

Abstract
IL-1β is a prototypical proinflammatory cytokine that plays a central role in the intestinal inflammation amplification cascade. Recent studies have indicated that a TNF-α- and IFN-γ-induced increase in intestinal epithelial paracellular permeability may be an important mechanism contributing to intestinal inflammation. Despite its central role in promoting intestinal inflammation, the role of IL-1β on intestinal epithelial tight junction (TJ) barrier function remains unclear. The major aims of this study were to determine the effect of IL-1β on intestinal epithelial TJ permeability and to elucidate the mechanisms involved in this process, using a well-established in vitro intestinal epithelial model system consisting of filter-grown Caco-2 intestinal epithelial monolayers. IL-1β (0–100 ng/ml) produced a concentration- and time-dependent decrease in Caco-2 transepithelial resistance. Conversely, IL-1β caused a progressive time-dependent increase in transepithelial permeability to paracellular marker inulin. IL-1β-induced increase in Caco-2 TJ permeability was accompanied by a rapid activation of NF-κB. NF-κB inhibitors, pyrrolidine dithiocarbamate and curcumin, prevented the IL-1β-induced increase in Caco-2 TJ permeability. To further confirm the role of NF-κB in the IL-1β-induced increase in Caco-2 TJ permeability, NF-κB p65 expression was silenced by small interfering RNA transfection. NF-κB p65 depletion completely inhibited the IL-1β-induced increase in Caco-2 TJ permeability. IL-1β did not induce apoptosis in the Caco-2 cell. In conclusion, our findings show for the first time that IL-1β at physiologically relevant concentrations causes an increase in intestinal epithelial TJ permeability. The IL-1β-induced increase in Caco-2 TJ permeability was mediated in part by the activation of NF-κB pathways but not apoptosis.
Interleukin-1 was one of the first cytokines to be discovered and has been shown to play a central role in the cytokine network, controlling important functions in the immune system during development, infection, inflammation, cell differentiation, tissue remodeling, and cell death (1, 2). It is a prototypical multifunctional cytokine playing a crucial role in the inflammatory process in various inflammatory conditions of the gut (1, 2), including inflammatory bowel disease (IBD),3 ischemic-reperfusion injury, various types of infectious enteritis, celiac disease, nonsteroidal anti-inflammatory drug NSAID-associated enteropathy, and irritable bowel syndrome (1, 3). IL-1 consists of three members: IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1ra) (2). IL-1α is present either in the cytosol or bound to the cell membrane and it is not secreted and is normally undetectable in blood circulation or in inflammatory fluids (1). In contrast, IL-1β is secreted into surrounding interstitial fluid and blood circulation during inflammation and mediates wide-ranging proinflammatory actions. IL-1ra is a specific antagonist of IL-1 and binds to the IL-1R1 preventing the binding of the active forms of IL-1 (IL-1α and IL-1β) (4).
IL-1β is involved in both the initiation and amplification of the inflammatory response leading to intestinal injury. IL-1β has been shown to play an important role in the pathogenesis of intestinal inflammation in IBD and in animal models of intestinal inflammation. IBD patients have elevated levels of IL-1β in their intestinal tissue (2, 5) and a correlation between increasing levels of IL-1β and the level of intestinal inflammation has been demonstrated (6). An imbalance between IL-1β and its antagonist IL-1ra exists in the intestinal mucosa of IBD patients, suggesting that the lack of anti-inflammatory forms of IL-1 to counteract the elevated levels of IL-1β may be an important pathogenic defect (7). Consistent with this possibility, administration of rIL-1ra prevented the intestinal inflammation in a rabbit model of colitis (8). Recent studies have also demonstrated an existence of IL-1β gene polymorphisms in IBD patients that determines the course and the severity of intestinal inflammation in these patients (9).
Intact intestinal epithelial tight junctions (TJ) are crucial for maintaining intestinal barrier function and preventing paracellular permeation of harmful luminal agents (10, 11). Defective intestinal TJ barrier has been proposed as a central pathogenic factor of Crohn’s disease (CD) and other inflammatory conditions, including nonsteroidal anti-inflammatory drug NSAID-associated enteropathy, postinfectious irritable bowel syndrome, alcoholic liver disease, and infectious diarrheal syndromes (12, 13). An impaired TJ barrier allows increased subepithelial exposure of the innate and acquired immune system to proinflammatory substances that are normally isolated to the intestinal lumen. Proinflammatory cytokines TNF-α and IFN-γ are significantly elevated in IBD and have been shown to cause an increase in intestinal epithelial TJ permeability (14, 15). It has been proposed that an important proinflammatory mechanism of proinflammatory cytokines is to induce a pathologic opening of the intestinal TJ barrier, allowing increased paracellular permeation of the toxic luminal agents (16).
Despite its central importance in the inflammatory process, the role of IL-1β on intestinal TJ barrier function remains unknown. The major aims of this study were to determine the effect of IL-1β on intestinal epithelial TJ permeability and to elucidate the intra-cellular mechanisms involved in the IL-1β effect, using a well-established in vitro intestinal epithelial model system consisting of filter-grown Caco-2 monolayers. Understanding the intracellular processes involved in the IL-1β modulation of intestinal TJ permeability could be important in developing new therapeutic approaches to induce retightening of the intestinal TJ barrier in various intestinal permeability disorders.
Materials and Methods
Materials
Cell culture medium (DMEM), trypsin, FBS, and other related reagents were purchased from Invitrogen Life Technologies. Glutamine, penicillin, streptomycin, and PBS were purchased from Invitrogen Life Technologies. Transwell permeable filters were purchased from Corning. IL-1β and IL-1β mAb were purchased from Pierce Endogen. Anti-occludin Ab, anti ZO-1 Ab, anti-claudin-1 Ab, and anti-NF-κB p65 Ab were obtained from Zymed Laboratories. Anti-IκB-α Ab, curcumin, pyrrolidine dithiocarbamate (PDTC), and cycloheximide were purchased from Sigma-Aldrich. HRP-conjugated secondary Abs for Western blot analysis were purchased from Zymed Laboratories. ELISA reagents were obtained from Active Motif. Cy-5 and Cy-3 Abs for immunostaining were purchased from Jackson ImmunoResearch Laboratories. Transfection reagents were obtained from Dharmacon. Small interfering RNA (siRNA) of NF-κB p65 was purchased from GenScript.
Cell cultures
Caco-2 cells (passage 20) were purchased from the American Type Culture Collection and maintained at 37°C in a culture medium composed of DMEM with 4.5 mg/ml glucose, 50 U/ml penicillin, 50 U/ml streptomycin, 4 mM glutamine, and 25 mM HEPES, and 10% FBS. The cells were kept at 37°C in a 5% CO2 environment. Culture medium was changed every 2 days. Caco-2 cells were subcultured after partial digestion with 0.25% trypsin and 0.9 mM EDTA in Ca2+- and Mg2+-free PBS. Caco-2 monolayers were cultured for 3–4 wk after seeding and only Caco-2 cells from passages 21 to 25 were used to maintain consistency.
Determination of epithelial monolayer resistance and paracellular permeability
An epithelial voltameter (World Precision Instruments) was used for measurements of the transepithelial electrical resistance (TER) of the filter-grown Caco-2 intestinal monolayers as previously reported (17). To study the time-course effects of IL-1β on TER, Caco-2 monolayers were treated with increasing doses ranging 1–100 ng/ml over a 72-h time period. The effect of IL-1β on Caco-2 paracellular permeability was determined using an established paracellular marker inulin. For determination of mucosal-to-serosal flux rates of inulin, Caco-2-plated filters having epithelial resistance of 400–500 Ω·cm2 were used. Known concentrations of permeability marker (2 μM) and its radioactive tracer were added to the apical solution. Low concentrations of permeability marker were used to ensure that negligible osmotic or concentration gradient was introduced.
Assessment of protein expression by Western blot analysis
To study the time-course effect of IL-1β on IκB-α and TJ protein expression, Caco-2 monolayers were treated with IL-1β (10 ng/ml) for varying time periods. At the end of the experimental period, Caco-2 monolayers were immediately rinsed with ice-cold PBS, and cells were lysed with lysis buffer (50 mM Tris·HCl (pH 7.5), 150 mM NaCl, 500 μM NaF, 2 mM EDTA, 100 μM vanadate, 100 μM PMSF, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 40 mM paranitrophenyl phosphate, 1 μg/ml aprotinin, and 1% Triton X-100) and scraped, and the cell lysates were placed in microfuge tubes. Cell lysates were centrifuged to yield a clear lysate. Supernatant was collected and protein measurement was performed using a Bio-Rad Protein Assay kit. Laemmli gel loading buffer was added to the lysate containing 10–20 μg of protein and boiled for 7 min, after which proteins were separated on an SDS-PAGE gel. Proteins from the gel were transferred to the membrane (Trans-Blot Transfer Medium, Nitrocellulose Membrane; Bio-Rad) overnight. The membrane was incubated for 2 h in blocking solution (5% dry milk in TBS-Tween 20 buffer). The membrane was incubated with appropriate primary Abs in blocking solution. The concentrations of primary Abs used were 1–2 μg/ml. After being washed in TBS-1% Tween 20 buffer, the membrane was incubated in appropriate secondary Abs and developed using the Santa Cruz Western Blotting Luminol Reagents (Santa Cruz Biotechnology) on the Kodak BioMax MS film (Fisher Scientific). The films were exposed for a period of time that ranged between 5 and 30 s.
Immunostaining of TJ proteins
Cellular localization of the TJ protein occludin was assessed by an immunofluorescent Ab-labeling technique as previously described (18). At the end of the experimental period, filter-grown Caco-2 monolayers were washed twice in cold PBS and were fixed with 2% paraformaldehyde for 20 min. Then, cells were permeabilized with 0.1% Triton X-100 in PBS at room temperature for 20 min. The Caco-2 monolayers were then incubated in blocking solution composed of BSA and normal donkey serum in PBS for 1 h. Cells were then labeled with primary Abs in blocking solution overnight at 4°C. After being washed with PBS, the filters were incubated in Cy-5-conjugated secondary Ab for 1 h at room temperature. Mowiol was used to mount the filters onto the coverslips. Immunolocalization of TJ proteins was visualized and images obtained using a Nikon fluorescence microscope equipped with a Hamamatsu digital camera in automatic mode (Hamamatsu Photonics). Images were processed with Wasabi software (Hamamatsu Photonics Deutschland).
RNA isolation and reverse transcription
Caco-2 cells (5 × 105) were seeded into transwell permeable inserts and grown to confluency. Filter-grown Caco-2 cells were then treated with IL-1β for desired time periods. At the end of the experimental period, Caco-2 monolayers were washed twice with ice-cold PBS. Total RNA was isolated using the Qiagen RNeasy kit according to the manufacturer’s protocol. Total RNA concentration was determined by absorbance at 260/280 nm using SpectraMax 190 (Molecular Devices). The reverse transcription (RT) was conducted using the GeneAmp Gold RNA PCR Core kit (Applied Biosystems). Two micrograms of total RNA from each sample were reverse transcribed into cDNA in a 40-μl reaction containing 1 × RT-PCR buffer, 2.5 mM MgCl2, 250 μM of each dNTP, 20 U RNase inhibitor, 10 mM DTT, 1.25 μM random hexamer, and 30 U multiscribe RT. The RT reactions were performed in a thermocycler (PTC-100; MJ Research) at 25°C for 10 min, 42°C for 30 min, and 95°C for 5 min.
Quantification of occludin mRNA expression using real-time PCR
The real-time PCRs were conducted using the ABI Prism 7900 sequence Detection System and the TaqMan Universal PCR Master Mix kit (Applied Biosystems) as previously described (19). Each real-time PCR contained 10 μl of RT reaction mix, 25 μl of 2× TaqMan universal PCR master mix, 0.2 μM probe, and 0.6 μM primers. Primer and probe design for the real-time PCR was made with Primer Express version 2 from Applied Biosystems. (The primers used in this study are as follows: occludin-specific primer pairs consisted of 5′-CCCCATCTGACTATGTGGAAAGA-3′ (forward), 5′-AAAACCGCTTGTCATTCACTTTG-3′ (reverse); probe specific for occludin consisted of FAM 5′-GACAGTCCCATGGCATAC TCTTCCAATG-3′ TAMRA; the internal control GAPDH-specific primer pairs consisted of 5′-CCACCCATGGCAAATTCC-3′ (forward), 5′-TG GGATTTCCATTGATGACCAG-3′ (reverse); probe specific for GAPDH consisted of JOE 5′-TGGCACCGTCAAGGCTGAGAACG-3′ TAMRA). All runs were performed according to the default PCR protocol (50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min). For each sample, real-time PCR were performed in triplicate, and the average threshold cycle was calculated. Standard curve was generated to convert the threshold cycle to copy numbers. Expression of occludin mRNA was normalized with GAPDH mRNA expression. The average copy number of occludin mRNA expression in control samples was set to 1.0. The relative expression of occludin mRNA in treated samples was determined as a fold increase compared with control samples.
Assessment of apoptosis and cell death
Caco-2 apoptosis was assessed by Annexin VFITC labeling as previously described (14). After the appropriate experimental treatment, Caco-2 cells were trypsinized and assessed for apoptosis using the Annexin VFITC Apoptosis Detection kit II from BD Biosciences/BD Pharmingen. Annexin VFITC was used to stain for the apoptotic cells and propidium iodide was used to stain the necrotic cells. Propidium iodide is a fluorescent vital dye that stains DNA. In live cells, propidium iodide does not cross the intact plasma membrane of cells. In necrotic or dead cells, plasma membrane becomes permeable to propidium iodide for staining of DNA. Apoptosis or necrosis was measured by flow cytometry using a FACScan flow cytometer (BD Biosciences). Fluorescence of all dyes was excited with the 488-nm line of an argon ion laser. Fluorescence emission was detected in the FL-1 channel (530 ± 15 nm) for cells labeled with Annexin VFITC and in the FL-2 channel (585 ± 21 nm) for cells labeled with propidium iodide; subsequently, an FL-1/FL-2 dot plot was generated. For each experimental sample, a total of 20,000 cells were counted for Annexin VFITC and propidium iodide stain.
Nuclear extracts and ELISA
Filter-grown Caco-2 cells were treated with 10 ng/ml IL-1β for 30 min. Cells were washed with ice-cold PBS, scraped, and centrifuged at 14,000 rpm for 30 s. The cell pellets were resuspended in 200 μl of buffer A (in millimoles: 10 HEPES-KOH, 1.5 MgCl2, 10 KCl, 0.5 DTT, and 0.2 PMSF (pH 7.9)), and incubated on ice for 15 min. After centrifugation at 14,000 rpm for 30 s, pelleted nuclei were resuspended in 30 μl of buffer C (in millimoles: 20 HEPES-KOH (25% glycerol), 420 NaCl, 1.5 MgCl2, 0.2 EDTA, 0.5 DTT, and 0.2 PMSF (pH 7.9)). After incubation on ice for 20 min, the lysates were centrifuged at 14,000 rpm for 20 min. Protein concentrations were determined using the Bradford method. The NF-κB DNA-binding activity assay was performed using Trans-AM ELISA-based kits from Active Motif according to the manufacturer’s protocol. In brief, the binding reactions contained 1 pM biotinylated probe (Integrated DNA Technologies) and 5 μg of nuclear extract in complete binding buffer with a total volume of 50 μl. After 30 min of incubation, the solution was transferred to an individual well on the plate and incubated for 1 h. Rabbit NF-κB p65 Ab (2 μg/ml) was added to the well to bind NF-κB p65 from the nuclear extract. After incubation for 1 h, NF-κB p65 Ab was removed, and 100 μl of anti-rabbit HRP-conjugated IgG were added to the well and incubated for 1 h. Subsequently, 100 μl of developing solution was added for 2–10 min, and 100 μl of stop solution were added. The absorbance at 450 nm was determined using the SpectraMax 190 (Molecular Devices).
siRNA of NF-κB p65
pRNATin-H1.2/Neo plasmid vector containing the siRNA of NF-κB p65 was purchased from GenScript. Caco-2 monolayers were transiently transfected using DharmaFect transfection reagent. Caco-2 cells were seeded into a six-well transwell plates and grown to confluency. Caco-2 monolayers were then washed with PBS and Opti-MEM medium was added to the apical and the basolateral compartments of each filter. The plasmid vector containing the siRNA of NF-κB p65 and DharmaFect reagent were preincubated in Opti-MEM. After 5 min of incubation, two solutions were mixed and incubated for another 20 min and the mixture was added to the apical compartment of each filter. After incubation for 3 h at 37°C, 500 μl of DMEM containing 10% FBS and no antibiotics were added to both sides of the filter to reach a 2.5% final concentration of FBS. The IL-1β experiments were conducted 96 h posttransfection. The siRNA-induced silencing the NF-κB p65 was confirmed by immunoblot of NF-κB p65.
Statistical analysis
Results are expressed as means ± SE. Statistical significance of differences between mean values was assessed with paired t test (Graph Pad Prizm 4.00 for Windows; GraphPad Software). A p value of <0.05 was used to indicate statistical significance. All experiments were repeated a minimum of three times to ensure reproducibility.
Results
IL-1β effect on Caco-2 intestinal epithelial TJ permeability
In the following studies, the IL-1β effect on Caco-2 TJ permeability was determined by measuring TER and epithelial permeability to paracellular marker inulin. Increasing concentrations of IL-1β (0–100 ng/ml) caused a dose-dependent decrease in Caco-2 TER after a 48-h treatment period (Fig. 1). The maximal drop in Caco-2 TER occurred at IL-1β concentration of 10 ng/ml and increasing concentrations above the 10 ng/ml dose did not produce a greater drop in TER. The time course of IL-1β (10 ng/ml) effect on Caco-2 TER is shown in Fig. 2A. IL-1β did not have a significant effect on Caco-2 TER during the first 6 h of treatment. There was a sharp time-dependent drop in Caco-2 TER between 12 and 24 h. The maximal drop in the TER was reached by 48 h of IL-1β treatment and the drop in the TER remained unchanged thereafter. Conversely, IL-1β (10 ng/ml) caused a time-dependent increase in Caco-2 paracellular permeability to inulin starting after 6 h of IL-1β treatment and the maximal increase in paracellular permeability was also reached by 48 h of treatment (Fig. 2B). Although IL-1β caused only a moderate drop in Caco-2 TER, it caused ~20-fold increase in paracellular permeability to inulin. The graph of Caco-2 TER vs paracellular permeability is shown in Fig. 2C. There was a linear relationship between the IL-1β-induced drop in TER and increase in paracellular permeability to inulin (r = 0.97), indicating that IL-1β-induced decrease in Caco-2 TER directly correlated with an increase in paracellular permeability. To con-firm that the IL-1β-induced drop in Caco-2 TER was due to direct effect of IL-1β, the effect of IL-1β-neutralizing Ab on the IL-1β-induced drop in Caco-2 TER was determined. Pretreatment with neutralizing IL-1β mAb (100 ng/ml) prevented the IL-1β-induced drop in Caco-2 TER (Fig. 1B), confirming a direct effect of IL-1β on Caco-2 TER.
FIGURE 1.
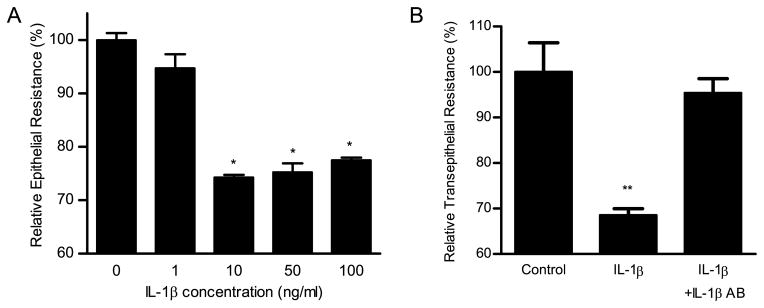
A, Effect of increasing concentration of IL-1β (0, 1, 10, 50, and 100 ng/ml) on Caco-2 TER. Filter-grown Caco-2 monolayers were treated with IL-1β for a 48-h experimental period. IL-1β produced a concentration-dependent decrease in Caco-2 TER. Data represent means ± SE of TER (n = 6). *, p < 0.0001 vs control. B, The effect of IL-1β mAb (100 ng/ml) on an IL-1β-induced drop in Caco-2 TER. IL-1β mAb pre-treatment prevented the IL-1β-induced drop in Caco-2 TER. Data represent means ± SE of TER (n = 6).**, p < 0.0001 vs control.
FIGURE 2.
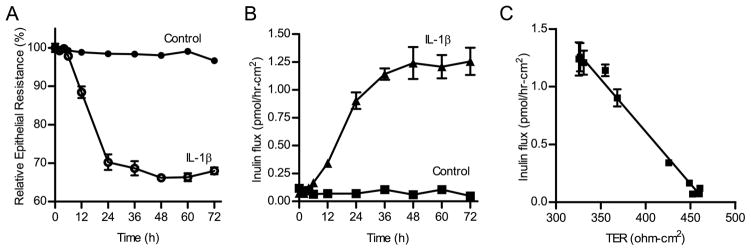
Time-course effect of IL-1β on Caco-2 TER and paracellular permeability. The effect of IL-1β (10 ng/ml) on Caco-2 TER and mucosal-to-serosal flux of paracellular marker, inulin were measured over a 72-h experimental period. A, Time-course effect of IL-1β on Caco-2 TER (n = 4). B, Time-course effect of IL-1β on transepithelial inulin flux (n = 3). C, Graph of TER vs inulin flux (r = 0.97).
IL-1β-induced increase in Caco-2 TJ permeability is mediated by NF-κB pathway
Because IL-1β is known to activate nuclear transcription factor NF-κB (20), we investigated the possibility that NF-κB activation may play a regulatory role in the IL-1β-induced increase in Caco-2 TJ permeability. IκB-α degradation is required for the activation of NF-κB (21). As shown in Fig. 3A, IL-1β (10 ng/ml) produced a rapid degradation of IκB-α in the filter-grown Caco-2 monolayers. By 10 min of IL-1β treatment, most of IκB-α had been degraded. The IL-1β effect on NF-κB activation was also confirmed by measuring NF-κB p65 expression in the cytoplasmic and nuclear fractions by immunoblot analysis. In the control Caco-2 monolayers, NF-κB p65 were present mostly in the cytoplasmic fraction with only minimal amount in the nuclear fraction (Fig. 3B). By 30 min of IL-1β treatment, most of NF-κB p65 had translocated into the nuclear fraction, and only small amount remained in the cytoplasmic fraction (Fig. 3B). The IL-1β effect on NF-κB activation was also confirmed by immunostaining of NF-κB p65. In the control Caco-2 monolayers, NF-κB p65 was localized mostly in the cytoplasm (Fig. 3C). Following IL-1β treatment, NF-κB p65 rapidly translocated into the nucleus and by 15–30 min, most of NF-κB p65 had translocated into the nucleus (Fig. 3D). Together, these findings indicated that IL-1β causes a rapid activation and cytoplasmic-to-nuclear translocation of NF-κB p65. To determine the role of NF-κB activation in the IL-1β-induced increase in Caco-2 TJ permeability, the effect of known NF-κB inhibitors curcumin and PDTC on IL-1β-induced drop in Caco-2 TER was examined. Curcumin (5 μM) and PDTC (100 μM) inhibited the IL-1β-induced activation of NF-κB p65 (as shown by the inhibition of nuclear translocation of NF-κB) (Fig. 3, E and F) and completely prevented the IL-1β-induced decrease in Caco-2 TER (Fig. 3G), suggesting that NF-κB activation was required for the IL-1β-induced increase in Caco-2 TJ permeability.
FIGURE 3.

Time-course effect of IL-1β on NF-κB activation in Caco-2 monolayers. Filter-grown Caco-2 monolayers were treated with IL-1β (10 ng/ml) for increasing time periods (0–60 min). A, The IκB-α protein expression was determined by Western blot analysis as described in Materials and Methods. B, Filter-grown Caco-2 monolayers were treated with IL-1β (10 ng/ml) for 30 min, and changes in the NF-κB p65 expression in the cytoplasmic and nuclear fractions were assayed by Western blot analysis. The effect of IL-1β (10 ng/ml) on NF-κB p65 cytoplasmic-to-nuclear translocation as determined by immunofluorescent Ab labeling as described in Materials and Methods. C, Control or untreated Caco-2 monolayers. D, Caco-2 monolayers treated for 30 min with IL-1β (10 ng/ml). Caco-2 monolayers pretreated with (E) curcumin (5 μM) or (F) PDTC (100 μM) before the 30-min treatment with IL-1β. IL-1β caused a rapid cytoplasmic-to-nuclear translocation of NF-κB p65 subunit. Original magnification, ×400. G, The effect of NF-κB inhibitors PDTC and curcumin on IL-1β-induced drop in Caco-2 TER. PDTC (100 μM) and curcumin (5 μM) significantly prevented the IL-1β-induced drop in Caco-2 TER (n = 6). *, p < 0.01 vs control. **, p < 0.01 vs IL-1β-treated monolayers.
Next, the binding of IL-1β-activated NF-κB to the DNA-binding site was determined by an ELISA-based DNA-binding assay. IL-1β treatment resulted in a marked (6-fold) increase in NF-κB p65 binding to the consensus κB sequence (5′-GGGACTTTCC-3′) on the oligonucleotide probe. The NF-κB inhibitors curcumin and PDTC prevented the IL-1β-induced increase in NF-κB binding to the DNA probe (Fig. 4A). To confirm the specificity of NF-κB binding to the DNA probe, a wild-type oligonucleotide containing the consensus κB-binding site was added in excess (100-fold higher concentration) as a competitive inhibitor for NF-κB binding. The addition of wild-type oligonucleotide inhibited the binding of NF-κB to the DNA probe. In contrast, addition of excess oligonucleotide containing a mutated NF-κB-binding motif (5′-CTCACTTTCC-3′) did not inhibit the NF-κB binding (Fig. 4B). These findings indicated that IL-1β-activated NF-κB binds to a κB site on DNA.
FIGURE 4.

ELISA-binding assay of IL-1β-activated NF-κB p65 binding to the oligonucleotide probe containing the κB-binding site. The binding of p65 to binding site was expressed as a concentration of NF-κB p65 (nanograms per milliliter) bound to the binding site. A, IL-1β caused a significant increase in NF-κB binding. NF-κB inhibitors PDTC (100 μM) and curcumin (5 μM) inhibited the IL-1β-induced NF-κB binding. B, The addition of wild-type (WT) oligonucleotide containing the consensus NF-κB-binding site in excess (100:1 ratio) as a competitive inhibitor prevented the binding of NF-κB to the DNA probe. In contrast, the addition of excess oligonucleotide containing a mutated NF-κB-binding motif did not inhibit the NF-κB binding. C, Effect of NF-κB p65 depletion by siRNA on the IL-1β-induced drop on Caco-2 TER. Caco-2 monolayers were transfected with NF-κB p65 siRNA for a 96-h time period as described in Materials and Methods. D, NF-κB p65 siRNA resulted in a marked depletion in NF-κB p65 protein expression. B, NF-κB p65 siRNA transfection prevented the IL-1β-induced drop in Caco-2 TER. *, p < 0.001 vs control; **, p < 0.001vs IL-1β treatment.
To further validate the requirement of NF-κB p65 in the IL-1β-induced increase in Caco-2 TJ permeability, NF-κB p65 expression was selectively silenced by siRNA transfection. siRNA interfere with the translation of mRNA and targets endogenous RNA for degradation in a sequence-specific manner, thereby inhibiting the function of a targeted gene (22). As shown in Fig. 4C, the transfection of filter-grown Caco-2 monolayers with NF-κB p65 siRNA resulted in a near-complete knockdown of NF-κB p65 protein expression. The NF-κB p65 silencing resulted in a complete inhibition of the IL-1β-induced drop in Caco-2 TER (Fig. 4D). These studies confirmed that NF-κB p65 was required for the IL-1β-induced increase in Caco-2 TJ permeability.
IL-1β effect on Caco-2 TJ permeability is not due to apoptosis
Apoptosis has been proposed as a mechanism of cytokine-induced increase in intestinal epithelial paracellular permeability (23). Because IL-1β is known to induce apoptosis in some cell types (24), in the following studies, we examined the possibility that the IL-1β-induced increase in Caco-2 TJ permeability may be due to apoptosis or cell death. IL-1β effect on Caco-2 cell apoptosis and necrosis was determined by flow cytometry by labeling the apoptotic cells with Annexin VFITC and necrotic cells with propidium iodide. During apoptosis, phosphatidylserine translocates from the cytoplasmic face of the plasma membrane to the cell surface. Annexin V has a strong, Ca2+-dependent affinity for phosphatidylserine and is used as a probe for detecting apoptosis. Cells undergoing apoptosis stain positively for Annexin VFITC as phosphatidylserine is located on the cell surface. In necrotic cells, propidium iodide is able to permeate across the cell membrane to label the DNA. The dot-plot of IL-1β effect on propidium iodide (necrosis) and Annexin VFITC (apoptosis) staining generated from flow cytometry analysis is shown in Fig. 5A. IL-1β treatment did not have significant effect on Caco-2 cell apoptosis (Fig. 5B) or necrosis (Fig. 5C), indicating that the IL-1β-induced increase in Caco-2 TJ permeability was not due to induction of cell apoptosis or cell death.
FIGURE 5.

The effect of IL-1β on Caco-2 cell apoptosis. The IL-1β effect on Caco-2 cell apoptosis and necrosis was assessed by Annexin VFITC and propidium iodide labeling as described in Materials and Methods. Flow cytometric analysis of Annexin VFITC and propidium iodide staining was performed. A, The dot plots represent a total of 20,000 events for each sample. Upper left panel, Necrotic cells; lower right panel, apoptotic cells. Caco-2 monolayers were treated with IL-1β (10 ng/ml) for 48 h. Subsequently, Caco-2 cells were trypsinized and labeled with Annexin VFITC (apoptosis) or propidium iodide (necrosis). Camptothecin was used as a positive control for apo-ptosis. IL-1β did not induce (B) apoptosis or (C) cell necrosis in IL-1β-treated Caco-2 cells.
IL-1β induces a protein-specific down-regulation of occludin protein and mRNA expression and disturbance in junctional localization
In the following studies, we examined the effect of IL-1β (10 ng/ml) on expression of cytoplasmic TJ protein ZO-1 and transmembrane proteins occludin and claudin-1. IL-1β did not have significant effect on ZO-1 protein expression over the 48-h treatment period, but caused a protein-specific decrease in occludin protein expression and an increase in claudin-1 protein expression (Fig. 6A). IL-1β also caused a disturbance in junctional localization of occludin as determined by immunofluorescent staining. In the control Caco-2 monolayers, occludin formed a dense peripheral band outlining the cellular junctions (Fig. 6B). After IL-1β treatment, the intensity of occludin staining at cell junctions was reduced and the junctional localization became irregular and distorted (arrows), correlating with a functional alteration in Caco-2 TJ barrier function. To examine whether the IL-1β-induced decrease in occludin protein expression was due in part to a decrease in mRNA transcription, the effect of IL-1β on occludin transcript expression was determined. IL-1β treatment (10 ng/ml) resulted in a significant decrease in occludin mRNA expression (Fig. 7A). These findings suggested that the IL-1β-induced decrease in occludin protein expression was due in part to a decrease in mRNA expression.
FIGURE 6.
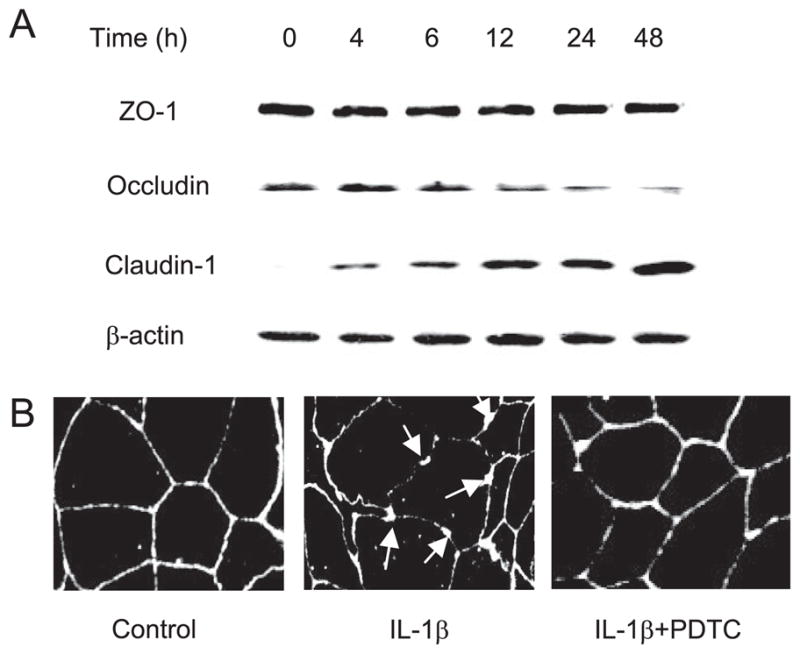
A, Time-course effect of IL-1β on TJ protein expression in Caco-2 monolayers. Filter-grown Caco-2 monolayers were treated with IL-1β (10 ng/ml) for increasing time periods (0–48 h). The protein expression was determined by Western blot analysis as described in Materials and Methods. IL-1β did not cause any change in the ZO-1 protein expression. IL-1β caused a progressive down-regulation in occludin protein expression and up-regulation in claudin-1 expression. B, Effect of IL-1β on occludin junctional localization. The junctional localization of occludin protein in filter-grown Caco-2 monolayers was assessed by immunofluorescent Ab labeling. IL-1β produced a significant disturbance in occludin junctional localization (arrows). PDTC prevented the IL-1β-induced alteration in junctional localization of occludin protein.
FIGURE 7.

A, Effect of IL-1β on occludin mRNA levels was assessed by real-time PCR. Filter-grown Caco-2 cells were treated with 10 ng/ml IL-1β for 16 h. B, Pretreatment with PDTC (100 μM) prevented the IL-1β-induced decrease in occludin mRNA levels. Data are represented as means of four replicates ± SE; *, p < 0.001 vs control. C, Effect of NF-κB inhibitor, PDTC (100 μM), and (D) NF-κB p65 depletion by siRNA on IL-1β-induced down-regulation of occludin protein expression. Caco-2 monolayers were treated with IL-1β for the 48-h experimental period. PDTC (100 μM) and NF-κB p65 siRNA significantly prevented the IL-1β-induced down-regulation in occludin protein expression.
To further examine the intracellular processes involved in the IL-1β-induced decrease in occludin protein expression, we examined the possibility that the IL-1β effect was due to NF-κB modulation of occludin transcript expression. The NF-κB inhibitor (PDTC) inhibited the IL-1β-induced decrease in occludin mRNA expression (Fig. 7B). Additionally, PDTC also prevented the IL-1β-induced decrease in occludin protein expression (Fig. 7C) and the disturbance in junctional localization (Fig. 6B). Moreover, NF-κB p65 silencing by siRNA also prevented the down-regulation of occludin protein expression (Fig. 7D). Together, these results suggested that IL-1β-induced NF-κB activation mediated the down-regulation of occludin transcript and protein expression.
Discussion
The defective intestinal epithelial TJ barrier has been proposed as an important pathogenic factor leading to the intestinal inflammation in CD and other inflammatory conditions (25). It has been proposed that the cytokine-induced increase in intestinal TJ permeability represents an important proinflammatory mechanism (14, 26). The cytokine-induced increase in intestinal TJ permeability allows increased paracellular permeation of luminal Ags, bacteria, and other toxic substances that promote inflammation (10, 11). The TJ barrier disruptive actions of TNF-α and IFN-γ have been well-established (27, 28). However, despite its central importance in mediating the inflammatory process in CD and other inflammatory conditions, the role of IL-1β on intestinal TJ barrier function has not been previously reported. In this study, we show for the first time that IL-1β at physiologically relevant concentrations (1–10 ng/ml) causes a significant increase in intestinal TJ permeability. Previous studies have shown that inflammatory cells secrete IL-1β in the concentration range of 1–25 ng/ml range (29). The basal levels of IL-1β secreted by inflammatory cells (including macrophages and monocytes) are in the concentration range of 1–5 ng/ml (7). Following endotoxin (1 μg/ml) stimulation, the activated macrophages and monocytes secreted IL-1β in the 10–25 ng/ml range (7). Interestingly, IL-1β (10 ng/ml) caused only a modest drop (~30% drop) in Caco-2 TER but produced a marked increase (20-fold increase) in paracellular permeability to inulin (m.w. = 5,000 g/mol), suggesting that IL-1β affects the paracellular permeation of larger molecules to a much greater extent than smaller ions.
The time course of IL-1β effect on Caco-2 TJ permeability suggested that the IL-1β effect was not due to acute intracellular signaling process but required protein synthesis. IL-1β did not affect Caco-2 TJ permeability within the first 6 h (Fig. 2). The initial increase in Caco-2 TJ permeability occurred between 6 and 12 h following IL-1β exposure; the increase in Caco-2 TJ permeability progressed to the maximal levels by 48 h. Moreover, we found that protein synthesis inhibitor cycloheximide and transcription inhibitor actinomycin D prevented the IL-1β-induced increase in Caco-2 TJ permeability (our unpublished data), confirming that the IL-1β-induced increase in Caco-2 TJ permeability required transcription and new protein synthesis. Consistent with our present findings, previous studies have suggested that TNF-α- and IFN-γ-induced modulation of Caco-2 TJ permeability was mediated by an increase in myosin L chain kinase (MLCK) transcription and protein synthesis (27, 30). The possibility that IL-1β effect may also be mediated by up-regulation of MLCK protein expression remains to be further explored.
In this study, we examined some of the intracellular mechanisms that mediate the IL-1β modulation of Caco-2 TJ barrier and TJ proteins. Because many of the proinflammatory effects of IL-1β are known to be mediated by nuclear transcription factor NF-κB (31), we investigated the possibility that IL-1β effect on Caco-2 TJ permeability was also mediated by NF-κB. IL-1β caused a rapid degradation of IκB-α and activation of NF-κB in Caco-2 cells (Fig. 3, A and B) and the inhibition of NF-κB activation by NF-κB inhibitors prevented the increase in Caco-2 TJ permeability (Fig. 3, E–G). The requirement of NF-κB in IL-1β-induced increase in TJ permeability was further validated by the silencing of NF-κB p65 expression in Caco-2 cells. NF-κB p65 depletion by NF-κB p65 siRNA transfection completely inhibited the IL-1β-induced increase in Caco-2 TJ permeability (Fig. 4D). In combination, these studies confirmed the requirement (but not sufficiency) of NF-κB in mediating the IL-1β-induced increase in Caco-2 TJ permeability. Additionally, our data also indicated that neutralizing mAb directed against IL-1β prevented the IL-1β-induced drop in Caco-2 TER, confirming that the TJ modulating effect was due to the presence of IL-1β.
Previous studies have shown that various intracellular mechanisms are involved in the cytokine-induced increase in intestinal TJ permeability. Earlier studies suggested that the TNF-α-induced increase in HT-29 intestinal epithelial TJ permeability may be due to a TNF-α-induced activation of apoptosis with a resultant formation of large paracellular gaps or openings between cells (32). Recent studies have suggested that the TNF-α effect on Caco-2 TJ barrier may be mediated by NF-κB activation and an increase in MLCK protein expression (27, 33). In contrast, IFN-γ-induced increase in Caco-2 TJ permeability did not involve NF-κB activation, but required an increase in MLCK expression (34). The IFN-γ-induced increase in T84 intestinal TJ permeability also appeared to be mediated in part by endocytosis of TJ proteins, but not apoptosis (34, 35). The IL-13- and IL-4-induced increase in HT-29 intestinal epithelial TJ permeability has been suggested to be mediated by apoptosis (36, 37). The IL-13 and IL-4 effect on the TJ barrier has also been suggested to be mediated in part by PI3K activation (38). Earlier studies have also suggested that the production of NO is a possible mechanism for the cytomix-induced increase in TJ permeability in Caco-2 cells (39). The cytomix (a mixture of TNF-α, IFN-γ, and IL-1β) has been shown to target the TJ proteins and the TJ permeability through both NO-dependent and -independent mechanisms. Subsequent in vivo experiments on endotoxemic mice demonstrated that LPS-induced-increase in inducible NO synthase activity may represent an important pathophysiological mechanism of increased TJ permeability in ileal and colonic epithelia (40). Thus, previous studies have indicated that different cytokines modulate the intestinal TJ barrier through distinct intracellular mechanisms.
Because apoptosis had been proposed as a mechanism modulating the TNF-α, IL-4, and IL-13 increase in intestinal TJ permeability (23, 41), we examined the possibility that the IL-1β induced increase in Caco-2 TJ permeability may also be due to an increase in cell apoptosis. IL-1β treatment did not induce apoptosis or necrosis in Caco-2 cells, suggesting that Caco-2 cell apoptosis or cell death was not a mechanism mediating the IL-1β-induced increase in Caco-2 TJ permeability.
Cytokines have been shown to modulate TJ protein expression in a protein-specific manner. TNF-α caused a down-regulation of ZO-1, occludin, and claudin-1 protein expression in Caco-2 cells (14). In contrast, IFN-γ caused a decrease in ZO-1 and occludin protein expression but caused an increase in claudin-1 expression in T-84 cells. IL-4 and IL-13 caused an increase in the expression of claudin-2 protein but did not affect claudin-3 or claudin-4 protein expression in T-84 cells (38). Interestingly, a cytomix consisting of TNF-α, IFN-γ, and IL-1β caused a decrease in ZO-1 and occludin protein expression followed by a decrease in ZO-1 and a slight decrease in occludin mRNA transcripts but an increase in the expression of claudin-1 protein in Caco-2 cells (39). In the present study, we found that IL-1β did not affect the ZO-1 protein expression but caused a marked down-regulation in occludin protein expression. Similar to IFN-γ, IL-1β also caused an up-regulation of claudin-1 protein expression. The precise role of TJ proteins in the modulation of intestinal TJ barrier function remains unclear. For example, previous studies suggested a correlation between occludin expression and TJ barrier function. Increased expression of occludin in MDCK cells correlated with an enhancement of TJ barrier function as evidenced by an increase in MDCK TER (42). Additionally, the expression of occludin in fibroblasts also resulted in an enhancement of adhesion or contact between cells (43). Moreover, a number of studies have indicated a correlation between an increase in epithelial TJ permeability and a decrease in occludin expression (44). In enteropathogenic Escherichia coli-induced increase in intestinal epithelial TJ permeability, the dissociation of occludin and ZO-1 binding has been postulated as an important mechanism leading to the increase in intestinal TJ permeability (45). It has been shown in earlier studies that LPS-treated mice had a disrupted intestinal barrier function and increased TJ permeability due to down-regulation in occludin protein expression and alteration in its cellular localization (40). In contrast, recent studies of occludin knockdown in in vivo and in vitro models suggested that occludin depletion or knockout did not affect the TJ barrier function (46, 47), arguing against a role for occludin in maintaining the TJ barrier function.
Although the precise role of occludin protein on TJ barrier function modulation remains to be further clarified, our results suggested that nuclear transcription factor NF-κB mediated the IL-1β-induced down-regulation of occludin mRNA and protein expression. The inhibition of NF-κB activation by NF-κB inhibitors prevented both IL-1β-induced decrease in occludin transcription process (Fig. 7, B and C). NF-κB inhibitor also prevented the alteration in occludin junctional localization (Fig. 6B). The depletion of NF-κB p65 by siRNA transfection also prevented the IL-1β-induced decrease in occludin expression (Fig. 7D). These results suggested that IL-1β-induced down-regulation of occludin protein expression was mediated by NF-κB modulation of occludin mRNA expression. As far as we are aware, this is the first study to show that NF-κB has a regulatory role on TJ protein expression. Although the precise mechanisms by which NF-κB regulates occludin protein expression remain unclear, based on our present findings we hypothesize that the IL-1β-induced decrease in occludin transcript and protein expression may be due to NF-κB-induced suppression of occludin promoter activity. Consistent with this hypothesis, recent studies have shown that NF-κB suppresses the promoter activity of various genes including choline acetyl-transferase (48), guanylate-binding protein (Gbp) and Ag-presenting protein (Tap) (49), caspase-8 and TNFR-associated factor-1 and -2 (50).
It is important to note that different cell types may react differently to IL-1β treatment. In this regard, previous studies have shown that IL-1β treatment (100 ng/ml) caused about a 40% drop in TER in the airway epithelial cells but caused only a small increase in epithelial permeability to FITC-labeled dextran (51). In an earlier study, IL-1β caused a small (nonstatistically significant) drop in T-84 intestinal epithelial TER (28). However, the dose of IL-1β used in that study was lower than the dose used in the present study. The dose of IL-1β used (when converted to nanograms per milliliter) was in the range of 0.1–1 ng/ml. When we assessed the effect of 10 ng/ml IL-1β on T-84 monolayers, there was a similar drop (~30%) in TER as in Caco-2 cells (our unpublished data). IL-1β also caused a similar decrease in TER in filter-grown MDCK cells (our unpublished data).
In conclusion, our results indicated for the first time that IL-1β causes an increase in intestinal epithelial TJ permeability. The IL-1β-induced increase in Caco-2 TJ permeability was mediated by NF-κB activation and inhibition of NF-κB activation or NF-κB p65 depletion with siRNA prevented the IL-1β increase in Caco-2 TJ permeability. IL-1β also caused a protein-specific down-regulation of occludin expression, a decrease in occludin mRNA expression and disturbance in its junctional localization. The IL-1β modulation of occludin was also regulated by NF-κB activation. Our data provide new insight into the role of IL-1β on intestinal TJ barrier function and provide supporting evidence that the elevated levels of IL-1β during intestinal inflammation may contribute to the intestinal TJ barrier defect observed in these patients.
Acknowledgments
We thank Dr. Dongmei Ye for the excellent assistance in respect to the molecular studies and Dr. Karol Dokladny for the excellent assistance in respect to the apoptosis studies.
Footnotes
This work was supported by a Veterans Affairs Merit Review Grant from the Veterans Affairs Research Service and by National Institute of Diabetes and Digestive and Kidney Diseases Grant RO 1-DK-64165-01 (to T.Y.M.).
Abbreviations used in this paper: IBD, inflammatory bowel disease; IL-1ra, IL-1 receptor antagonist; TJ, tight junction; CD, Crohn’s disease; siRNA, small interfering RNA; TER, transepithelial electrical resistance; RT, reverse transcription; MLCK, myosin L chain kinase; PDTC, pyrrolidine dithiocarbamate.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 2.Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. Role of interleukin 1 in inflammatory bowel disease—enhanced production during active disease. Gut. 1990;31:686–689. doi: 10.1136/gut.31.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spiller RC, Jenkins D, Thornley JP, Hebden JM, Wright T, Skinner M, Neal KR. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut. 2000;47:804–811. doi: 10.1136/gut.47.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease: a novel mechanism of chronic intestinal inflammation. J Immunol. 1995;154:2434–2440. [PubMed] [Google Scholar]
- 5.Isaacs KL, Sartor RB, Haskill S. Cytokine messenger RNA profiles in inflammatory bowel disease mucosa detected by polymerase chain reaction amplification. Gastroenterology. 1992;103:1587–1595. doi: 10.1016/0016-5085(92)91182-4. [DOI] [PubMed] [Google Scholar]
- 6.Reinecker HC, Steffen M, Doehn C, Petersen J, Pfluger I, Voss A, Raedler A. Proinflammatory cytokines in intestinal mucosa. Immunol Res. 1991;10:247–248. doi: 10.1007/BF02919700. [DOI] [PubMed] [Google Scholar]
- 7.Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, Thompson RC, Dinarello CA. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest. 1990;86:972–980. doi: 10.1172/JCI114799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cominelli F, Nast CC, Duchini A, Lee M. Recombinant interleukin-1 receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology. 1992;103:65–71. doi: 10.1016/0016-5085(92)91096-m. [DOI] [PubMed] [Google Scholar]
- 9.Nemetz A, Nosti-Escanilla MP, Molnar T, Kope A, Kovacs A, Feher J, Tulassay Z, Nagy F, Garcia-Gonzalez MA, Pena AS. IL1B gene polymorphisms influence the course and severity of inflammatory bowel disease. Immunogenetics. 1999;49:527–531. doi: 10.1007/s002510050530. [DOI] [PubMed] [Google Scholar]
- 10.Ma TY. Intestinal epithelial barrier dysfunction in Crohn’s disease. Proc Soc Exp Biol Med. 1997;214:318–327. doi: 10.3181/00379727-214-44099. [DOI] [PubMed] [Google Scholar]
- 11.Madara JL. Loosening tight junctions: lessons from the intestine. J Clin Invest. 1989;83:1089–1094. doi: 10.1172/JCI113987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMeo MT, Mutlu EA, Keshavarzian A, Tobin MC. Intestinal permeation and gastrointestinal disease. J Clin Gastroenterol. 2002;34:385–396. doi: 10.1097/00004836-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 14.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-α -induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 15.Stevens C, Walz G, Singaram C, Lipman ML, Zanker B, Muggia A, Antonioli D, Peppercorn MA, Strom TB. Tumor necrosis factor-α, interleukin-1β, and interleukin-6 expression in inflammatory bowel disease. Dig Dis Sci. 1992;37:818–826. doi: 10.1007/BF01300378. [DOI] [PubMed] [Google Scholar]
- 16.McKay DM, Singh PK. Superantigen activation of immune cells evokes epithelial (T84) transport and barrier abnormalities via IFN-γ and TNF α: inhibition of increased permeability, but not diminished secretory responses by TGF-β2. J Immunol. 1997;159:2382–2390. [PubMed] [Google Scholar]
- 17.Ma TY, Hoa NT, Tran DD, Bui V, Pedram A, Mills S, Merryfield M. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am J Physiol. 2000;279:G875–G885. doi: 10.1152/ajpgi.2000.279.5.G875. [DOI] [PubMed] [Google Scholar]
- 18.Ma TY, Tran D, Hoa N, Nguyen D, Merryfield M, Tarnawski A. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc Res Tech. 2000;51:156–168. doi: 10.1002/1097-0029(20001015)51:2<156::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 19.Cheung PY, Chan CW, Wong W, Cheung TL, Kam KM. Evaluation of two real-time polymerase chain reaction pathogen detection kits for Salmonella spp. in food. Lett Appl Microbiol. 2004;39:509–515. doi: 10.1111/j.1472-765X.2004.01609.x. [DOI] [PubMed] [Google Scholar]
- 20.Delhalle S, Blasius R, Dicato M, Diederich M. A beginner’s guide to NF-κB signaling pathways. Ann NY Acad Sci. 2004;1030:1–13. doi: 10.1196/annals.1329.002. [DOI] [PubMed] [Google Scholar]
- 21.Karin M, Delhase M. The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 22.Aronin N. Target selectivity in mRNA silencing. Gene Ther. 2006;13:509–516. doi: 10.1038/sj.gt.3302726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gitter AH, Bendfeldt K, Schmitz H, Schulzke JD, Bentzel CJ, Fromm M. Epithelial barrier defects in HT-29/B6 colonic cell monolayers induced by tumor necrosis factor-α. Ann NY Acad Sci. 2000;915:193–203. doi: 10.1111/j.1749-6632.2000.tb05242.x. [DOI] [PubMed] [Google Scholar]
- 24.Mandrup-Poulsen T. Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol. 2003;66:1433–1440. doi: 10.1016/s0006-2952(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 25.Welcker K, Martin A, Kolle P, Siebeck M, Gross M. Increased intestinal permeability in patients with inflammatory bowel disease. Eur J Med Res. 2004;9:456–460. [PubMed] [Google Scholar]
- 26.Nakamura M, Saito H, Kasanuki J, Tamura Y, Yoshida S. Cytokine production in patients with inflammatory bowel disease. Gut. 1992;33:933–937. doi: 10.1136/gut.33.7.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-α modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 28.Madara JL, Stafford J. Interferon-γ directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83:724–727. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwamoto GK, Monick MM, Burmeister LF, Hunninghake GW. Interleukin 1 release by human alveolar macrophages and blood monocytes. Am J Physiol. 1989;256:C1012–C1015. doi: 10.1152/ajpcell.1989.256.5.C1012. [DOI] [PubMed] [Google Scholar]
- 30.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-γ and tumor necrosis factor-α synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 32.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 33.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-α modulation of intestinal epithelial tight junction barrier. Am J Physiol. 2006;290:G496–G504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- 34.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-γ induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923–933. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- 36.Murata T, Noguchi PD, Puri RK. IL-13 induces phosphorylation and activation of JAK2 Janus kinase in human colon carcinoma cell lines: similarities between IL-4 and IL-13 signaling. J Immunol. 1996;156:2972–2978. [PubMed] [Google Scholar]
- 37.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 38.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85:1139–1162. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 39.Han X, Fink MP, Delude RL. Proinflammatory cytokines cause NO-dependent and -independent changes in expression and localization of tight junction proteins in intestinal epithelial cells. Shock. 2003;19:229–237. doi: 10.1097/00024382-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Han X, Fink MP, Yang R, Delude RL. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 2004;21:261–270. doi: 10.1097/01.shk.0000112346.38599.10. [DOI] [PubMed] [Google Scholar]
- 41.Gitter AH, Bendfeldt K, Schulzke JD, Fromm M. Leaks in the epithelial barrier caused by spontaneous and TNF-α -induced single-cell apoptosis. FASEB J. 2000;14:1749–1753. doi: 10.1096/fj.99-0898com. [DOI] [PubMed] [Google Scholar]
- 42.Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem. 2000;78:85–96. [PubMed] [Google Scholar]
- 43.Van Itallie CM, Anderson JM. Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci. 1997;110(Pt. 9):1113–1121. doi: 10.1242/jcs.110.9.1113. [DOI] [PubMed] [Google Scholar]
- 44.Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–1049. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simonovic I, Rosenberg J, Koutsouris A, Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol. 2000;2:305–315. doi: 10.1046/j.1462-5822.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- 46.Yu AS, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Lynch RD, Schneeberger EE. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol. 2005;288:C1231–C1241. doi: 10.1152/ajpcell.00581.2004. [DOI] [PubMed] [Google Scholar]
- 47.Xia W, Wong CH, Lee NP, Lee WM, Cheng CY. Disruption of Sertoligerm cell adhesion function in the seminiferous epithelium of the rat testis can be limited to adherens junctions without affecting the blood-testis barrier integrity: an in vivo study using an androgen suppression model. J Cell Physiol. 2005;205:141–157. doi: 10.1002/jcp.20377. [DOI] [PubMed] [Google Scholar]
- 48.Toliver-Kinsky T, Wood T, Perez-Polo JR. Nuclear factor κB/p49 is a negative regulatory factor in nerve growth factor-induced choline acetyl-transferase promoter activity in PC12 cells. J Neurochem. 2000;75:2241–2251. doi: 10.1046/j.1471-4159.2000.0752241.x. [DOI] [PubMed] [Google Scholar]
- 49.Wei L, Sandbulte MR, Thomas PG, Webby RJ, Homayouni R, Pfeffer LM. NFκB negatively regulates interferon-induced gene expression and anti-influenza activity. J Biol Chem. 2006;281:11678–11684. doi: 10.1074/jbc.M513286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 51.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13:3218–3234. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]