

Abstract
A defective intestinal epithelial tight junction (TJ) barrier has been proposed as an important pathogenic factor contributing to the intestinal inflammation of Crohn's disease. Glucocorticoids are first-line therapeutic agents for the treatment of moderate to severe Crohn's disease. Glucocorticoid treatment has been shown to induce retightening of the intestinal TJ barrier defect in Crohn's disease patients. However, the mechanisms that mediate the glucocorticoid therapeutic action on intestinal TJ barrier function remain unknown. The aim of this study was to elucidate the mechanism of glucocorticoid modulation of the intestinal epithelial TJ barrier using an in vitro model system. Filter-grown Caco-2 intestinal epithelial cells were used as an in vitro model to examine the effects of glucocorticoids on basal intestinal epithelial TJ barrier function and on TNF-α-induced disruption of the TJ barrier. Glucocorticoids (prednisolone and dexamethasone) did not have a significant effect on baseline Caco-2 TJ barrier function but prevented the TNF-α-induced increase in Caco-2 TJ permeability. The glucocorticoid protective effect against the TNF-α-induced increase in Caco-2 TJ permeability required activation of the glucocorticoid receptor (GR) complex. The activation of the GR complex resulted in GR complex binding to the glucocorticoid response element (GRE) site on DNA and activation of a GR-responsive promoter. Glucocorticoids inhibited the TNF-α-induced increase in myosin light chain kinase (MLCK) protein expression, a key process mediating the TNF-α increase in intestinal TJ permeability. The glucocorticoid inhibition of the TNF-α-induced increase in MLCK protein expression was due to the binding of the GR complex to a GRE binding site on the MLCK promoter region suppressing the TNF-α-induced activation. Glucocorticoids inhibit the TNF-α-induced increase in Caco-2 TJ permeability. The prednisolone protective action was mediated by binding of activated GR complex to the GRE site on the MLCK promoter, suppressing the TNF-α-induced increase in MLCK gene activity, protein expression, and subsequent opening of the intestinal TJ barrier.
Keywords: prednisolone, inflammation, tumor necrosis factor-α, glucocorticoid receptor, glucocorticoid response element, Crohn's disease
The intrinsic intestinal epithelial barrier consists of the apical plasma membrane and the tight junctions (TJ) of intestinal epithelial cells (12, 20). The plasma membrane (consisting of a lipid bilayer) acts as a transcellular barrier while the inter-cellular TJs provide the paracellular barrier function against the transepithelial permeation of water soluble molecules from the intestinal lumen (8, 11). A defective intestinal TJ barrier has been proposed as an important pathogenic factor leading to inflammation in Crohn's disease (CD) and other inflammatory diseases, including alcoholic liver disease, celiac disease, postinfectious irritable bowel syndrome, and various infectious diarrheal syndromes (1, 12, 20, 29, 32). CD is a chronic idiopathic inflammatory bowel disease characterized by recurrent inflammation of the gastrointestinal tract (26). It is well established that patients with CD have a defect in the intestinal TJ barrier as evidenced by an increase in intestinal permeability (8–11, 38). Clinical studies have shown that the intestinal TJ barrier defect in CD patients predates the onset of active disease and is a positive predictive factor for early recurrence of the disease following treatment (2, 11, 41). A direct correlation exists between normalization of intestinal permeability and clinical improvement of active disease following medical therapy (23, 33, 40). It is also well recognized that environmental factors (e.g., NSAID ingestion and bacterial or viral infection) that cause disruption of the intestinal TJ barrier induce exacerbation of CD (11), whereas therapeutic agents that induce retightening of the intestinal TJ barrier or decrease the luminal antigenic load lead to resolution of active inflammation (12, 23, 24, 33, 40). Thus it has been proposed that an important therapeutic strategy in CD is to induce retightening of the leaky intestinal TJ barrier during active inflammation (7–11).
Glucocorticoids (GCs) are first-line therapeutic agents for the treatment of moderate-to-severe active CD (28, 30). GC treatment has been shown to result in clinical improvement in ~60–90% of treated patients with more severe disease (26, 28). In clinical intestinal permeability studies, GC treatment of patients with active CD results in a significant decrease in intestinal permeability in the majority of treated patients (23, 40). The GC-induced normalization of intestinal permeability was a positive predictive factor of prolonged remission of the disease (40). Furthermore, those patients with a persistent elevation in intestinal permeability following GC therapy had high rates of early relapse (40). Although it is well established that GC therapy leads to normalization of intestinal permeability (23, 40, 41), it remains unclear whether the GC-induced normalization of the intestinal TJ barrier function is due to the anti-inflammatory effects of GCs leading to healing of the inflamed mucosal epithelial surface or due to a direct TJ barrier “tightening” effects of GCs.
The central role of cytokines in the intestinal inflammation of CD has been established by studies showing elevation of the proinflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1β, and interferon-γ during active inflammation and the therapeutic efficacy of anti-TNF-α antibody in the treatment of active inflammation (27, 35). Previous studies have demonstrated that TNF-α at clinically relevant concentrations (1–10 ng/ml) causes an increase in intestinal epithelial TJ permeability (6, 16). It has been suggested that the elevated levels of TNF-α present in CD patients are responsible for the observed increases in intestinal permeability seen in these patients (6, 7, 16). Consistent with this possibility, anti-TNF-α treatment of patients with active CD results in normalization of intestinal permeability (33). On the basis of these studies, we hypothesized that a potential mechanism of GC modulation of intestinal TJ barrier function during active inflammation may be to attenuate the TNF-α-induced increase in intestinal TJ permeability.
The major aim of this study was to investigate the role of GCs in the modulation of intestinal epithelial TJ barrier function and to elucidate the intracellular mechanisms involved, by using an in vitro intestinal epithelial model system consisting of filter-grown Caco-2 monolayers. The use of an in vitro intestinal epithelial monolayer system allowed us to directly assess the effects of GCs (and their mechanisms of action) on intestinal TJ barrier function, in the absence of humoral, vascular, immunological, or other systemic factors. In this study, we examined the effects of GCs on basal intestinal TJ barrier function and on the TNF-α-induced increase in intestinal TJ permeability. The results of our study demonstrate for the first time that GCs have a direct protective action against the TNF-α-induced increase in intestinal TJ permeability. Additionally, our data provide new insight into the intracellular mechanisms that mediate prednisolone's protective effect against the TNF-α-induced disturbance of intestinal TJ barrier function.
MATERIALS AND METHODS
Materials
Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Atlanta, GA). Trypsin, penicillin, streptomycin, and phosphate-buffered saline solution (PBS) were purchased from Invitrogen (Carlsbad, CA). TNF-α was purchased from R&D Systems (Minneapolis, MN). Dulbecco's modified Eagle's medium (DMEM), GCs, RU-486, and antibodies for Western blot analysis were purchased from Sigma (St. Louis, MO). ELISA reagents were obtained from Active Motif (Carlsbad, CA). Transwell permeable filters were purchased from Corning (Corning, NY). Primers were from Integrated DNA Technologies (Coralville, IA). Luciferase assay reagents were from Promega (Madison, WI). Transfection and cloning reagents were from Invitrogen. Secreted embryonic alkaline phosphatase (SEAP) assay reagents were obtained from Clontech (Palo Alto, CA).
Cell cultures
Caco-2 cells were purchased from the American Type Culture Collection (Rockville, MD) at passages 18 to 20. The cells were maintained in a culture medium composed of DMEM with 4.5 mg/ml glucose, 50 U/ml penicillin, 50 U/ml streptomycin, and 10% FBS. The cells were kept at 37°C in a 5% CO2 environment. Culture medium was changed every 2 days. The cells were subcultured by partial digestion with 0.25% trypsin and 0.9 mmol/l EDTA in Ca2+-free and Mg2+-free PBS every 7 days.
Determination of Caco-2 epithelial monolayer resistance and paracellular permeability
The effect of TNF-α on Caco-2 monolayer epithelial electrical resistance was measured by epithelial volt-ohmmeter (World Precision Instruments, Sarasota, FL) as previously described (16). For resistance measurements, both apical and basolateral sides of the epithelia were bathed in regular growth medium. Each experiment was repeated at least 3 times in quadruplicate to ensure reproducibility. The transepithelial electrical resistance (TER) was reported after subtraction of the resistance value of the filters alone and was in the range of 400–500 Ω·cm2. Filter-grown Caco-2 cells were treated with selected GCs 1 h before the addition of TNF-α. When relevant, cells were treated with RU-486 30 min before the addition of prednisolone. The effect of TNF-α and prednisolone on Caco-2 monolayer paracellular permeability was determined by use of the extracellular marker inulin (16). [14C]inulin (Perkin-Elmer, Boston, MA) was added to the apical compartment, and the basolateral chamber was sampled at desired time points to assess the transepithelial flux rates. Low concentrations of inulin were used to ensure negligible osmotic gradients. Measurement of [14C]inulin was by a Packard (Perkin-Elmer) liquid scintillation counter. All experiments were repeated three to five times to ensure reproducibility.
Immunofluorescent protein localization
Cellular localization of GC receptor (GR) protein in filter-grown Caco-2 cells was assessed by immunofluorescent antibody labeling as previously described (14, 16). Caco-2 cells were grown on Transwell filters, fixed with 2% formaldehyde, and permeabilized with 0.1% Triton X-100. After being blocked in 1% BSA/5% NDS for 1 h, the Caco-2 monolayers were then labeled with rabbit anti-GR antibody (Oncogene, Carpinteria, CA); this was followed by incubation with secondary Cy3 goat anti-rabbit antibody (Jackson Immunolaboratories, West Grove, PA). Mowiol-Na gallate was used to mount the filters onto coverslips. The slides were imaged on a Nikon (Melville, NY) fluorescent microscope with an Orca-Hamamatsu (Bridgewater, NJ) camera.
ELISA-based transcription factor binding assay
Assessment of GR binding to a consensus GC response element (GRE) DNA oligonucleotide was determined by using the TransAM GR activation assay kit from Active Motif. Nuclear proteins were extracted per manufacturer's protocol with the Active Motif nuclear extract kit. Equal amounts of nuclear protein (Bradford assay) were loaded (5–10 μg) according to the manufacturer's protocol. Nuclear proteins were placed in the respective wells and exposed to plate-bound oligonucleotide (containing a consensus GRE sequence: GGTACAnnnTGTTCT), followed by washes and addition of a rabbit primary antibody to GR. After a further wash, the oligonucleotide-bound protein was incubated with an horseradish peroxidase-conjugated anti-rabbit secondary antibody, washed again, and developed. Absorbance for the ELISA assay was determined on a SpectraMax 190 plate reader at 450 nm (Molecular Devices, Sunnyvale, CA).
Transfection of DNA constructs
DNA constructs of promoters were transiently transfected into Caco-2 cells by use of the transfection reagent Lipofectamine 2000 (Life Technologies). Caco-2 cells grown on Transwell six-well filters were transfected with 1 μg of pGL3 basic plasmid [containing the 2,019 kb 5′ promoter region upstream of the myosin light chain kinase (MLCK) gene (MYLK: NM_053025) (42) and a firefly luciferase reporter gene] or 1 μg of pGRE-SEAP (Clontech). Renilla luciferase vector (pRL-TK, Promega) was cotransfected with each plasmid construct as an internal control. Cells (5 × 105 cells/filter) were seeded into a six-well Transwell plate and grown to confluency. Caco-2 monolayers were then washed with PBS twice, and 1.0 ml of Opti-MEM medium was added to the apical compartment of each filter and 1.5 ml was added to the basolateral compartment of each filter; 1 μg of each plasmid construct and 0.25 μg of pRL-TK or 2 μl of Lipofectamine 2000 were preincubated in 250 μl of Opti-MEM, respectively. After 5 min of incubation, the two solutions were mixed and incubated for another 20 min, and the mixture was added to the apical compartment of each filter. After an incubation for 3 h at 37°C, 500 μl of DMEM containing 10% FBS without antibiotics were added to both sides of the filter to reach a 2.5% final concentration of FBS. Subsequently, media were replaced with normal Caco-2 growth medium 16 h after transfection. The experiments were carried out 48 h after transfection.
Luciferase assay
After the TNF-α treatment (7 h), Caco-2 cells were washed twice with 1 ml of ice-cold PBS, followed by the addition of 400 μl of 1× passive lysis buffer, incubated at room temperature for 15 min, scraped, transferred into an Eppendorf tube, and centrifuged for 15 s at 13,000 RPM in a microcentrifuge. Luciferase activity was determined using the dual luciferase assay kit (Promega); 20 μl of the supernatant were used for each assay. Luciferase values were determined by Lumat LB 9507 (EG&G Berthold, Oak Ridge, TN). The values of reporter luciferase activities were then divided by those of Renilla luciferase activities to normalize for differences in transfection efficiencies. The average activity value of the control samples was set to 1.0. The luciferase activity of the MLCK promoter in treated samples was determined relative to the control samples.
SEAP assay
SEAP activity was determined by using the Great EsCAPE chemiluminescence kit from Clontech, following the manufacturer's protocol. In brief, in cells transfected with pGRE-SEAP, 110 μl of medium were collected from the apical surface 24 h after treatment with selected test reagents. The medium was centrifuged at 5,000 rpm for 1 min, and 100 μl were collected. The medium was mixed 1:3 with supplied dilution buffer and heated to 65°C for 30 min. Assay buffer was added and then CPSD developing solution. Luminescence was determined on a Veritas microplate luminometer (Turner Biosystems, Sunnyvale, CA).
Site-directed mutagenesis
The four core base pairs (GTCC) of the GRE (GTGGCAGCAGATGTCCTGA) in the MLCK promoter were mutated by using the GeneTailor Site-Directed Mutagenesis System (Invitrogen). Briefly, primers were generated that included a 4-bp mutation TGAA flanked by a wild-type sequence on either side. A PCR reaction produced a new complete copy of the plasmid containing the mutation coded for by the primers. The mutation (GTCC → TGAA) was selected by substituting purines for pyrimidines and vice versa. The linear PCR product was subsequently transformed into DH5α-T1 Escherichia coli, which circularized the PCR product and digested any remaining parent plasmid. DNA sequence was verified by DNA services at University of New Mexico.
Assessment of protein expression by Western blot analysis
Caco-2 cells were pretreated with prednisolone for 1 h and then treated in combination with TNF-α for 48 h. Cells were washed twice with ice-cold PBS and collected with rubber policemen. After a centrifugation for 1 min at 14,000 rpm, the supernatant was discarded and the pellet was resuspended in triple-detergent lysis buffer and rotated end over end for 15 min. Protein level of the lysate was determined by Lowry assay. Equal amounts of protein (10 μg) were loaded with 2× SDS-gel loading buffer (Bio-Rad). The samples were boiled for 5 min and then run on a 6% PAGE gel at 120 V. The gel was transferred to a nitrocellulose membrane and probed with primary antibody (mouse anti-MLCK, Sigma) and secondary rabbit anti-mouse IgG2b antibody conjugated to horseradish peroxidase (Zymed). The gels were exposed on Kodak film using Luminol reagent (Santa Cruz, Santa Cruz, CA).
Statistical analysis and software
The values of experimental data were expressed as means ± SE and analyzed by an unpaired t-test (Graph Pad Prizm 4.00 for Windows, GraphPad Software, San Diego, CA). P values of <0.05 were considered significant. All experiments were repeated at least three times to ensure reproducibility. Promoter inspection for transcription factor binding sites was performed by using Genomatix (Genomatix software, Munich, Germany).
RESULTS
Effect of GCs on the Caco-2 intestinal epithelial TJ barrier
In the following studies, we examined the effect of GCs on Caco-2 intestinal epithelial TJ barrier function by measuring TER and paracellular permeability (14–16). Prednisolone treatment (1–5 μM) did not have a significant effect on baseline Caco-2 TER over the 48-h experimental period (Fig. 1A). Similar to the previous report, TNF-α caused a significant drop in TER at the 24- and 48-h time periods (Fig. 1B) (16). In our previous report, we showed that TNF-α (10 ng/ml) caused a time-dependent decrease in Caco-2 TER that began at 12 h and was sustained over at least 96 h (16). Prednisolone (5 μM) significantly reduced the TNF-α-induced drop in Caco-2 TER (Fig. 1C). Prednisolone inhibition of the TNF-α-induced drop in TER was dose dependent with maximal inhibition occurring at a dose of 5 μM (data not shown). An equipotent dose (1 μM) of dexamethasone (a more potent GC) produced a similar inhibition of the TNF-α-induced drop in Caco-2 TER (P < 0.01), indicating that the barrier protective effect of prednisolone extended to other GCs. Dexamethasone also did not affect the baseline Caco-2 TER. Next, the effect of prednisolone on the TNF-α-induced increase in Caco-2 paracellular permeability was determined using the extracellular marker inulin (5,000 g/mol molecular weight). TNF-α (10 ng/ml) treatment produced an approximately eightfold increase in mucosal-to-serosal inulin flux, and prednisolone (5 μM) completely prevented the TNF-α-induced increase in paracellular permeability (Fig. 2). Consistent with the epithelial resistance studies, prednisolone alone did not have any significant effect on the transepithelial flux of inulin. These results indicate that prednisolone does not affect the basal Caco-2 TJ barrier function but prevents the TNF-α-induced increase in TJ permeability.
Fig. 1.

Effect of prednisolone on the baseline and TNF-α-induced decrease in Caco-2 transepithelial electrical resistance (TER). A: time course of prednisolone effect on baseline Caco-2 TER. Filter-grown intestinal epithelial cells were treated with prednisolone (●,1 μM; ◯,5 μM) or vehicle (∎) over a 48-h experimental period. B: time course of TNF-α effect on TER. Filter-grown Caco-2 monolayers were treated with TNF-α or vehicle over a 24- and 48-h period (*P < 0.01). C: effect of prednisolone on the TNF-α-induced decrease in Caco-2 TER. Epithelial monolayers were treated with vehicle (control), TNF-α (10 ng/ml), or TNF-α+ prednisolone (5 μM) for 48 h. *P < 0.01 compared with control; **P < 0.01 compared with TNF-α.
Fig. 2.
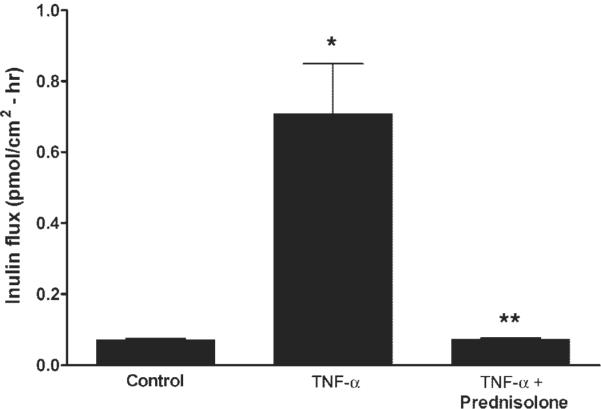
Effect of TNF-α and prednisolone on Caco-2 paracellular permeability. The paracellular marker [14C]inulin was added to the apical chamber of filter-grown Caco-2 cells. After 48 h, the basolateral compartment was sampled over a 1-h interval. Inulin permeability was determined in groups treated with vehicle (control), TNF-α (10 ng/ml), or TNF-α+ prednisolone (5 μM). *P < 0.01 compared with control; **P < 0.01 compared with TNF-α.
Prednisolone activation of GR is required for the prednisolone protective effect against the TNF-α-induced increase in Caco-2 TJ permeability
To delineate the intracellular processes involved in the GC protective effect against the TNF-α-induced increase in Caco-2 TJ permeability, we examined the effect of prednisolone on activation of GR complex in Caco-2 cells. In untreated Caco-2 cells, GR complex was present mostly in the cytoplasm in its inactive state (Fig. 3). Prednisolone stimulation of Caco-2 cells resulted in a rapid activation and cytoplasmic-to-nuclear translocation (within minutes) of activated GC-GR complexes as shown by immunostaining (Fig. 3). Correlating with the nuclear translocation of GR complex, prednisolone treatment resulted in an increase in binding of activated GR complexes to the GRE binding site on DNA, as determined by an ELISA-based DNA binding assay (Fig. 4A). In the GRE binding assay, we utilized an oligonucleotide probe containing a consensus GRE (GGTACAnnnTGTTCT) binding motif to assess the binding of GR complex to the GRE DNA site. As shown in Fig. 4A, prednisolone (5 μM) caused a significant increase in the binding of translocated GR complexes to the oligonucleotide probe. In subsequent studies, we also examined the effect of prednisolone on activation of a GR-responsive promoter. The plasmid vector used contained three tandem copies of a consensus GRE (GGTACAnnnTGTTCT) fused to a TATA promoter with SEAP as the reporter gene. As shown in Fig. 4B, prednisolone (5 μM) treatment resulted in an increase in GR-responsive promoter activity in the transfected Caco-2 cells as determined by SEAP reporter expression. Together, these findings suggested that the prednisolone-induced activation of GR complex leads to an increase in binding to the GRE site on DNA and activation of GR-responsive promoter activity.
Fig. 3.
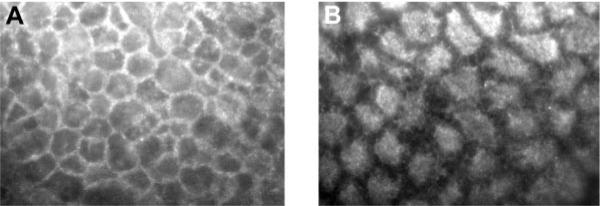
Prednisolone effect on glucocorticoid (GC) receptor (GR) localization. Filter-grown Caco-2 monolayers were immunolabeled as described in MATERIALS AND METHODS for determination of cytoplasmic to nuclear translocation of GR. A: control monolayers. B: Caco-2 cells after incubation with prednisolone (5 μM) for 30 min (magnification, ×400).
Fig. 4.

Prednisolone effect on GR binding to DNA and promoter activation. A: effect of prednisolone on GR binding to a consensus GC response element (GRE) DNA binding site using an ELISA-based GR DNA binding assay as described in MATERIALS AND METHODS. Cells were treated with vehicle (control) or prednisolone (5 μM for 30 min) or pretreated with RU-486 (0.5 μM for 30 min) followed by prednisolone treatment. *P < 0.01 compared with control; **P < 0.05 compared with prednisolone. B: effect of prednisolone on GRE-responsive secreted embryonic alkaline phosphatase (SEAP)-promoter activity. Caco-2 intestinal epithelial cells (transfected with pGRE-SEAP) were treated with vehicle (control), prednisolone (5 μM), or prednisolone + RU-486 (1 μM) for 24 h. *P < 0.05 compared with control; **P < 0.05 compared with prednisolone.
To further determine the role of GR complex activation in the prednisolone protective effect against the TNF-α-induced increase in Caco-2 TJ permeability, we examined the effect of the known GC antagonist RU-486 (which binds to GR and induces a conformational change in the receptor) on the prednisolone-induced protective effect (21). As shown in Fig. 4, RU-486 [at doses previously shown to inhibit the GR complex activation in other cell types (1 μM)] inhibited the prednisolone-induced increase in GR complex binding to GRE and the upregulation of GR-responsive promoter activity (21). These findings indicated that RU-486 inhibits the prednisolone-induced GR complex binding to GRE and subsequent modulation of gene activity in Caco-2 cells. We next examined the effect of RU-486 on prednisolone inhibition of the TNF-α-induced drop in Caco-2 TER. RU-486 completely prevented the protective effect of prednisolone on the TNF-α-induced drop in Caco-2 TER (Fig. 5A). To confirm that the early activation of GR complex was indeed important in the prednisolone protection against the TNF-α-induced drop in TER that occurs later (12–24 h), prednisolone was removed from the incubation solution after 6 h of exposure (before the observed drop in TER). TNF-α exposure for only 6 h still induced a similar decrease in Caco-2 TER at 24 h as continuous exposure. Prednisolone treatment for 6 h also resulted in a similar protection against the TNF-α-induced drop in TER. These findings suggested that early GR complex activation was required for the late prednisolone protective action against the TNF-α-induced increase in Caco-2 TJ permeability.
Fig. 5.

A: effect of RU-486 on prednisolone protection of the TNF-α-induced drop in Caco-2 TER. Filter-grown epithelial monolayers were treated with vehicle (control), TNF-α (10 ng/ml), TNF-α + prednisolone (5 μM), or TNF-α, prednisolone, and RU-486 (1 μM) over a 48-h period. B: effect of short exposure (6 h) time of TNF-α and prednisolone on Caco-2 TER. Caco-2 cells were incubated with vehicle, TNF-α 10 ng/ml, or TNF-α+ prednisolone (5 μM) for 6 h and then replaced with regular medium. The effect of 6-h exposure on Caco-2 TER was determined at 24 h. *P < 0.01 compared with control; **P < 0.01 compared with TNF; ***P < 0.01 compared with prednisolone.
Prednisolone protection against the TNF-α-induced increase in Caco-2 TJ permeability is mediated by GR complex inhibition of the TNF-α-induced increase in MLCK promoter activity
Previous studies have shown that the TNF-α-induced increase in Caco-2 TJ permeability was mediated by a TNF-α-induced increase in MLCK protein expression and that inhibition of TNF-α-induced increase in MLCK protein expression prevents the TNF-α increase in Caco-2 TJ permeability (13, 42). The time course of increase in MLCK expression mirrors that of the permeability increase, with both beginning at ~12 h and reaching their maximum effect by 48 h (13). In the following studies, we examined the possibility that the prednisolone protective effect was also due to inhibition of TNF-α-induced increase in MLCK protein expression. As previously reported, TNF-α (10 ng/ml) caused an increase in MLCK protein expression (Fig. 6). Prednisolone (5 μM) treatment inhibited the TNF-α-induced increase in MLCK expression (Fig. 6). Prednisolone, by itself, did not affect MLCK protein expression. These results suggested that the prednisolone protective effect on the TNF-α increase in Caco-2 TJ permeability was due to inhibition of the TNF-α-induced increase in MLCK protein expression. To investigate the mechanism of prednisolone inhibition of MLCK protein expression, we examined the effect of prednisolone on the TNF-α-induced increase in MLCK promoter activity. In the following studies, we used the recently described 2,091-bp MLCK promoter region (42) (NM 182493) to assess the role of prednisolone on MLCK promoter activity. As shown in Fig. 7, TNF-α treatment resulted in a significant increase in MLCK promoter activity (13). Prednisolone, by itself, did not affect the basal activity of the MLCK promoter. Prednisolone (5 μM) prevented the TNF-α increase in MLCK promoter activity (Fig. 7). The GC antagonist RU-486 (1 μM) prevented the prednisolone inhibition of the TNF-α increase in MLCK promoter activity (Fig. 7), indicating that GR complex activation was required for the prednisolone inhibition of MLCK promoter activity.
Fig. 6.

Effect of TNF-α and prednisolone on MLCK protein expression. Caco-2 myosin light chain kinase (MLCK) expression was assessed by Western blotting; bands are localized at ~210 kDa. Experimental groups were treated with vehicle (control), TNF-α (10 ng/ml), or TNF-α+ prednisolone (5 μM) for 48 h.
Fig. 7.
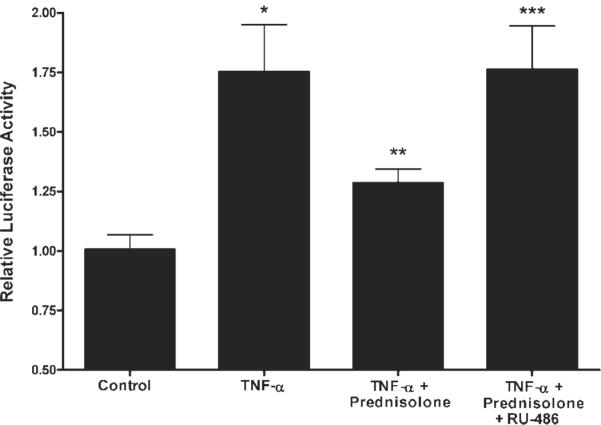
Effect of prednisolone on the TNF-α-induced upregulation of MLCK promoter activity. The pGL3 vector (containing the full-length MLCK promoter with a luciferase reporter) was transfected into Caco-2 cells as described in MATERIALS AND METHODS. Cells were treated with vehicle (control), TNF-α (10 ng/ml), TNF-α+ prednisolone (5 μM), or TNF-α, prednisolone, and RU-486 (1 μM)for6h.*P < 0.01 compared with control; **P < 0.05 compared with TNF-α; ***P < 0.05 compared with TNF-α+ prednisolone.
To investigate the molecular mechanism involved in the prednisolone inhibition of the TNF-α-induced increase in MLCK promoter activity, we identified a GRE (GTGGCAGCAGATGTCCTGA) site on the MLCK promoter region 1,757 bp upstream of the translational start site using Genomatix/ Promoter Inspector Software. We then studied the regulatory role of the GRE site on MLCK promoter activity by creating a mutant MLCK promoter with mutation of the four core base pairs within the GRE motif as predicted by Genomatix/Promoter Inspector Software (from GTCC to TGAA) by site-directed mutagenesis. Mutation of the GRE site was verified by DNA sequencing. TNF-α treatment resulted in a similar increase in promoter activity of the GRE-mutated MLCK promoter as in the wild type (Fig. 8). However, in the GRE-mutated MLCK promoter, prednisolone no longer inhibited the TNF-α-induced increase in promoter activity. These results suggested that the prednisolone inhibition of the TNF-α increase in MLCK promoter activity was regulated by GR complex binding to the GRE site on the MLCK promoter region.
Fig. 8.
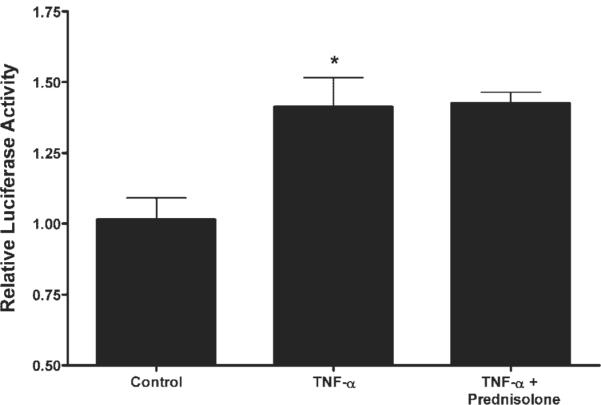
Effect of site-directed mutation of the GRE region on TNF-α and prednisolone modulation of the MLCK promoter. The mutant MLCK promoter containing the mutation of the GRE region was generated and then transfected into filter-grown Caco-2 cells as described in MATERIALS AND METHODS. Transfected Caco-2 cells were treated with vehicle (control), TNF-α (10 ng/ml), or TNF-α+ prednisolone (5 μM)for6h.*P < 0.01 compared with control. No significant difference between TNF-α and TNF-α+ prednisolone groups.
DISCUSSION
Previous studies have shown that GC treatment leads to normalization of intestinal permeability in CD patients with active disease (40). The normalization of intestinal permeability correlated directly with clinical improvement of the disease and was a positive predictor of prolonged clinical remission (23, 40). Moreover, CD patients having persistently elevated intestinal permeability had early recurrences of the disease (23, 33, 36, 40). A number of studies have shown that TNF-α,at the concentrations present in CD patients, causes an increase in intestinal epithelial TJ permeability (12, 16, 22). The elevated TNF-α levels in CD patients have been proposed as an important factor contributing to the increase in intestinal permeability in these patients (6, 16). Consistent with this possibility, intravenous infusion of anti-TNF-α antibody has been shown to result in normalization of intestinal permeability in CD patients (33, 34). Although clinical studies have shown that GC therapy induces a retightening of the intestinal TJ barrier in CD patients, an important question remains as to whether the normalization of intestinal barrier dysfunction is due to primary TJ barrier-promoting effects of GCs or is secondary to the healing of the inflamed mucosal epithelial surface as a result of the immunosuppressive effects of GCs. In this study, we used an in vitro intestinal epithelial model system to study the direct effects of GCs on intestinal epithelial TJ barrier function in the absence of immunological, humoral, vascular, or other systemic factors that could be affected by GC treatment. Our data indicated that, although GCs do not affect basal intestinal epithelial TJ barrier function, GCs inhibit the TNF-α-induced disturbance of the TJ barrier. Thus our findings provide new evidence indicating that the observed intestinal barrier enhancing effects of GCs in CD patients could be related to their protective action against the TNF-α-induced disruption of the intestinal TJ barrier. On the basis of our data, we advance the novel hypothesis that, in addition to their well known immune suppressive effects, GCs also attenuate intestinal inflammation by inhibiting the TNF-α-induced increase in intestinal permeability.
In this study, we also elucidated the mechanisms involved in the TJ barrier protective action of GCs. Since GRs are known to mediate various GC actions, we examined the role of GR complex in mediating the prednisolone protective action. GR is a ubiquitously expressed transcriptional factor that is normally present in the cytoplasm in an inactive form in association with chaperone proteins (31). Upon ligand binding by GC, GR dissociates from chaperone proteins, dimerizes, and translocates to the nucleus as a ligand-GR complex and regulates target gene transcription (31). Our data demonstrated that prednisolone causes an activation and rapid translocation of activated GR complex into the Caco-2 nucleus (Fig. 3). The nuclear translocation of GR complex was accompanied by an increase in binding of the GR complex to the GRE on DNA and activation of a GR-responsive promoter (Fig. 4). Presumably, the inhibition of GR complex activation by the GC antagonist RU-486 prevented the prednisolone-induced inhibition of the TNF-α increase in Caco-2 TJ permeability (Fig. 5). RU-486 inhibition of prednisolone's protective action was also preceded by inhibition of GRE binding and GR responsive promoter activation. These results indicated that the prednisolone-induced GR complex activation that occurs within minutes was required for the TJ barrier-protective effect of prednisolone that occurs within 24 h. Additionally, these findings suggested that the GR complex modulation of target gene expression was a likely mechanism mediating the TJ barrier-protective effect of prednisolone. On the basis of these results, we explored the possibility that the prednisolone protective effect was due to GR complex modulation of MLCK promoter activity.
It is well established that MLCK plays a central role in the pharmacological and physiological regulation of intestinal TJ permeability (12, 13, 16, 42). Previous studies have shown that various pharmacological and physiological agents, including cytochalasin, ethanol, low extracellular Ca2+, Na+-glucose transport, and bacteria, induce an increase in intestinal TJ permeability requiring MLCK activation (12, 14, 17, 18, 25, 37). Activated MLCK catalyzes the phosphorylation of MLC and stimulates the energy-driven contraction of the perijunctional actomyosin ring. This increase in tension presumably leads to mechanical tension generated retraction of TJ complexes and concurrent opening of the intestinal TJ barrier (12, 15, 19). In support of a direct cytoskeletal-TJ protein interaction, molecular studies have demonstrated protein-protein binding of actin microfilaments and ZO-1 proteins (5). By successive carboxyl-terminal truncation of ZO-1 protein, it has previously been shown that a 220-amino acid region (1151–1371) on ZO-1 protein directly binds to F-actin filaments (using a ZO-1/F-actin pull-down assay) (5).
In regard to the role of MLCK in mediating the cytokine-induced increase in intestinal TJ permeability, we have previously shown that the TNF-α increase in intestinal TJ permeability was mediated by an increase in MLCK protein synthesis and activity (13, 16). In those studies, the TNF-α-induced increase in Caco-2 TJ permeability was preceded by an increase in MLCK protein synthesis and activity, and inhibition of MLCK protein synthesis or MLCK activity prevented the TNF-α increase in TJ permeability (13, 16). Those studies concluded that the increase in MLCK protein expression was required for the TNF-α increase in Caco-2 TJ permeability. Similarly, other investigators have shown that the increase in Caco-2 TJ permeability induced by an interferon-γ and TNF-α combination was also mediated by an increase in MLCK protein expression (39). The mechanism of the increase in MLCK expression was shown to result from an increase in MLCK promoter activity and gene transcription (13, 42). An important advance in this area was the recent identification of a 2-kb functionally active MLCK promoter region (Genbank accession no. NM_053025) that allowed investigations into the molecular regulation of the MLCK gene (42).
In the present study, the molecular mechanisms involved in GR complex modulation of the Caco-2 TJ barrier was also examined by determining the role of GR complex in the regulation of MLCK protein expression. Since the TNF-α-induced increase in intestinal TJ permeability is mediated by an increase in MLCK gene transcription and protein expression (13), the effect of prednisolone on the TNF-α increase in MLCK gene activity and protein expression was evaluated. Our data indicated that prednisolone inhibited both the TNF-α-induced increase in MLCK promoter activity and protein expression (Figs. 6 and 7). Furthermore, our studies suggested that the prednisolone effect was mediated by GR complex activation and subsequent GR complex-mediated inhibition of MLCK promoter activity. Mutation of the core GRE binding region (at 1,757 bp from the translational start site) on the MLCK promoter resulted in a complete loss of MLCK promoter response to GC treatment, demonstrating the regulatory role of the GRE in MLCK promoter activity (Fig. 8). These results suggested that the prednisolone activated GR complex inhibited MLCK protein expression by inhibiting the TNF-α-induced upregulation of MLCK promoter activity. Furthermore, these studies identified the GRE site as the regulatory site of GC modulation of MLCK promoter activity.
The clinical relevance of MLCK protein in the intestinal TJ barrier defect and intestinal inflammation of CD has been suggested by recent human studies showing that enterocyte MLCK expression is increased in intestinal tissues obtained from patients with CD (3). In these studies, a direct correlation between increased enterocyte MLCK expression and disease activity was demonstrated (3). The increase in MLCK expression directly correlated with an increase in MLCK activity as measured by MLC phosphorylation (3). Further support for the role of MLCK expression in intestinal TJ barrier defect and intestinal inflammation was also demonstrated in a murine intestinal inflammation model system. In this model, anti-CD3 antibody-induced intestinal inflammation required an increase in intestinal permeability (4). The inhibition of MLCK activity by pharmacological inhibitors prevented the anti-CD3 antibody-induced increase in intestinal permeability and the subsequent development of intestinal inflammation (4). Furthermore, in MLCK knockout mice, anti-CD3 antibody did not cause an increase in intestinal permeability or intestinal inflammation. These studies indicated that the MLCK-mediated increase in intestinal TJ permeability was a prerequisite for the development of intestinal inflammation. In the context of our present data showing that GCs protect against the TNF-α-induced increase in intestinal TJ permeability, we propose that the GC barrier protective effect in CD patients may also be due to GR complex-mediated inhibition of MLCK protein expression.
In conclusion, our findings demonstrated for the first time that GCs prevent the TNF-α-induced increase in intestinal epithelial TJ permeability. Our findings suggested that the GC protective effect against the TNF-α-induced disruption of the intestinal TJ barrier was mediated by GC-induced activation and nuclear translocation of GR complex, binding of the GR complex to the GRE site on DNA, and modulation of target gene expression. Specifically, our data indicated that MLCK promoter was targeted by the GR complex. GC-activated GR complex inhibited the TNF-α-induced increase in MLCK gene activation and MLCK protein expression. The GR complex suppression of MLCK promoter activity was regulated by the GRE site on the promoter. The proposed scheme of GC inhibition of the TNF-α-induced increase in intestinal TJ permeability is outlined in Fig. 9. Thus our present data provide novel insight into the mechanisms of GC protection against the TNF-α-induced increase in intestinal TJ permeability.
Fig. 9.

Proposed mechanism of GC modulation of the TNF-α-induced increase in intestinal epithelial tight junction (TJ) permeability.
Acknowledgments
GRANTS This study was supported by a Veterans Affairs Merit Review grant, research funds from the University of New Mexico, and the National Institute of Diabetes and Digestive and Kidney Diseases Grant RO1-DK-64165-01 (to T. Y. Ma).
REFERENCES
- 1.Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol Gastrointest Liver Physiol. 1995;269:G467–G475. doi: 10.1152/ajpgi.1995.269.4.G467. [DOI] [PubMed] [Google Scholar]
- 2.Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol. 2000;35:1163–1169. doi: 10.1080/003655200750056637. [DOI] [PubMed] [Google Scholar]
- 3.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 4.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J. 2002;16:1835–1837. doi: 10.1096/fj.02-0121fje. [DOI] [PubMed] [Google Scholar]
- 6.Gibson PR. Increased gut permeability in Crohn's disease: is TNF the link? Gut. 2004;53:1724–1725. doi: 10.1136/gut.2004.047092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollander D. Crohn's disease, TNF-alpha, and the leaky gut. The chicken or the egg? Am J Gastroenterol. 2002;97:1867–1868. doi: 10.1111/j.1572-0241.2002.05895.x. [DOI] [PubMed] [Google Scholar]
- 8.Hollander D. The intestinal permeability barrier. A hypothesis as to its regulation and involvement in Crohn's disease. Scand J Gastroenterol. 1992;27:721–726. doi: 10.3109/00365529209011172. [DOI] [PubMed] [Google Scholar]
- 9.Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 10.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, Krugliak P, Rotter JI. Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology. 1989;97:927–931. doi: 10.1016/0016-5085(89)91499-6. [DOI] [PubMed] [Google Scholar]
- 11.Ma TY. Intestinal epithelial barrier dysfunction in Crohn's disease. Proc Soc Exp Biol Med. 1997;214:318–327. doi: 10.3181/00379727-214-44099. [DOI] [PubMed] [Google Scholar]
- 12.Ma TY, Anderson JM. Tight junctions and the intestinal barrier. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. 4th ed. Elsevier Science & Technology; Philadelphia, PA: 2006. [Google Scholar]
- 13.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-α modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 14.Ma TY, Hoa NT, Tran DD, Bui V, Pedram A, Mills S, Merryfield M. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am J Physiol Gastrointest Liver Physiol. 2000;279:G875–G885. doi: 10.1152/ajpgi.2000.279.5.G875. [DOI] [PubMed] [Google Scholar]
- 15.Ma TY, Hollander D, Tran LT, Nguyen D, Hoa N, Bhalla D. Cytoskeletal regulation of Caco-2 intestinal monolayer paracellular permeability. J Cell Physiol. 1995;164:533–545. doi: 10.1002/jcp.1041640311. [DOI] [PubMed] [Google Scholar]
- 16.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 17.Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 1999;276:G965–G974. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- 18.Ma TY, Tran D, Hoa N, Nguyen D, Merryfield M, Tarnawski A. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc Res Tech. 2000;51:156–168. doi: 10.1002/1097-0029(20001015)51:2<156::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 19.Madara JL. Intestinal absorptive cell tight junctions are linked to cytoskeleton. Am J Physiol Cell Physiol. 1987;253:C171–C175. doi: 10.1152/ajpcell.1987.253.1.C171. [DOI] [PubMed] [Google Scholar]
- 20.Madara JL. Loosening tight junctions. Lessons from the intestine. J Clin Invest. 1989;83:1089–1094. doi: 10.1172/JCI113987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahajan DK, London SN. Mifepristone (RU486): a review. Fertil Steril. 1997;68:967–976. doi: 10.1016/s0015-0282(97)00189-1. [DOI] [PubMed] [Google Scholar]
- 22.Marano CW, Lewis SA, Garulacan LA, Soler AP, Mullin JM. Tumor necrosis factor-alpha increases sodium and chloride conductance across the tight junction of CACO-2 BBE, a human intestinal epithelial cell line. J Membr Biol. 1998;161:263–274. doi: 10.1007/s002329900333. [DOI] [PubMed] [Google Scholar]
- 23.Miehsler W, Puspok A, Oberhuber T, Vogelsang H. Impact of different therapeutic regimens on the outcome of patients with Crohn's disease of the upper gastrointestinal tract. Inflamm Bowel Dis. 2001;7:99–105. doi: 10.1097/00054725-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Moriez R, Salvador-Cartier C, Theodorou V, Fioramonti J, Eutamene H, Bueno L. Myosin light chain kinase is involved in lipopolysaccharide-induced disruption of colonic epithelial barrier and bacterial translocation in rats. Am J Pathol. 2005;167:1071–1079. doi: 10.1016/S0002-9440(10)61196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philpott DJ, McKay DM, Sherman PM, Perdue MH. Infection of T84 cells with enteropathogenic Escherichia coli alters barrier and transport functions. Am J Physiol Gastrointest Liver Physiol. 1996;270:G634–G645. doi: 10.1152/ajpgi.1996.270.4.G634. [DOI] [PubMed] [Google Scholar]
- 26.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 27.Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–1610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- 28.Rutgeerts PJ. Conventional treatment of Crohn's disease: objectives and outcomes. Inflamm Bowel Dis. 2001;7(Suppl 1):S2–S8. doi: 10.1002/ibd.3780070503. [DOI] [PubMed] [Google Scholar]
- 29.Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. 2003;125:413–420. doi: 10.1016/s0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 30.Scholmerich J. Review article: systemic and topical steroids in inflammatory bowel disease. Aliment Pharmacol Ther. 2004;20(Suppl 4):66–74. doi: 10.1111/j.1365-2036.2004.02059.x. [DOI] [PubMed] [Google Scholar]
- 31.Schoneveld OJ, Gaemers IC, Lamers WH. Mechanisms of glucocorticoid signalling. Biochim Biophys Acta. 2004;1680:114–128. doi: 10.1016/j.bbaexp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Spiller RC, Jenkins D, Thornley JP, Hebden JM, Wright T, Skinner M, Neal KR. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut. 2000;47:804–811. doi: 10.1136/gut.47.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, Rutgeerts P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. 2002;97:2000–2004. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 34.Suenaert P, Bulteel V, Vermeire S, Noman M, Van Assche G, Rutgeerts P. Hyperresponsiveness of the mucosal barrier in Crohn's disease is not tumor necrosis factor-dependent. Inflamm Bowel Dis. 2005;11:667–673. doi: 10.1097/01.mib.0000168371.87283.4b. [DOI] [PubMed] [Google Scholar]
- 35.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 36.Tibble JA, Bjarnason I. Non-invasive investigation of inflammatory bowel disease. World J Gastroenterol. 2001;7:460–465. doi: 10.3748/wjg.v7.i4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol Cell Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 38.Ukabam SO, Clamp JR, Cooper BT. Abnormal small intestinal permeability to sugars in patients with Crohn's disease of the terminal ileum and colon. Digestion. 1983;27:70–74. doi: 10.1159/000198932. [DOI] [PubMed] [Google Scholar]
- 39.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wild GE, Waschke KA, Bitton A, Thomson AB. The mechanisms of prednisone inhibition of inflammation in Crohn's disease involve changes in intestinal permeability, mucosal TNFalpha production and nuclear factor kappa B expression. Aliment Pharmacol Ther. 2003;18:309–317. doi: 10.1046/j.1365-2036.2003.01611.x. [DOI] [PubMed] [Google Scholar]
- 41.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet. 1993;341:1437–1439. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 42.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-α modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496–G504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]