [PMC free article] [PubMed]
- Mani RS, Chinnaiyan AM. Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010;11:819–29. doi: 10.1038/nrg2883. [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–75. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastus NC, Boyd LK, Mao X, Stankiewicz E, Kudahetti SC, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70:9544–8. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yang L, Tanasa B, Hutt K, Ju BG, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–83. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]