

Abstract
Background & Aims
Defects in the intestinal epithelial tight junction (TJ) barrier contribute to intestinal inflammation. Tumor necrosis factor (TNF)-α induced increase in intestinal TJ permeability contributes to the intestinal TJ barrier defect in inflammatory disorders. We investigated the mechanisms by which TNF-α induces occludin depletion and increase in intestinal TJ permeability.
Methods
We assessed intestinal TJ barrier function using intestinal epithelial model systems: filter-grown Caco-2 monolayers and recycling perfusion studies of mouse small intestine.
Results
TNF-α caused a rapid increase in expression of miR-122a in enterocytes, in cultured cells and intestinal tissue. The over-expressed miR-122a bound to a binding motif at the 3′-untranslated region of occludin mRNA to induce its degradation; mRNA degradation depleted occludin from enterocytes, resulting in increased intestinal TJ permeability. Transfection of enterocytes with an anti-sense oligoribonucleotide against miR-122a blocked the TNF-α-induced increase in enterocyte expression of miR-122a, degradation of occludin mRNA, and increase in intestinal permeability. Overexpression of miR-122a in enterocytes using pre-miR-122a was sufficient to induce degradation of occludin mRNA and increase in intestinal permeability.
Conclusions
TNF-α regulates intestinal permeability by inducing miR-122a-mediated degradation of occludin mRNA. These studies demonstrate the feasibility of therapeutically targeting miR-122a in-vivo to preserve the intestinal barrier.
Keywords: Intestine, inflammatory bowel disease, Crohn’s disease, colitis, mouse model, cytokine
Introduction
Defective intestinal epithelial tight junction (TJ) barrier, characterized by an increase in intestinal permeability, has been shown to be an important pathogenic factor contributing to the development of intestinal inflammation1–5. In various inflammatory diseases of the gut, including Crohn’s disease, ulcerative colitis, celiac disease, and number of infectious diarrheal syndromes, the disturbance in intestinal TJ barrier allows increased antigenic penetration, leading to an inflammatory response1, 2. The enhancement of the intestinal TJ barrier prevents the development of intestinal inflammation or leads to a more rapid resolution of the inflammatory disease3–5. The intracellular mechanisms that mediate the disturbance in TJ barrier in intestinal permeability disorders remain largely undefined.
The studies from our laboratory and others have shown that an increase in myosin light chain kinase expression and activity may be an important mechanism contributing to the increase in intestinal TJ permeability in various inflammatory conditions of the gut6, 7. However, the role of transmembrane TJ protein depletion in intestinal permeability disorders remains unclear. Previous clinical studies have shown occludin levels to be markedly decreased in intestinal permeability disorders, including in Crohn’s disease, ulcerative colitis, and celiac disease8–10; and, it has been proposed that the decrease in intestinal occludin level contributes to the increase in intestinal permeability. The mechanisms that lead to occludin depletion and the role of occludin depletion as a cause of increase in intestinal permeability remain unclear.
Tumor necrosis factor-α (TNF-α) is an essential mediator of inflammation in the gut11, 12. TNF-α levels are markedly elevated in patients with inflammatory conditions, including Crohn’s disease, ulcerative colitis and celiac disease. Anti-TNF-α therapy has been shown to be effective in inducing remission in patients with severe active Crohn’s disease and ulcerative colitis11, 13 and refractory celiac disease14. In addition to direct immune activation, it has been shown that an important pro-inflammatory action of TNF-α is to cause an increase in intestinal TJ permeability, which allows increased intestinal penetration of luminal antigens6, 7, 15, 16. Previous studies have shown that the enhancement of intestinal TJ barrier prevents the cytokine mediated development of intestinal inflammation and diarrhea3, 16–18. In this regard, understanding the mechanisms that mediate the TNF-α induced increase in intestinal TJ permeability is important in developing future therapeutic strategies to prevent the disturbance in TJ barrier function in various inflammatory conditions.
The major aim of this study was to elucidate the mechanisms that mediate the TNF-α induced increase in intestinal TJ permeability using an in-vitro (consisting of filter-grown Caco-2 monolayers) and an in-vivo (re-cycling perfusion of mouse intestine) intestinal model systems. Herein, we describe a novel mechanism in which microRNA-122a (miR-122a) plays a central role in the regulation of intestinal TJ permeability by targeting occludin mRNA degradation. Our data show that TNF-α induces a rapid increase in miR-122a expression in enterocytes; miR-122a binds to the non-coding region of occludin mRNA and induces occludin mRNA degradation and occludin depletion, leading to an increase in intestinal permeability.
Results
TNF-α induced increase in Caco-2 intestinal epithelial TJ permeability correlates with occludin depletion
Transmembrane TJ proteins including occludin and claudins localize at the TJ strands and have a role in TJ barrier function19, 20. In the following studies, the TNF-α effect on transmembrane protein expression was determined in filter-grown Caco-2 intestinal epithelial monolayers. TNF-α at physiologically relevant concentration (10 ng/ml)7, 21 produced a progressive decrease in occludin protein expression starting at about 12 h (Fig. 1a). The drop in occludin expression reached the trough level by 24–48 h. TNF-α also caused an increase in claudin-2 and -8 expression and did not affect claudin-1, -3 or -5 expression (Fig. 1a). These data suggested that the TNF-α induced depletion of occludin protein was specific to occludin and did not apply to other TJ proteins. The TNF-α effect on Caco-2 TJ permeability was assessed using a commonly utilized paracellular marker inulin (MW = 5,000 g/mol)22. TNF-α produced a time-dependent increase in mucosal-to-serosal flux of paracellular marker inulin (Fig. 1b). By 48 h, TNF-α treatment resulted in about 10-fold increase in Caco-2 paracellular permeability to inulin (Fig. 1b). As shown in Fig. 1c, there was a linear relationship between TNF-α induced depletion of occludin and an increase in inulin flux, with a relative correlation coefficient of 0.99. These results suggested a possible cause-and-effect relationship between occludin depletion and an increase in Caco-2 TJ permeability to inulin.
Figure 1.
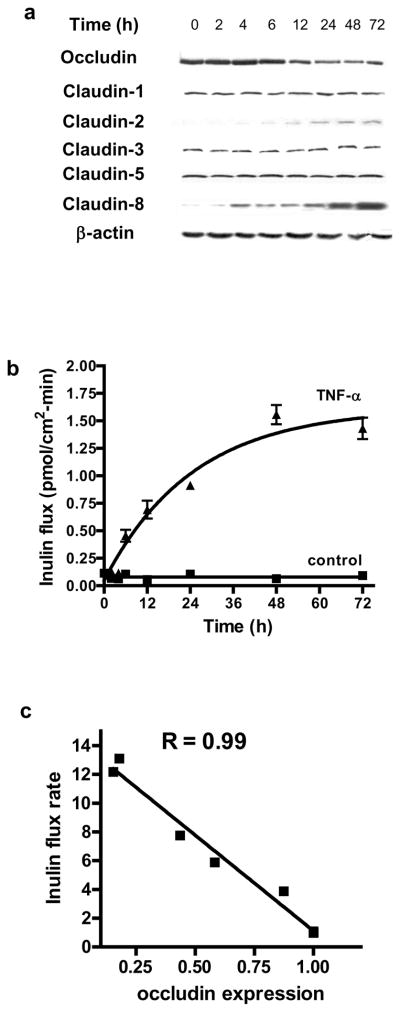
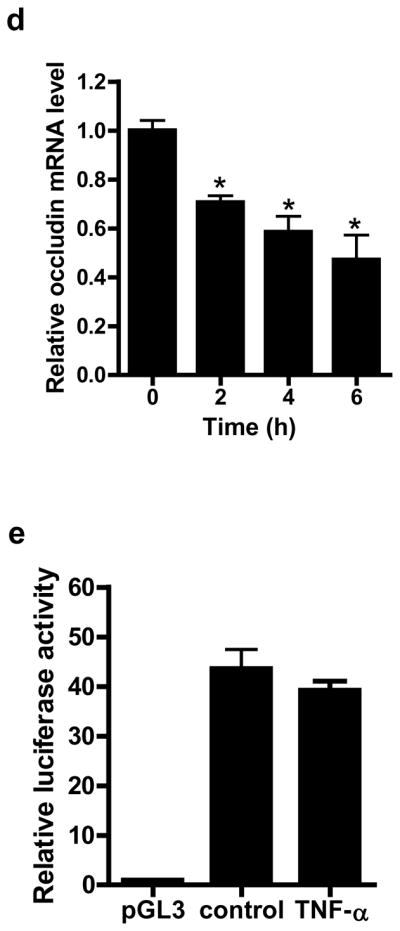
Time course of TNF-α effect on TJ protein expression, paracellular permeability, occludin mRNA transcription, and promoter activity in filter-grown Caco-2 monolayers. (a) Filter-grown Caco-2 monolayers were treated with TNF-α (10 ng/ml) over 72 h experimental period. The time-course of TJ protein expression was determined. (b) Time course of TNF-α effect on mucosal-to-serosal inulin flux. Inulin flux of 1 is equal to 0.114 pmol/cm2-min. (c) Graph of occludin expression versus inulin flux. The correlation coefficient of occludin expression and inulin flux was 0.99. (d) TNF-α effect on occludin mRNA expression as determined by real-time PCR. (e) TNF-α effect on occludin promoter activity as determined by luciferase assay. * p < 0.005 vs control.
TNF-α induced depletion of occludin is due to accelerated mRNA degradation
In the following studies, the intracellular mechanisms that mediated the TNF-α induced depletion of occludin was investigated. First, the possibility that occludin depletion was due to accelerated protein degradation was examined. TNF-α treatment did not affect Caco-2 occludin half-life as assessed by [35S]-methionine pulse-chase studies (data not shown), suggesting that TNF-α did not accelerate occludin degradation. TNF-α caused a progressive decrease in occludin mRNA level (Fig. 1d), suggesting a rapid mRNA degradation process. The possibility that the decrease in occludin mRNA may be due in part to a decrease in occludin gene activity was also examined. In these studies, occludin promoter region was cloned into a pGL-3 basic vector, and transfected into filter-grown Caco-2 monolayers. The TNF-α effect on occludin promoter activity was then assessed by determining luciferase (reporter gene) activity. TNF-α treatment did not have significant effect on luciferase activity (Fig. 1e), indicating that occludin promoter activity was not affected by TNF-α. Together, these results suggested that TNF-α did not affect occludin promoter activity or protein degradation but induced a rapid degradation of occludin mRNA.
MiR-122a binding to occludin 3′ untranslated region (3′UTR) regulates occludin mRNA expression
Recent studies have shown microRNAs to play an integral role in the post-transcriptional regulation of mRNAs23, 24. In the following studies, the possibility that miRNAs may be involved in the post-transcriptional regulation of occludin mRNA was examined. Using 3 different bioinformatics algorithms (Mirbase, UCSC, and PicTar), miRNA binding sites on 3′UTR of occludin mRNA were determined23, 24. The PicTar algorithm, which has the highest accuracy and sensitivity among the bioinformatics algorithms currently in use predicted three potential miRNA binding motifs on occludin 3′UTR: miR-122a, miR-200b, and miR-200c. The PicTar algorithm predicted with very high likelihood (98 % probability) that miR-122a had functional activity; miR-200b (86 %) and miR-200c (91 %) were predicted with lower probabilities. MiR-122a, miR-200b, and miR-200c were expressed in resting Caco-2 cells. TNF-α treatment produced a rapid and dramatic increase in miR-122a expression (Fig. 2a). By 2 h, there was about an eighty-fold increase in miR-122a level (Fig. 2a). In contrast, TNF-α had only minimal effect on miR-200b and miR-200c expression (data not shown). In combination with the computational data, these results suggested a high probability that miR-122a was involved in the post-transcriptional regulation of occludin mRNA; and thus, miR-122a was selected for further analysis.
Figure 2.


TNF-α effect on miR-122a expression, and effect of miR-122a over-expression on occludin 3′UTR. (a) Time course of TNF-α (10 ng/ml) effect on miR-122a expression as analyzed by real-time PCR. (b) MiR-122a binding site on occludin 3′UTR in various mammalian species. (c) Occludin 3′UTR region was cloned into pMIR-REPORT vector in 5′ to 3′ direction, and the effect of pre-miR-122a transfection on occludin 3′UTR was determined by luciferase assay. (d) MiR-122a binding site was deleted by site-directed mutagenesis, and effect of pre-miR-122a transfection on deletion construct was determined. * p < 0.01 vs control; ** p < 0.001 vs WT transfected with pre-miR-122a.
The miR-122a binding sequence was located between 171–195 bps down-stream of occludin stop codon in 3′UTR and a complete match in binding sequence was present (Fig. 2b). The blast analysis revealed that the miR-122a binding site was highly conserved among mammalian species, further supporting a functional role of miR-122a (Fig. 2b). MiRNA induces mRNA degradation by binding to the complementary site on 3′UTR23. In the following studies, the effect of miR-122a on occludin 3′UTR was determined. In these studies, occludin 3′UTR was cloned into pMir-Report plasmid vector and transfected into filter-grown Caco-2 monolayers. To simulate TNF-α induced over-expression of miR-122a, filter-grown Caco-2 monolayers were transfected with pre-miR-122a. Pre-miR-122a transfection resulted in a 50 to 70 fold increase in miR-122a expression (Fig. 3a). Pre-miR-122a co-transfection with occludin 3′UTR plasmid vector inhibited the luciferase (reporter gene) activity (Fig. 2c). The deletion of the miR-122a binding sequence on 3′UTR prevented the pre-miR-122a inhibition of luciferase activity (Fig. 2d), suggesting that miR-122a inhibits the luciferase activity by binding to its complementary sequence on 3′UTR.
Figure 3.

Effect of pre-miR-122a transfection induced miR-122a over-expression on occludin mRNA and protein expression and paracellular permeability in filter-grown Caco-2 monolayers. (a) Effect of pre-miR-122a transfection on miR-122a expression. (b) Effect of pre-miR-122a transfection on occludin mRNA expression. (c) Effect of pre-miR-122a transfection on occludin protein expression. (d) Effect of pre-miR-122a transfection on Caco-2 paracellular permeability to inulin. * p < 0.01 vs control.
MiR-122a expression induces occludin mRNA degradation and increase in Caco-2 TJ permeability
In the following series of studies, we tested the hypothesis that the TNF-α increase in miR-122a expression causes an increase in Caco-2 TJ permeability. In these studies, miR-122a was expressed to the similar level induced by TNF-α, by transfecting pre-miR-122a (Fig. 3a). The miR-122a over-expression caused a degradation of occludin mRNA and decrease in occludin protein level (Fig. 3b, 3c). The increase in miR-122a expression caused a similar increase in inulin flux rate as the TNF-α treatment (Fig. 3d). These data indicated that the over-expression of miR-122a expression was sufficient to cause occludin mRNA degradation and an increase in Caco-2 TJ permeability.
Anti-sense inhibition of miR-122a prevented the TNF-α induced increase in Caco-2 TJ permeability
In the following studies, the requirement of miR-122a in TNF-α induced increase in Caco-2 TJ permeability was examined. For these studies, anti-sense oligoribonucleotide was used to inhibit miR-122a expression. The transfection of 2′-0-methyl-modified anti-sense oligoribonucleotide (M-ASO) directed against miR-122a resulted in an inhibition of TNF-α increase in miR-122a expression (Fig. 4a). The M-ASO inhibition of miR-122a completely prevented the TNF-α induced increase in inulin flux (Fig. 4b). The M-ASO inhibition of miR-122a also prevented the TNF-α induced decrease in occludin mRNA and protein expression (Fig. 4c, 4d). These results indicated that the increase in miR-122a expression was required for the TNF-α induced degradation of occludin mRNA and increase in Caco-2 TJ permeability.
Figure 4.

Effect of anti-sense oligoribonucleotide (M-ASO) inhibition of miR-122a on TNF-α modulation of miR-122a, inulin flux, and occludin expression in Caco-2 monolayers. (a) M-ASO was transfected into filter-grown Caco-2 monolayer and the effect of M-ASO transfection on TNF-α induced increase in miR-122a expression was determined. (b) Effect of M-ASO transfection on TNF-α induced increase in inulin flux. (c) Effect of M-ASO transfection on TNF-α induced decrease in occludin mRNA expression. (d) Effect of M-ASO transfection on TNF-α induced decrease in occludin protein expression. * p < 0.01 vs control. ** p < 0.001 vs TNF-α treatment.
TNF-α causes an increase in mouse enterocyte expression of miR-122a and an increase in intestinal permeability in-vivo
We next examined the involvement of miR-122a in TNF-α induced increase in mouse intestinal permeability in-vivo. In these studies, the TNF-α effect on mouse intestinal permeability was determined by recycling perfusion of isolated segment (6 cm) of small intestine in-vivo, using FITC-labeled dextran (10 Kd) as a paracellular marker17. Intraperitoneal administration of TNF-α (5 μg)25 at doses used in previous in-vivo studies produced a 20- to 30-fold increase in miR-122a expression in the intestinal tissue at 24 h experimental period (Fig. 5a) and a corresponding decrease in intestinal tissue occludin mRNA and protein level (Fig. 5b). TNF-α also caused an increase in small intestinal permeability to FITC-dextran (Fig. 5c). Since intestinal tissue consists of variety of cell types that could disproportionately affect tissue miR-122a or occludin mRNA level, laser capture microdissection (LCM) was used to isolate pure population of intestinal epithelial cells from the intestinal mucosal surface26. The enterocytes were captured by 7.5 μM diameter laser beam (pulsed at duration of .5 msec)26. TNF-α caused an increase in miR-122a (Fig. 5d) expression and a decrease in occludin mRNA level (Fig. 5e) in the enterocytes. These in-vivo studies indicated that the TNF-α induced increase in intestinal permeability was associated with an increase in enterocyte miR-122a expression and a decrease in occludin level.
Figure 5.
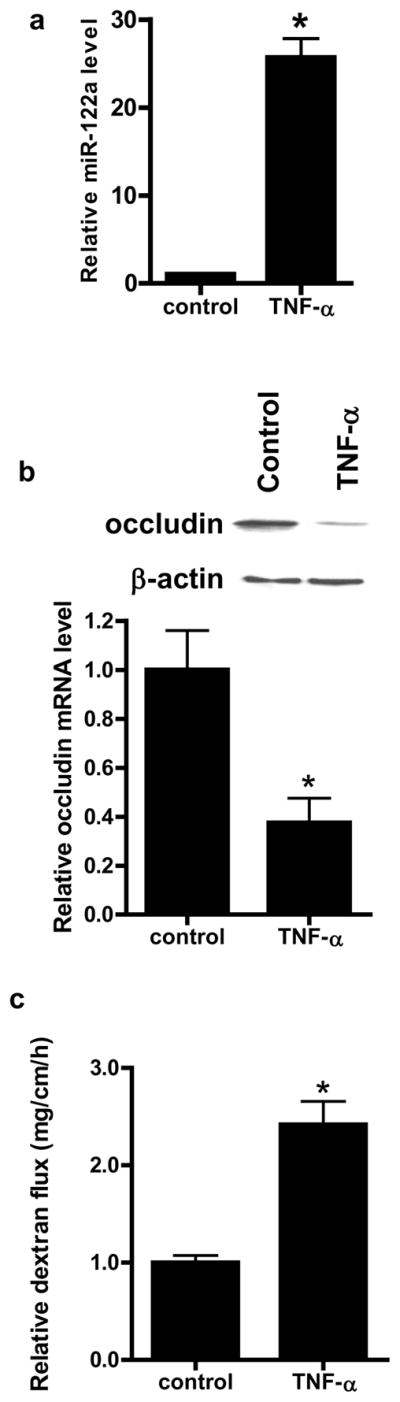
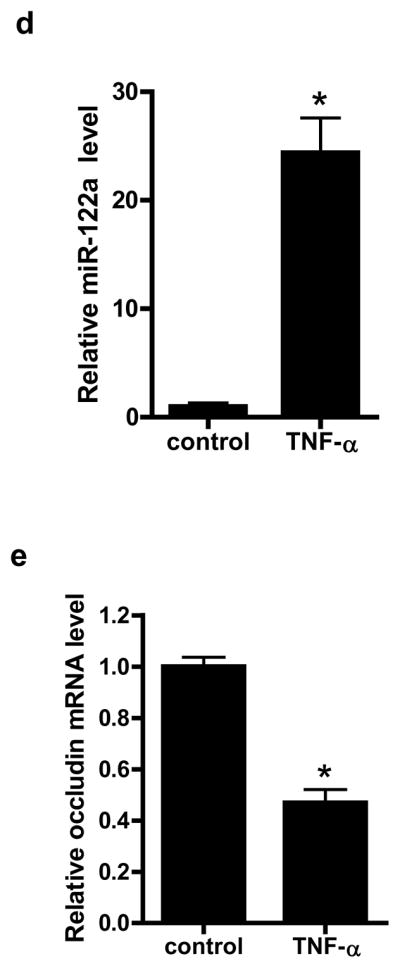
Effect of in-vivo administration of TNF-α on intestinal expression of miR-122a and occludin, and mouse intestinal permeability after 24 h treatment period. (a) Effect of introperitoneal (i.p.) TNF-α (5 μg) on miR-122a expression in small intestinal tissue. (b) Effect of i.p. TNF-α on occludin mRNA and protein expression in small intestinal tissue. (c) Effect of i.p. TNF-α on luminal-to-serosal FITC-dextran (MW = 10,000 g/mol) flux. (d) Effect of i.p. TNF-α on mouse enterocyte miR-122a expression. Mouse enterocytes were isolated using LCM. (e) Effect of i.p. TNF-α on mouse enterocyte occludin mRNA expression. * p < 0.05 vs control.
MiR-122a over-expression is sufficient to cause an increase in mouse small intestinal permeability in-vivo
To determine whether miR-122a over-expression in-vivo is sufficient to induce an increase in intestinal permeability, mouse intestinal mucosal surface was transfected with pre-miR-122a as decribed in the Methods. In brief, about 6 cm segment of small intestinal mucosal surface was transfected with pre-miR-122a in-vivo, and the effect of pre-miR-122a transfection on intestinal permeability determined 3 days after transfection (see Fig. 6a for the diagram of the experimental set-up). Pre-miR-122a transfection in-vivo resulted in an increase in miR-122a expression in mouse enterocytes (Fig. 6b) and an increase in mouse intestinal permeability (Fig. 6c). The pre-miR-122a transfection also caused a decrease in occludin mRNA and protein level (Fig. 6d, 6e). These results indicated that miR-122a over-expression was sufficient to cause an increase in intestinal permeability.
Figure 6.

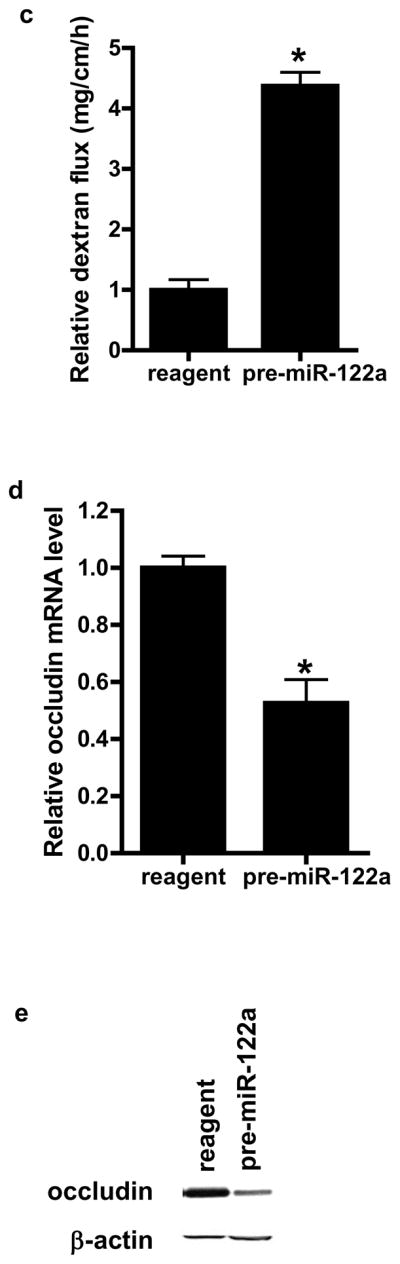
The effect of miR-122a over-expression on mouse small intestinal permeability in-vivo. (a) Experimental design for pre-miR-122a transfection in-vivo. (b) Mouse small intestinal mucosal surface were transfected with pre-miR-122a in-vivo. Effect of pre-miR-122a transfection on mouse enterocyte miR-122a level in-vivo. Enterocytes were isolated from the mucosal surface by LCM. (c) Effect of pre-miR-122a transfection on mouse intestinal permeability in-vivo. (d) Effect of pre-miR-122a transfection on enterocyte occludin mRNA level. (e) Effect of pre-miR-122a transfection on intestinal tissue occludin protein level. * p < 0.01 vs control.
Anti-sense inhibition of miR-122a expression inhibits the TNF-α induced increase in intestinal permeability in-vivo
To provide the proof-of-concept that miR-122a can be targeted in-vivo to preserve the intestinal epithelial barrier, we examined the effect of M-ASO inhibition of miR-122a on TNF-α induced increase in mouse intestinal permeability. In these studies, mouse intestinal mucosal surface was transfected with M-ASO directed against miR-122a in-vivo. M-ASO transfection in-vivo inhibited the TNF-α induced increase in miR-122a expression in mouse enterocytes and prevented the TNF-α induced increase in intestinal permeability (Fig. 7a, 7b). M-ASO transfection also prevented the decrease in enterocyte occludin mRNA expression (Fig. 7c). These results indicated that M-ASO inhibition of miR-122a expression inhibits the TNF-α induced increase in intestinal permeability.
Figure 7.
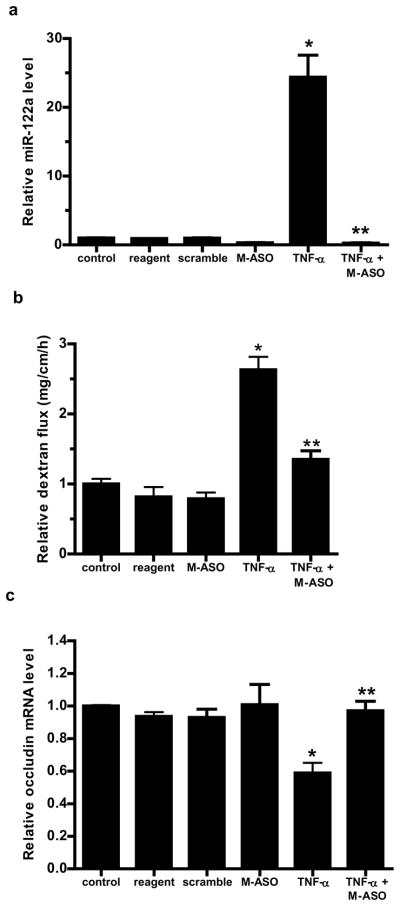

The effect of M-ASO inhibition of miR122a on mouse small intestinal permeability in-vivo and the schematic of miR-122a regulation of the intestinal epithelial TJ barrier. (a) Effect of M-ASO transfection on TNF-α induced increase in mouse enterocyte miR-122a expression was determined. Mouse intestinal mucosal surface was transfected with M-ASO or a scramble control in-vivo. (The “scramble” group was transfected with the transfecting reagent and a scramble control having the same nucleotide composition as M-ASO but not matching any known mRNA and miRNA sequences. The “reagent” group was transfected with transfecting reagent alone.) (b) Effect of M-ASO transfection on TNF-α induced increase in mouse intestinal permeability. (c) Effect of M-ASO transfection on TNF-α induced decrease in mouse enterocyte occludin mRNA expression. (d) Schematic of miR-122a regulation of intestinal TJ permeability. TNF-α induces an increase in miR-122a expression; miR-122a binds to 3′UTR of occludin mRNA, leading to occludin mRNA degradation, protein depletion and occludin depletion induced increase in TJ barrier permeability. * p < 0.01 vs control. ** p < 0.01 vs TNF-α treatment.
Discussion
Defective intestinal epithelial TJ barrier, leading to increased antigenic penetration, is an important pathogenic mechanism contributing to the development of intestinal inflammation in various intestinal permeability disorders1–3. Previous studies indicated that the enhancement of the TJ barrier prevents the development of intestinal inflammation3–5. Elucidation of the intracellular mechanisms that lead to intestinal TJ barrier disruption is essential in developing future therapeutic strategies to induce re-tightening of the TJ barrier3. Herein we describe a novel, physiologically relevant mechanism of intestinal TJ barrier regulation. Our data show that miR-122a plays a central role in the TNF-α modulation of intestinal TJ barrier. TNF-α caused a rapid increase in miR-122a level in filter-grown Caco-2 monolayers; and anti-sense inhibition of miR-122a prevented the increase in Caco-2 TJ permeability, indicating that the increase in miR-122a was required for the increase in TJ permeability. The in-vivo intestinal perfusion studies also confirmed that the TNF-α induced increase in mouse intestinal permeability was mediated by an increase in enterocyte miR-122a expression; and that over-expression of miR-122a was sufficient to induce an increase in mouse intestinal permeability. These studies demonstrated that miR-122a expression was an essential intracellular process mediating the TNF-α induced increase in intestinal TJ permeability.
MiRNAs are small RNA molecules that bind to the non-coding region of mRNA and regulate mRNA activity by inducing mRNA degradation or suppressing mRNA activity23, 24. MiRNAs are an essential component of transcriptional output of genomes of both animals and plants and they serve as an important regulator of biological response23, 24. MiRNAs regulate thousands of protein-coding genes23, 24; however, the vast majority of miRNA regulated protein-coding genes have yet to be identified. Our data show that miR-122a regulates intestinal TJ permeability by targeting occludin mRNA degradation. The bioinformatics data indicated a perfect complementary match with miR-122a binding sequence on occludin 3′UTR, predicting 98 % likelihood of functional activity. TNF-α caused a dramatic increase (~80-fold increase) in miR-122a expression. The 3′UTR studies suggested that miR-122a binding to the binding motif on 3′UTR suppressed occludin 3′UTR. Moreover, TNF-α induced increase in miR-122a expression caused a rapid degradation of occludin mRNA and depletion of occludin. The anti-sense inhibition of miR-122a prevented the TNF-α induced degradation of occludin mRNA, depletion of occludin, and increase in intestinal TJ permeability in-vitro and in-vivo, confirming that the miR-122a induced degradation of occludin mRNA was responsible for the increase in intestinal permeability.
The possibility that miR-122a may affect other transmembrane proteins was also considered but bioinformatics analysis did not reveal any miR-122a binding sequence on 3′UTR of claudins or JAM family of proteins. Our data also indicated that the TNF-α induced depletion of occludin was protein specific and did not extend to other transmembrane TJ proteins. TNF-α caused an increase in claudin-2 and claudin-8 expression and did not affect expression of claudin-1, claudin-3, or claudin-5. Since increase in claudin-8 expression causes an enhancement in TJ barrier27, the increase in claudin-8 levels could not have contributed to the observed increase in TJ permeability. Claudin-2 has been shown to be an important component of the TJ pore pathway, which is responsible for flux of ions and small solutes having molecular radius < 4 Å28. The pore pathway is functional under physiological conditions. Although unlikely, the possibility that the increase in claudin-2 expression may have contributed to the increase in inulin flux (molecular radius = 15 Å) was also considered. However, siRNA induced silencing of claudin-2 expression did not affect the TNF-α induced increase in inulin flux (data not shown), indicating that claudin-2 expression was not involved in the increase in inulin flux.
Occludin was the first transmembrane TJ protein to be discovered29. Previous studies have firmly established the role of occludin in TJ barrier function. Earlier studies have shown occludin to be localized at the TJ strands and that over-expression of occludin in fibroblasts conferred an adhesive property to these cells19, 30. Occludin over-expression in MDCK cells resulted in an increase in TJ strand and barrier function31; and decrease in occludin level correlated with a loss of TJ barrier function32. Although occludin knock-out mice did not appear to have obvious defect in TJ barrier function33, the studies were limited to a single time point in mature mice and compensatory or adaptive changes could not be ruled out. More recent studies have shown that occludin knock-down in intestinal epithelial cells in-vitro and in-vivo leads to an increase in intestinal TJ permeability via a non-restrictive or “leak” pathway and that occludin depletion leads to a preferential increase in flux of macromolecules34.
A noteworthy technological advance of the present study is the successful transfection of mouse enterocytes with pre-miR-122a in-vivo to induce over-expression of miR-122a and cause an increase in intestinal permeability. These studies also show the feasibility of selectively transfecting mouse enterocytes with anti-sense oligoribonucleotide in-vivo to inhibit the miR-122a mediated degradation of occludin mRNA and increase in intestinal permeability. As far as we are aware, this is the first report demonstrating the feasibility of enterocyte transfection of miRNA and M-ASO in-vivo to affect physiological activity.
In conclusion, in this report we describe a novel, physiologically relevant mechanism of intestinal epithelial TJ barrier regulation in vitro and in-vivo (Fig. 7d). Our data show for the first time that the TNF-α induced increase in intestinal epithelial TJ permeability was regulated by an increase in miR-122a expression. TNF-α induces a rapid increase in miR-122a expression in enterocytes; miR-122a binds to occludin 3′UTR and induces a degradation of occludin mRNA, leading to occludin depletion in enterocytes and an increase in TJ permeability. Our results also show the feasibility of targeting enterocyte miR-122a expression in-vivo to modulate intestinal permeability. Since defective intestinal TJ barrier has been shown to be an important pathogenic factor contributing to the development of intestinal inflammation3, elucidation of the intracellular and molecular targets that mediate intestinal TJ barrier has important future therapeutic implications. Mir-122a, as described here, represents a potential therapeutic target to induce re-tightening of intestinal TJ barrier in intestinal permeability disorders.
MATERIALS and METHODS
Cell cultures
Caco-2 cells were maintained in a culture medium as previously described7. The cells were subcultured by partial digestion with 0.25 % trypsin and 0.9 mmol/l EDTA in Ca2+-free and Mg2+-free PBS.
Cloning
Construction of occludin promoter reporter was carried out using the pGL-3 basic luciferase reporter vector. The primers used and PCR condition were as previously described35. Construction of occludin 3′UTR reporter was carried out using the pMIR-REPORT reporter vector. The primers used for cloning occludin 3′UTR are: ocln3′UTR1 5′-CGAGCTCAAGGCTGATGCCAAGTTGTTTGAG-3′; ocln3′UTR2 5′-CCCAAGCTTACAGAGGAGGCTGGTAGATCATCA-3′. Deletion construct of miR-122a binding site was performed by using the GeneTailor Site-Directed Mutagenesis System (Invitrogen) following manufacturer’s protocol. The sequence was confirmed by DNA services at University of New Mexico.
Transfection of DNA constructs and luciferase assay
Caco-2 cells were transfected using lipofectamine 2000. The experiments were carried out 48 h after transfection. Luciferase activity was determined using the duel luciferase assay kit (Promega) as previously described36.
Western blot
Occludin protein expression from Caco-2 cells and mouse tissue was assessed by western blot as previously described15. 10 μg of protein from each sample was loaded into a SDS-PAGE gel. The gel was transblotted against anti-occludin antibody, and developed using the Santa Cruz Western Blotting Luminol Reagents (Santa Cruz Biotechnology) on the Kodak BioMax MS film (Fisher Scientific).
Determination of Caco-2 paracellular permeability
Caco-2 paracellular permeability was determined using an established paracellular marker inulin15. Known concentrations of permeability marker (2 μM) and its radioactive tracer were added to the apical solution. Low concentrations of permeability marker were used to ensure that negligible osmotic or concentration gradient was introduced.
RNA isolation and reverse transcription
Total RNA was isolated using the miRNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Total RNA concentration was determined by absorbance at 260/280 nm using SpectrraMax 190 (Molecular Devices). The reverse transcription (RT) was performed using the GeneAmp Gold RNA PCR core kit (Applied Biosystems, Foster city, CA) as previously described36.
Quantification of gene expression using real-time PCR
The real-time PCRs were performed using an ABI Prism 7900 sequence detection system (Applied Biosystems) as previously described36. TaqMan universal PCR master mix kit was used for determine occludin expression, and power SYBR green PCR master mix kit was used to determine miRNA expression. GAPDH was used as an internal control. Primer sets used for occludin and GAPDH are as previously described35. Primer sets for miR-122a, miR-200b, and miR-200c were purchased from Applied Biosystems. For each sample, real-time PCR reactions were performed in triplicate, and the average threshold cycle (Ct) was calculated. Series dilution of RT reaction was used to cover different RNA expression level. Standard curve was generated to convert the Ct to copy numbers. The average copy number in control samples was set to 1.0. The relative expression in treated samples was determined as a fold increase compared with control samples.
Determination of mouse intestinal permeability in-vivo
TNF-α effect on intestinal permeability in an in-vivo mouse model system was established using a re-cycling intestinal perfusion method17. TNF-α (5 μg)25 was injected intraperitoneally into the mouse for 24 h, a 6 cm segment of mouse small intestine was isolated and cannulated with a small-diameter plastic tube (in an anesthetized mouse) and continuously perfused with 5 ml Krebs-phosphate saline buffer for a 2 h perfusion period. An external recirculating pump was used to recirculate the perfusate at a constant flow rate (0.75 ml/min). The body temperature of the mouse was maintained at 37 °C with a temperature-controlled warming blanket. The intestinal permeability was assessed by measuring luminal-to-serosal flux rate of paracellular probe, FITC-labeled dextran (MW = 10,000 g/mol). The water absorption was determined by using a non-absorbable marker sodium ferrocyanide or by measuring the difference between initial and final volume of the perfusate17.
In-vivo transfection of miR-122a precursor and antisense
MiR-122a effect on mouse small intestinal TJ permeability was determined using the perfusion model described above. In these studies, with the abdominal cavity open, a 6 cm segment of mouse small intestine was isolated and cannulated with plastic tubing. The transfection solution (0.5 ml) consisting of pre-miR-122a (25 nM) or M-ASO (25 nM) or scramble control and lipofectamine (50 μl) was injected through a 30-gauge needle into the lumen of small intestine and the small intestine was cannulated for 1 h. The small intestine was then placed back into the abdominal cavity, and the abdominal cavity was closed with sutures. The mouse was allowed to recover for three days before the permeability studies.
Laser capture microdissection
Frozen mouse tissue sections were fixed with 75 % ethanol for 30 sec; haematoxylin stained for 20 sec; and dehydrated with 75 % ethanol, 30 sec; 95 % ethanol, 30 sec; 100 % ethanol, 30 sec; xylene, 5 min. After dehydration, sections were air dried for 5 min. The arcturus PixCell II system (Molecular Devices, Sunnyvale, CA) was used for microdissection. The sections were captured using a 7.5 μM diameter laser beam typically at 80 to 100 mV power with pulse duration of 0.5 to 1.0 ms. On average, about 500 shots were taken per cap, and approximately 1000 cells were obtained per cap. Microdissection caps were inserted into 0.5 ml microcentrifuge tubes containing 350 μl of lysis buffer and total RNA was isolated.
Statistical Analysis
The values of experimental data were expressed as the mean ± S.E., and analyzed using paired t test (Graph Pad Prizm 4.00 for Windows, GraphPad Software, San Diego, CA). P values of < 0.05 were considered significant. All experiments were repeated at least three times to ensure reproducibility.
Acknowledgments
Grant support: This research project was supported by a Veterans Affairs (VA) Merit Review grant from the VA Research Service and National Institute of Diabetes and Digestive and Kidney Diseases Grant RO 1-DK-64165–01 and RO 1-DK-081429 (to T. Y. Ma).
Abbreviations
- Ct
threshold cycle
- LCM
laser capture microdissection
- M-ASO
2′-0-methyl-modified anti-sense oligoribonucleotide
- TJ
tight junction
- TNF-α
Tumor necrosis factor-α
Footnotes
Author contributions: Dongmei Ye designed and conducted experiments and wrote the manuscript, and Shuhong Guo conducted the in-vivo experiments. Rana Al-Sadi conducted western-blot and Caco-2 permeability studies. Thomas Ma evaluated the results, supervised this study and wrote the manuscript.
Disclosures: This is no conflict of interest for all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ma TY, Anderson JM. Tight junction and intestinal barrier. In: Johnson LR, editor. Textbook of Gastrointestinal Physiology. 2006. pp. 1559–94. [Google Scholar]
- 2.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 3.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–8. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–9. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 5.Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol. 2000;35:1163–9. doi: 10.1080/003655200750056637. [DOI] [PubMed] [Google Scholar]
- 6.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 7.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–30. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 8.Ciccocioppo R, Finamore A, Ara C, Di Sabatino A, Mengheri E, Corazza GR. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am J Clin Pathol. 2006;125:502–11. doi: 10.1309/DTYR-A91G-8R0K-TM8M. [DOI] [PubMed] [Google Scholar]
- 9.Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermuller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–28. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]
- 10.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study. Group N Engl J Med. 1997;337:1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 12.Van Deventer SJ. Tumour necrosis factor and Crohn’s disease. Gut. 1997;40:443–8. doi: 10.1136/gut.40.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJ, Present D, Sands BE, Colombel JF. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–76. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 14.Gillett HR, Arnott ID, McIntyre M, Campbell S, Dahele A, Priest M, Jackson R, Ghosh S. Successful infliximab treatment for steroid-refractory celiac disease: a case report. Gastroenterology. 2002;122:800–5. doi: 10.1053/gast.2002.31874. [DOI] [PubMed] [Google Scholar]
- 15.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–76. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 16.Schwarz BT, Wang F, Shen L, Clayburgh DR, Su L, Wang Y, Fu YX, Turner JR. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology. 2007;132:2383–94. doi: 10.1053/j.gastro.2007.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–15. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrier L, Mazelin L, Cenac N, Desreumaux P, Janin A, Emilie D, Colombel JF, Garcia-Villar R, Fioramonti J, Bueno L. Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology. 2003;125:795–804. doi: 10.1016/s0016-5085(03)01057-6. [DOI] [PubMed] [Google Scholar]
- 19.Fujimoto K. Freeze-fracture replica electron microscopy combined with SDS digestion for cytochemical labeling of integral membrane proteins. Application to the immunogold labeling of intercellular junctional complexes. J Cell Sci. 1995;108 (Pt 11):3443–9. doi: 10.1242/jcs.108.11.3443. [DOI] [PubMed] [Google Scholar]
- 20.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 21.Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet. 1992;339:89–91. doi: 10.1016/0140-6736(92)90999-j. [DOI] [PubMed] [Google Scholar]
- 22.Ma TY, Hollander D, Erickson RA, Truong H, Krugliak P. Is the small intestinal epithelium truly “tight” to inulin permeation? Am J Physiol. 1991;260:G669–76. doi: 10.1152/ajpgi.1991.260.5.G669. [DOI] [PubMed] [Google Scholar]
- 23.Krutzfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet. 2006;38 (Suppl):S14–9. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- 24.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38 (Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 25.Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–94. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George MD, Wehkamp J, Kays RJ, Leutenegger CM, Sabir S, Grishina I, Dandekar S, Bevins CL. In vivo gene expression profiling of human intestinal epithelial cells: analysis by laser microdissection of formalin fixed tissues. BMC Genomics. 2008;9:209. doi: 10.1186/1471-2164-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem. 2003;278:17350–9. doi: 10.1074/jbc.M213286200. [DOI] [PubMed] [Google Scholar]
- 28.Van Itallie CM, Holmes J, Bridges A, Anderson JM. Claudin-2-dependent changes in noncharged solute flux are mediated by the extracellular domains and require attachment to the PDZ-scaffold. Ann N Y Acad Sci. 2009;1165:82–7. doi: 10.1111/j.1749-6632.2009.04052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Itallie CM, Anderson JM. Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci. 1997;110 (Pt 9):1113–21. doi: 10.1242/jcs.110.9.1113. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy KM, Skare IB, Stankewich MC, Furuse M, Tsukita S, Rogers RA, Lynch RD, Schneeberger EE. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109 (Pt 9):2287–98. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 32.Li D, Mrsny RJ. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J Cell Biol. 2000;148:791–800. doi: 10.1083/jcb.148.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, Saitou M, Tsukita S, Fromm M. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669:34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma TY. Occludin Regulates Macromolecule Flux across the Intestinal Epithelial Tight Junction Barrier. Am J Physiol Gastrointest Liver Physiol. 2011 doi: 10.1152/ajpgi.00055.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dokladny K, Ye D, Kennedy JC, Moseley PL, Ma TY. Cellular and molecular mechanisms of heat stress-induced up-regulation of occludin protein expression: regulatory role of heat shock factor-1. Am J Pathol. 2008;172:659–70. doi: 10.2353/ajpath.2008.070522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye D, Ma TY. Cellular and molecular mechanisms that mediate basal and tumour necrosis factor-alpha-induced regulation of myosin light chain kinase gene activity. J Cell Mol Med. 2008;12:1331–46. doi: 10.1111/j.1582-4934.2008.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]