

Abstract
The mechanisms responsible for coronary pressure-flow autoregulation, a critical physiologic phenomenon that maintains coronary blood flow relatively constant in the presence of changes in perfusion pressure, remain poorly understood. This investigation tested the hypothesis that voltage-sensitive K+ (KV) and Ca2+ (CaV1.2) channels play a critical role in coronary pressure-flow autoregulation in vivo. Experiments were performed in open-chest, anesthetized Ossabaw swine during step changes in coronary perfusion pressure (CPP) from 40 to 140 mmHg before and during inhibition of KV channels with 4-aminopyridine (4AP, 0.3 mM, ic) or CaV1.2 channels with diltiazem (10 μg/min, ic). 4AP significantly decreased vasodilatory responses to H2O2 (0.3–10 μM, ic) and coronary flow at CPPs = 60–140 mmHg. This decrease in coronary flow was associated with diminished ventricular contractile function (dP/dT) and myocardial oxygen consumption. However, the overall sensitivity to changes in CPP from 60 to 100 mmHg (i.e. autoregulatory gain; Gc) was unaltered by 4-AP administration (Gc = 0.46 ± 0.11 control vs. 0.46 ± 0.06 4-AP). In contrast, inhibition of CaV1.2 channels progressively increased coronary blood flow at CPPs > 80 mmHg and substantially diminished coronary Gc to −0.20 ± 0.11 (P < 0.01), with no effect on contractile function or oxygen consumption. Taken together, these findings demonstrate that (1) KV channels tonically contribute to the control of microvascular resistance over a wide range of CPPs, but do not contribute to coronary responses to changes in pressure; (2) progressive activation of CaV1.2 channels with increases in CPP represents a critical mechanism of coronary pressure-flow autoregulation.
Keywords: Autoregulation, Coronary blood flow, Potassium channel, Calcium channel, Swine
Introduction
Coronary pressure-flow autoregulation is an essential mechanism by which the coronary circulation maintains constant blood flow in the presence of alterations in perfusion pressure. The autoregulatory capacity of the coronary vascular bed is particularly important during flow-limiting stenosis where if absent, hypoperfusion can rapidly diminish cardiac function [22, 64]. Alternatively, lack of vascular responses to elevations in perfusion pressure can lead to increases in coronary vascular volume, myocardial stiffness, and oxygen consumption (MVO2), i.e. Gregg Phenomenon [2, 24]. However, despite the importance of coronary pressure-flow autoregulation, the mechanisms underlying this phenomenon remain poorly understood.
Previous investigations of coronary autoregulation have focused primarily on the contribution of myocardial tissue pressure, local metabolic and myogenic mechanisms [3, 14, 16, 21, 22, 32]. Although support for tissue pressure can be found in encapsulated organs such as the kidney [30, 33], evidence in the heart is limited as similar increases in intramyocardial pressure occur in the presence and absence of pressure-flow autoregulation [18, 21]. In contrast, more prominent implications for local metabolic control have been identified as studies by the Feigl laboratory support that ~23 % of the changes in coronary conductance that occur with alterations in perfusion pressure are mediated by the synergistic effects of CO2 and O2 [9]. Other studies suggest that additional metabolites such as nitric oxide (NO), hydrogen peroxide (H2O2), and adenosine could also be involved, albeit at lower perfusion pressures [10, 57, 60, 68]. However, inhibition or catalytic degradation of these metabolites has failed to significantly alter coronary responses to changes in perfusion pressure (i.e. autoregulatory closed-loop gain). Subsequent data show that blockade of end effector KATP channels, which contribute to vasodilation in response to adenosine, also does not influence coronary autoregulatory capacity [61]. We propose that voltage-sensitive K+ (KV) channels may play a more prominent role as these channels contribute to the control of coronary blood flow at rest, in response to brief episodes of cardiac ischemia and during increases in MVO2 [4, 5, 15, 54–56]. However, the contribution of KV channels to coronary pressure-flow autoregulation has not been investigated.
In addition to metabolic mechanisms, myogenic vasoconstriction is likely critical for mitigating pressure-induced increases in coronary blood flow [40, 48]. Data from isolated vessel preparations indicate that coronary responses to increases in intraluminal pressure activate an endothelium-independent [39], mechanosensitive mechanism that results in graded decreases in smooth muscle membrane potential [12, 29]. Stretching vascular smooth muscle cells also induces an ~50 % increase in intracellular [Ca2+] that has been attributed to extracellular influx via voltage-gated (CaV1.2) Ca2+ channels [13]. Importantly, the extent to which CaV1.2 channels contribute to changes in coronary vasomotor tone in response to alterations in coronary perfusion pressure in vivo has not been determined.
Accordingly, the purpose of this investigation was to test the following hypotheses: (1) vasoactive metabolites produced in response to changes in perfusion pressure modulate coronary vascular resistance and autoregulatory capacity via a KV channel-dependent mechanism; (2) progressive activation of CaV1.2 channels in response to elevations in perfusion pressure is critical for pressure-flow autoregulation in the coronary circulation.
Methods
This investigation was approved by the Institutional Animal Care and Use Committee in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85–23, Revised 2011). Animals utilized for this study were Ossabaw swine (n = 12) weighing 30–60 kg. Following completion of experimental protocols, hearts were fibrillated and excised as recommended by the American Veterinary Medical Association Guide on Euthanasia (June 2007).
Surgical preparation
Swine were initially sedated with telazol (5 mg/kg, sc), zylazine (2.2 mg/kg, sc) and ketamine (3.0 mg/kg, sc). Following endotracheal intubation and venous access, anesthesia was maintained with morphine (3.0 mg/kg, sc) and α-chloralose (100 mg/kg, iv). The animals were mechanically ventilated (Harvard respirator) with room air supplemented with oxygen. Catheters were placed into the right femoral artery and vein for systemic hemodynamic measurements and administration of supplemental anesthetic, heparin, and sodium bicarbonate, respectively. The left femoral artery was catheterized to supply blood to an extracorporeal perfusion system used to perfuse the left anterior descending (LAD) coronary artery at controlled pressures. Arterial blood gases were analyzed periodically throughout the experimental protocol and adjustments made as needed to maintain blood gas parameters within normal physiological limits. A left lateral thoracotomy was performed to expose the heart, and the LAD was isolated and cannulated distal to its first major diagonal branch following heparin administration (500 U/kg, iv). Coronary perfusion pressure (CPP) was regulated by a servo-controlled roller pump and coronary blood flow was continuously measured by an in-line Transonic Systems flow transducer (Ithaca, New York, USA). A catheter was also inserted into the interventricular coronary vein for venous sampling of blood draining the LAD perfusion territory. Left ventricular contractile function was measured with a Millar Mikro-Tip manometer (Millar Instruments, Inc. Houston, TX). Data were continuously recorded on IOX data acquisition software from Emka Technologies (Falls Church, VA).
Experimental protocol
Following a stabilization period (~20 min post cannulation), H2O2 (0.3–10 μM) was infused into the LAD perfusion circuit before and during administration of the KV channel antagonist 4-aminopyridine (4AP; 0.3 mM, i.c.) with CPP held constant at 100 mmHg (n = 5). Pressure-flow autoregulation was assessed by 10 mmHg increment changes in CPP from 140 mmHg to 40 mmHg before or during intracoronary infusion of 4AP (0.3 mM, n = 7) or the CaV1.2 channel antagonist diltiazem (10 μg/min, n = 5). Arterial and coronary venous blood samples were collected simultaneously once hemodynamic variables were stable at each CPP. Blood samples were analyzed with an Instrumentation Laboratories automatic blood gas analyzer (GEM Premier 3000) and CO-oximeter (682) system. MVO2 (μl O2/min/g) was calculated by multiplying coronary blood flow by the arterial coronary venous difference in oxygen content. As previously reported [2], closed-loop autoregulatory gain (Gc) was calculated using the following formula: Gc = 1 − [(ΔF/F)/(ΔP/P)]. Changes in flow and pressure were assessed relative to control responses at CPP = 100 mmHg. All drugs were dissolved in saline, adjusted to physiologic pH, and infused at a constant, continuous rate directly into the coronary circulation.
Statistical analyses
Data are presented as mean ± SE for n swine. Statistical comparisons were made using a one-way or two-way (Factor A: drug treatment; Factor B: pressure) repeated measures analysis of variance (ANOVA) as appropriate (Sigma Stat 11.0 Software). If statistical differences (P < 0.05) in these analyses were noted, a Student–New-man–Keuls multiple comparison test was performed.
Results
Contribution of KV channels to H2O2-mediated coronary vasodilation
Consistent with previous studies [55], intracoronary administration of H2O2 (0.3–10 μM) dose-dependently increased coronary blood flow from 0.52 ± 0.03 ml/min/g, under control conditions, to 1.40 ± 0.04 ml/min/g at the highest dose of H2O2 (P < 0.001, n = 5). Inhibition of coronary KV channels with 4AP (0.3 mM) at CPP = 100 mmHg decreased coronary blood flow, indices of cardiac contractile function (dP/dTmax and dP/dTmin), and MVO2. Mean aortic pressure and heart rate were unaffected by 4AP (Table 1). Coronary vasodilation in response to exogenous H2O2 was markedly depressed by the administration of 4AP (Fig. 1, P < 0.05).
Table 1.
Hemodynamic, cardiac and blood gas parameters during variable coronary perfusion pressures with and without 4AP or Diltiazem
| CPP | 140 | 130 | 120 | 110 | 100 | 90 | 80 | 70 | 60 | 50 | 40 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | |||||||||||
| Systolic blood pressure (mmHg) | |||||||||||
| Control | 134 ± 9 | 133 ± 9 | 132 ± 8 | 132 ± 8 | 132 ± 8 | 131 ± 8 | 130 ± 8 | 129 ± 8 | 128 ± 8 | 125 ± 7 | 119 ± 6 |
| 4AP | 132 ± 9 | 134 ± 9 | 134 ± 9 | 130 ± 10 | 133 ± 10 | 131 ± 11 | 128 ± 11 | 128 ± 12 | 125 ± 11 | 120 ± 9 | 115 ± 8 |
| Diltiazem | 103 ± 8*,† | 100 ± 8*,† | 99 ± 8*,† | 97 ± 7*,† | 95 ± 7*,† | 97 ± 8*,† | 97 ± 9*,† | 95 ± 9*,† | 93 ± 8*,† | 93 ± 9*,† | 82 ± 6*,† |
| Diastolic blood pressure (mmHg) | |||||||||||
| Control | 83 ± 6 | 82 ± 6 | 82 ± 7 | 82 ± 7 | 82 ± 7 | 81 ± 7 | 81 ± 7 | 81 ± 6 | 80 ± 6 | 81 ± 5 | 78 ± 5 |
| 4AP | 77 ± 4 | 77 ± 5 | 84 ± 5 | 79 ± 4 | S3 ± 5 | 80 ± 5 | 79 ± 5 | 79 ± 6 | 76 ± 5 | 76 ± 4 | 74 ± 4 |
| Diltiazem | 69 ± 5 | 66 ± 5 | 66 ± 5† | 65 ± 4*,† | 63 ± 4*,† | 63 ± 4*,† | 63 ± 4*,† | 62 ± 4*,† | 61 ± 4*,† | 60 ± 4*,† | 54 ± 4*,† |
| Mean aortic pressure (mmHg) | |||||||||||
| Control | 108 ± 8 | 107 ± 8 | 106 ± 7 | 106 ± 7 | 106 ± 7 | 105 ± 7 | 105 ± 7 | 104 ± 7 | 103 ± 6 | 102 ± 6 | 97 ± 5 |
| 4AP | 101 ± 5 | 104 ± 7 | 107 ± 6 | 104 ± 7 | 106 ± 7 | 104 ± 7 | 102 ± 8 | 101 ± 8 | 98 ± 7 | 96 ± 6 | 93 ± 6 |
| Diltiazem | 86 ± 6* | 83 ± 7*,† | 82 ± 6*,† | 81 ± 6*,† | 79 ± 5*,† | 80 ± 6*,† | 80 ± 6*,† | 78 ± 6*,† | 76 ± 6*,† | 76 ± 7*,† | 74 ± 7*,† |
| Heart Rate (beats/min) | |||||||||||
| Control | 50 ± 5 | 51 ± 5 | 51 ± 6 | 51 ± 6 | 52 ± 6 | 53 ± 7 | 61 ± 12 | 53 ± 7 | 53 ± 7 | 56 ± 6 | 61 ± 5 |
| 4AP | 49 ± 7 | 46 ± 7 | 46 ± 6 | 48 ± 7 | 48 ± 6 | 49 ± 7 | 48 ± 7* | 48 ± 7 | 48 ± 7 | 49 ± 6 | 52 ± 6* |
| Diltiazem | 79 ± 8*,† | 80 ± 7*,† | 80 ± 6*,† | 81 ± 6*,† | 81 ± 7*,† | 77 ± 6*,† | 76 ± 6*,† | 77 ± 6*,† | 79 ± 6*,† | 76 ± 6*,† | 81 ± 6*,† |
| Coronary blood flow (ml/min/g) | |||||||||||
| Control | 0.76 ± 0.05 | 0.64 ± 0.04 | 0.58 ± 0.03 | 0.53 ± 0.03 | 0.50 ± 0.03 | 0.48 ± 0.03 | 0.45 ± 0.03 | 0.43 ± 0.03 | 0.39 ± 0.03 | 0.32 ± 0.04 | 0.23 ± 0.04 |
| 4AP | 0.55 ± 0.03* | 0.43 ± 0.03* | 0.44 ± 0.04* | 0.43 ± 0.03* | 0.39 ± 0.04* | 0.38 ± 0.05* | 0.37 ± 0.05* | 0.34 ± 0.04* | 0.31 ± 0.03* | 0.26 ± 0.02* | 0.17 ± 0.03* |
| Diltiazem | 1.48 ± 0.18*,† | 1.30 ± 0.12*,† | 1.15 ± 0.09*,† | 0.99 ± 0.08*,† | 0.86 ± 0.06*,† | 0.70 ± 0.05*,† | 0.60 ± 0.03† | 0.51 ± 0.03† | 0.44 ± 0.03 | 0.37 ± 0.03 | 0.22 ± 0.03 |
| Myocardial oxygen consumption (μl O2/min/g) | |||||||||||
| Control | 69 ± 8 | 68 ± 8 | 66 ± 8 | 64 ± 7 | 64 ± 7 | 60 ± 7 | 58 ± 6 | 56 ± 6 | 51 ± 5 | 44 ± 5 | 32 ± 5 |
| 4AP | 59 ± 5* | 54 ± 4* | 51 ± 5* | 52 ± 5* | 48 ± 4* | 48 ± 6* | 47 ± 6* | 44 ± 5* | 41 ± 4* | 34 ± 3* | 26 ± 4 |
| Diltiazem | 72 ± 17 | 68 ± 16 | 63 ± 16 | 64 ± 13 | 62 ± 11 | 56 ± 9 | 53 ± 7 | 50 ± 7 | 50 ± 6 | 40 ± 7 | 28 ± 5 |
| Coronary venous PO2 | |||||||||||
| Control | 27 ± 3.0 | 24 ± 2.3 | 21 ± 2.3 | 19 ± 2.6 | 18 ± 2.0 | 18 ± 2.0 | 17 ± 2.0 | 17 ± 1.2 | 16 ± 1.3 | 15 ± 0.7 | 15 ± 1.0 |
| 4AP | 23 ± 2.2* | 22 ± 1.7 | 21 ± 1.9 | 21 ± 1.5 | 19 ± 1.3 | 19 ± 1.4 | 19 ± 1.3 | 18 ± 1.5 | 17 ± 1.1 | 14 ± 0.9 | 14 ± 0.9 |
| Diltiazem | 44 ± 5.3*,† | 42 ± 5.3*,† | 40 ± 5.1*,† | 37 ± 4.5*,† | 33 ± 3.9*,† | 31 ± 3.0*,† | 29 ± 2.6*,† | 26 ± 1.7*,† | 24 ± 1.6*,† | 21 ± 1.0† | 19 ± 1.2 |
| dP/dT Max (mmHg/s) | |||||||||||
| Control | 1,501 ± 114 | 1,501 ± 115 | 1,527 ± 121 | 1,532 ± 115 | 1,569 ± 113 | 1,551 ± 111 | 1,561 ± 106 | 1,559 ± 111 | 1,534 ± 1115 | 1,393 ± 130 | 1,135 ± 99 |
| 4AP | 1,362 ± 115 | 1,334 ± 121* | 1,351 ± 128* | 1,349 ± 125* | 1,258 ± 127* | 1,254 ± 110* | 1,228 ± 123* | 1,242 ± 127* | 1,200 ± 127* | 1,144 ± 135* | 999 ± 82 |
| Diltiazem | 1,476 ± 239 | 1,491 ± 264 | 1,473 ± 265 | 1,477 ± 270 | 1,454 ± 251 | 1,456 ± 265 | 1,425 ± 270 | 1,426 ± 293 | 1,389 ± 279 | 1,365 ± 267 | 1,065 ± 130 |
| dP/dT Min (mmHg/s) | |||||||||||
| Control | −1,448 ± 159 | −1,455 ± 148 | −1,429 ± 148 | −1,359 ± 167 | −1,359 ± 150 | −1,338 ± 137 | −1,306 ± 130 | −1,276 ± 127 | −1,136 ± 134 | −1,016 ± 155 | −922 ± 146 |
| 4AP | −1,177 ± 132* | −1,148 ± 135* | −1,129 ± 133* | −1,086 ± 133* | −1,124 ± 155* | −1,079 ± 156* | −1,058 ± 147* | −1,058 ± 140* | − 987 ± 131 | − 879 ± 127 | −744 ± 114 |
| Diltiazem | −1,330 ± 117 | −1,271 ± 110 | −1,292 ± 116 | −1,290 ± 125 | −1,278 ± 127 | −1,305 ± 156 | −1,265 ± 171 | −1,212 ± 154 | −1,185 ± 139 | −1,138 ± 169 | −786 ± 165 |
Values are mean ± SE for Control, 4AP (n = 7), and diltiazem (n = 5)
P < 0.05 versus control same CPP;
P < 0.05 versus 4AP same CPP
Fig. 1. Role of KV channels in H2O2-mediated coronary vasodilation.
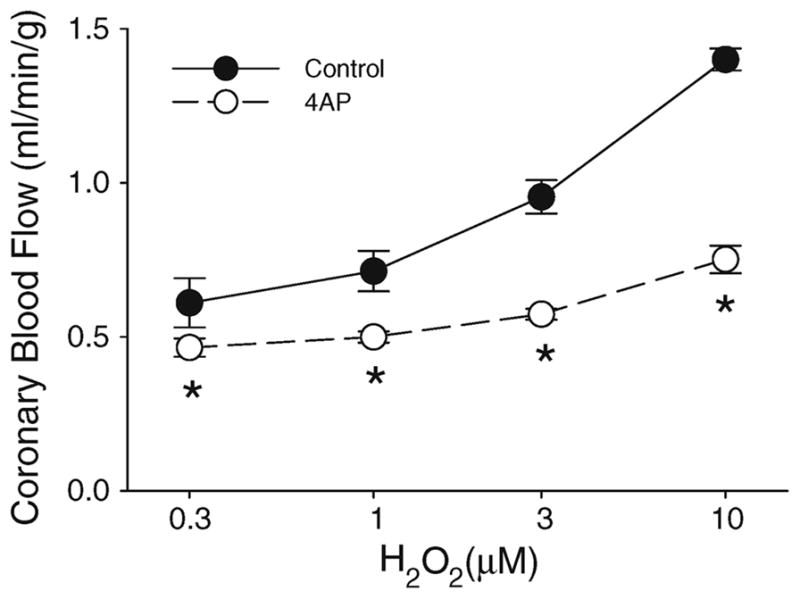
Intracoronary administration of H2O2 dose-dependently increased coronary blood flow. Coronary vasodilation in response to exogenous H2O2 was markedly depressed by the administration of 4AP. Figure shows grouped average traces for n = 5 swine; *P < 0.05 versus control, same concentration of H2O2
Coronary vascular response to changes in perfusion pressure
Effects of alterations in CPP on systemic hemodynamics are listed in Table 1. Modest changes in coronary blood flow (0.11 ± 0.02 ml/min/g) were noted over a CPP range of 60–100 mmHg, without significant effects on cardiac contractile function or MVO2 (n = 7). Determination of Gc indicated an autoregulatory capacity of 0.46 ± 0.11 in untreated hearts over a CPP range of 60–100 mmHg (Gc = 1 being perfect). However, Gc was significantly reduced to −0.43 ± 0.29 at CPPs ranging from 100 to 140 mmHg and −0.52 ± 0.42 at CPPs ranging from 40 to 60 mmHg (Fig. 2d). Reductions in coronary blood flow below CPP 60 mmHg (Fig. 2a) were associated with diminished dP/dTmax, dP/dTmin, MVO2, and mean aortic pressure (Table 1).
Fig. 2.

Role of KV channels in coronary pressure-flow autoregulation. a Inhibition of coronary KV channels with 4AP significantly attenuated coronary blood flow at CPPs from 60 to 140 mmHg. 4AP administration did not affect the change in coronary blood flow at CPPs < 120 mmHg (b), the balance between coronary blood flow and MVO2 (c) or autoregulatory gain (d). Figures show grouped average traces for n = 7 swine. *P < 0.05 versus control, same CPP
KV channels in coronary pressure-flow autoregulation
Relative to untreated control conditions, inhibition of coronary KV channels with 4AP significantly attenuated coronary blood flow ~20 % at CPPs ranging from 60 to 140 mmHg (Fig. 2a, P < 0.05). Intracoronary 4AP administration had no effect on mean aortic pressure at CPPs >50 mmHg (Table 1, n = 7). The reductions in coronary blood flow were associated with diminished cardiac contractile function as evidenced by the ~10–20 % decrease in dP/dTmax and dP/dTmin over a wide range of CPPs (Table 1, P < 0.05). These changes in contractile function were also accompanied by decreases in MVO2 at CPPs ranging from 50 to 140 mmHg (Table 1, P < 0.05). However, 4AP administration did not alter the relationship between coronary blood flow and MVO2 (Fig. 2c). Importantly, inhibition of KV channels did not affect the overall change in coronary blood flow at CPPs <120 mmHg (Fig 2b) as flow only varied 0.08 ± 0.01 ml/min/g over CPPs of 60–100 mmHg (P = 0.53 vs. untreated control). Calculation of Gc over this range of CPPs also revealed essentially identical Gcs for both control (0.46 ± 0.11) and 4AP (0.46 ± 0.06) treated conditions (Fig. 2d, P = 0.99). Administration of 4AP also failed to significantly influence Gc over CPPs ranging from 40 to 60 mmHg (P = 0.78) or 100 to 140 mmHg (P = 0.46). Alternatively, pressure-induced increases in coronary blood flow were attenuated by 4AP at CPPs of 130–140 mmHg (Fig. 2b), while coronary zero flow pressure (Pzf) was unaffected (average = 23 ± 2 mmHg).
CaV1.2 channels in coronary pressure-flow autoregulation
Inhibition of coronary CaV1.2 channels with diltiazem (10 μg/min) progressively increased coronary blood flow (Fig. 3a, P < 0.05) and significantly increased the change in blood flow to 0.42 ± 0.06 ml/min/g over CPPs ranging from 60 to 100 mmHg (Fig. 3b; P < 0.01 vs. untreated control). Diltiazem administration reduced mean aortic pressure and increased heart rate at all CPPs (Table 1, P < 0.05). However, this vasodilatory effect was not associated with alterations in indices of cardiac contractile function or MVO2 (Table 1). Thus, diltiazem-mediated increases in coronary blood flow were independent of changes in MVO2 (Fig. 3c). Importantly, inhibition of coronary CaV1.2 channels markedly reduced Gc to −0.20 ± 0.11 over a CPP range of 60–100 mmHg, i.e. essentially abolished pressure-flow autoregulation (Fig. 3d, P < 0.01). Diltiazem did not affect Gc at CPPs ranging from 40 to 60 mmHg (P = 0.79), 100 to 140 mmHg (P = 0.47), or significantly alter Pzf relative to untreated controls (average = 20 ± 1 mmHg).
Fig. 3.

Role of CaV1.2 channels in coronary pressure-flow autoregulation. Inhibition of coronary CaV1.2 channels with diltiazem (10 μg/min) significantly increased coronary blood flow at CPPs >80 mmHg (a) and the change in blood flow over a wide range of CPPs (b). Coronary vasodilation in response to diltiazem was independent of MVO2 (c) and abolished pressure-flow autoregulation within CPPs ranging from 60 to 100 mmHg (d). Figures show grouped average traces for n = 5 swine. *P < 0.05 versus control
Discussion
First identified in the coronary circulation by Eckel [16], coronary autoregulation refers to the intrinsic ability of the heart to maintain constant blood flow despite changes in arterial perfusion pressure [33]. Although numerous studies have focused on delineating the interdependent relationship between coronary blood flow and CPP [23, 51, 65], the underlying mechanisms responsible for coronary pressure-flow autoregulation have not been clearly defined. Given the important role for voltage-dependent KV and CaV1.2 channels in the control of smooth muscle membrane potential and coronary vascular resistance [50, 63, 66], we hypothesized that these specific channels may also serve as critical end-effectors in modulating the vascular responses to alterations in CPP. Findings from this investigation demonstrate that KV channels tonically contribute to the control of microvascular resistance over a wide range of CPPs, but do not contribute to coronary responses to changes in perfusion pressure within the autoregulatory range. In contrast, progressive activation of CaV1.2 channels with increases in CPP represents a critical mechanism of coronary pressure-flow autoregulation.
Role of H2O2 and KV channels in coronary pressure-flow autoregulation
Experiments performed in this study were designed to test the hypothesis that vasoactive metabolites produced in response to changes in perfusion pressure modulate coronary vascular resistance and autoregulatory capacity via a KV channel-dependent mechanism. The rationale for this hypothesis is based on earlier studies by our group which demonstrated that activation of KV channels is critical to the regulation of coronary blood flow [4, 6, 15] and that several key vasoactive metabolites (e.g. adenosine, NO, H2O2) mediate coronary vasodilation predominantly via KV channels [5, 15, 54, 55]. Consistent with prior studies in dogs [55], the present findings indicate that H2O2 induces marked coronary vasodilation via activation of KV channels in swine (Fig. 1). We propose this dilator effect is due to direct activation of smooth muscle KV channels as Rogers et al. [54] found that H2O2 dose-dependently activates 4AP sensitive K+ current in coronary smooth muscle cells and that denudation has no effect on H2O2-induced dilation of isolated coronary arterioles [55]. These data are in contrast with alternative studies that documented H2O2 is a key endothelium-derived hyperpolarizing factor that mediates coronary vasodilation via a BKCa channel-dependent mechanism [44, 69].
To examine the role of KV channels in coronary pressure-flow autoregulation, we performed experiments in anesthetized, open-chest swine in which CPP was varied from 40 to 140 mmHg via a servo-controlled, extracorporeal perfusion circuit. In these studies, we demonstrated that the inhibition of KV channels significantly reduced coronary blood flow over a wide range of CPPs (Fig. 2a). However, 4AP administration did not affect the change in coronary blood flow with alterations in perfusion pressure at CPPs < 120 mmHg (Fig. 2b) or influence the overall autoregulatory capacity (Gc) of the coronary circulation (Fig. 2d). Thus, given that 4AP abolished H2O2-mediated coronary vasodilation, our findings do not support a prominent role for KV channel-dependent pathways, such as H2O2, in modulating coronary vascular responses to changes in perfusion pressure within the autoregulatory range. This conclusion differs from that of Yada et al. [68] who suggested that H2O2, in cooperation with NO and adenosine, plays an important role in coronary pressure-flow autoregulation in vivo in dogs. However, other investigations fail to support a prominent role for NO or adenosine in coronary autoregulation [37, 38, 60, 61]. Closer examination of the data presented by Yada et al. also indicates that the significant reductions in coronary blood flow at lower CPPs in the presence of L-NAME and/or catalase did not significantly alter the coronary pressure-flow relationship; i.e. closed-loop autoregulatory gain. Interestingly, our findings with KV channel inhibition are similar with those of Stepp et al. [61] who determined that blockade of KATP channels with glibenclamide produced a tonic decrease in coronary blood flow, but did not significantly influence the autoregulatory capability of the coronary circulation. Taken together, these data indicate that neither KV channels, KATP channels, nor the upstream vasodilatory factors known to converge on these channels, are necessary to support a metabolic component of coronary pressure-flow autoregulation [42]. In contrast, the limitation of pressure-induced increases in coronary flow by 4AP supports earlier studies implicating KV channels as an important negative-feedback mechanism that limits myogenic constriction, especially at high perfusion pressures (CPP > 120 mmHg) [1, 17, 67].
Role of CaV1.2 channels in coronary pressure-flow autoregulation
Functioning as a predominant mediator of extracellular Ca2+ influx, CaV1.2 channels constitutively contribute to the control of coronary microvascular resistance. This is evidenced in vivo by the marked, dose-dependent increases in coronary blood flow observed in response to CaV1.2 channel blockade [36]. Findings from the present study are the first to demonstrate that inhibition of coronary CaV1.2 channels results in a progressive increase in coronary blood flow as CPP is elevated (Fig. 3a). Thus, administration of diltiazem significantly augmented pressure-induced changes in coronary blood flow (Fig. 3b), resulting in responses that would be predicted in a maximally dilated bed; i.e. passive vasculature [28]. Importantly, the dose of diltiazem used (10 μg/min) did not produce maximal dilation (coronary flow = 0.86 ± 0.06 ml/min/g at CPP = 100 mmHg) but did markedly diminish coronary autoregulatory capacity as Gc was reduced to −0.20 ± 0.11 within the autoregulatory range of 60–100 mmHg (Fig. 3d), i.e. inhibition of CaV1.2 channels essentially abolished pressure-flow autoregulation (see “Limitations of the study” below). This observed decrement of pressure-flow auto-regulation supports that increasing activation of CaV1.2 channels with elevations in CPP is a central mechanism underlying the intrinsic ability of the coronary circulation to maintain blood flow constant with changes in perfusion pressure. Our findings are consistent with earlier in vitro myogenic studies demonstrating pressure-induced increases in membrane potential and arteriolar wall [Ca2+] are dependent on CaV1.2 channels [12, 13]. Experiments in isolated, pressurized arterioles indicate a functional myogenic component also exists in the human and porcine coronary microcirculation [34, 41, 45, 49]. However, evidence for the involvement of other mechanosensitive, nonselective cation channels in myogenic vasoconstriction has also been reported [12, 29]. Consequently, it is important to point out that the present data cannot distinguish between a strictly myogenic versus metabolic-induced activation of CaV1.2 channels. Regardless, the present data demonstrate that CaV1.2 channel-dependent pathways represent a critical mechanism of coronary pressure-flow autoregulation in vivo.
Relationship between coronary blood flow, CPP, and MVO2
Coronary blood flow is dependent on CPP and MVO2 [59], and coronary pressure can arguably influence metabolism via the “Gregg Phenomenon” [24, 58]. Although the Gregg effect is more pronounced in poorly autoregulating hearts (~55 % increase in MVO2 over CPP range of 60–120 mmHg [2]), modest changes in MVO2 are detected in hearts with effective pressure-flow autoregulation (~20 % increase in MVO2 over CPP range 60–120 mmHg [2, 17]). Thus, changes in arterial pressure and/or myocardial metabolism can influence the overall level of myocardial perfusion. In this study, administration of both 4AP and diltiazem altered these determinants of coronary blood flow. In particular, 4AP-mediated reductions in coronary flow were accompanied by reductions in cardiac function and MVO2, while diltiazem decreased arterial pressure and reflexively increased heart rate (Table 1). Changes in arterial pressure did not directly influence CPP as these experiments were conducted in a cannulated, extracorporeal perfused preparation. However, to account for these drug-induced alterations, we plotted coronary blood flow relative to its respective MVO2 at CPPs ranging from 40 to 140 mmHg. These plots indicate that 4AP did not significantly affect the balance between coronary blood flow and MVO2 as CPP was changed over this range of perfusion pressures (Fig. 2c). Alternatively, blockade of coronary CaV1.2 channels resulted in a progressive increase in coronary blood flow at a given level of MVO2 (Fig. 3c). Thus, the increases in coronary blood flow induced by diltiazem were not mediated by significant increases in MVO2, i.e. reflect pressure-mediated increases in coronary flow, not metabolic vasodilation. Regardless of the experimental condition, our findings support a strong interdependent relationship between CPP, MVO2, and coronary blood flow. To evaluate the individual contribution of each of these factors, additional examination of the interrelationship between coronary blood flow, MVO2, and CPP was assessed by 3-dimensional analysis. These data further support that inhibition of KV channels did not significantly affect the relationship between coronary blood flow, MVO2, and CPP (Fig. 4a). In contrast, increases in coronary blood flow observed with elevations in CPP and MVO2 were significantly augmented following diltiazem administration (Fig. 4b), further confirming a prominent role for CaV1.2 channels in coronary pressure-flow autoregulation.
Fig. 4.
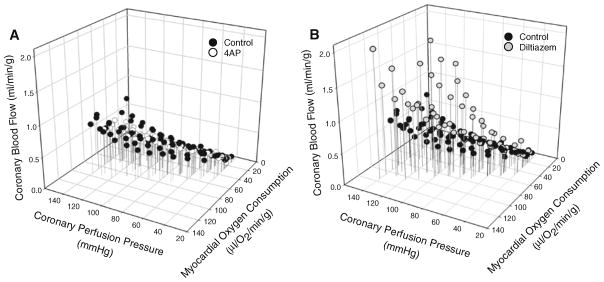
Effects of KV and CaV1.2 channel inhibition on 3-dimensional analysis of coronary pressure-flow autoregulation. Inhibition of KV channels reduced coronary blood flow and MVO2 but did not affect pressure-induced changes in coronary flow (a). Blockade of coronary CaV1.2 channels significantly increased coronary blood flow as CPP and MVO2 were elevated (b). Figures show individual data for n = 7 (control, 4AP) or n = 5 (diltiazem) swine
Limitations of the study
Conclusions regarding the role of KV and CaV1.2 channels in coronary pressure-flow autoregulation are confounded by specific effects of both 4AP and diltiazem on cardiac function and MVO2. In the case of 4AP, we found that inhibition of KV channels significantly decreased coronary blood flow and that these reductions in flow were accompanied by decreases in left ventricular dP/dT and MVO2 (Table 1). Such data present a circular argument as to whether 4AP-mediated decreases in coronary flow resulted in diminished contractile performance and subsequent decreases in MVO2, or alternatively if 4AP initially decreased contractile function which then led to reductions in MVO2 and coronary blood flow. Importantly, when 4AP administration is initiated, we typically find that coronary blood flow falls within a matter of seconds, which contrasts with reductions in left ventricular dP/dT that typically occur within ~2 min of 4AP administration. Accordingly, we conclude that 4AP resulted in rapid (tonic) coronary vasoconstriction that was followed by commensurate reductions in contractile function and MVO2, responses consistent with the characteristics of myocardial hibernation [11]. The present findings are in contrast with earlier studies [56] regarding the effect of 4AP on the balance between coronary blood flow and myocardial metabolism as 4AP failed to significantly decrease coronary venous PO2 at CPPs <140 mmHg. We propose this discrepancy is related to the higher levels of MVO2s reported in these studies at rest (70–100 μl O2/min/g) and during increases in metabolism (up to ~400 μl O2/min/g), relative to the much lower levels of MVO2 reported in our current preparation (~50 μl O2/min/g at rest). This point is supported by the significant reduction in coronary venous PO2 in the presence of 4AP when MVO2 was elevated to ~60 μl O2/min/g at CPP = 140 mmHg (Table 1).
Earlier studies have demonstrated that administration of coronary vasodilator agents significantly impair coronary pressure-flow autoregulation [16, 28]. In the present study, inhibition of CaV1.2 channels resulted in a ~70 % increase in baseline coronary flow at CPP = 100 mmHg. This effect of diltiazem significantly complicates interpretation as to whether decreased coronary autoregulatory capacity following diltiazem administration was due to the inhibition of CaV1.2 channels alone or simply to its vasodilator influence. This matter is further complicated by the fact that compounds induce vasodilation via activation of coronary K+ channels [35], which hyperpolarizes smooth muscle and inhibits CaV1.2 channels [7, 20, 27, 31, 62]. Thus, studies which demonstrate reductions in coronary autoregulation in the presence of vasodilators actually support our present conclusion regarding the role of CaV1.2 channels in pressure-flow autoregulation. Importantly, the dose of diltiazem used in the present study is selective for CaV1.2 channels and increased coronary blood flow from 0.50 to 0.86 ml/min/g, i.e. an average flow that is within the “normal range” of baseline coronary blood flow [19]. Despite these confounding effects, inhibition of CaV1.2 channels has been shown to produce marked, dose-dependent increases in baseline coronary blood flow [36], which supports a prominent role for these channels in the regulation of coronary vasomotor tone.
Summary and implications
We have identified a tonically active role for KV channels in the control of coronary microvascular resistance over wide range of CPPs (60–140 mmHg). However, our data indicate that these channels, and the vasodilatory pathways known to converge on them (adenosine, NO, H2O2), are not required for coronary responses to changes in perfusion pressure within the autoregulatory range. Although KV channels are not necessary for coronary pressure-flow autoregulation, it is possible that alternative pathways/channels are activated to compensate for KV channel inhibition. Alternatively, our findings do support that KV channels serve as a negative-feedback mechanism that limits myogenic constriction, especially at high perfusion pressures (CPP>120 mmHg). These findings are important in light of recent studies implicating impaired KV channel activity in a variety of disease states including hypercholesterolemia, hypertension, and hyperglycemia [8, 25, 26, 43, 46, 47].
Data from this study are the first to demonstrate that inhibition of CaV1.2 channels with diltiazem essentially abolishes the ability of the coronary circulation to maintain blood flow constant with alterations in perfusion pressure. Although our findings support a critical role for CaV1.2 channels in pressure-flow autoregulation, the mechanism by which CaV1.2 channels are activated (myogenic vs. metabolic) requires further investigation. Delineating mechanisms of coronary CaV1.2 channel activation is important given findings supporting elevated CaV1.2 channel function and associated activators (e.g. PKC) during hypertension and metabolic syndrome [36, 49, 52, 53]. We propose that administration of CaV1.2 channel blockers to such patients would likely prove beneficial, despite effects on autoregulatory capacity, as these agents would act to increase coronary blood flow, reduce left ventricular afterload, and improve the balance between myocardial oxygen delivery and metabolism.
Acknowledgments
This work was supported by AHA/NIH grants 10PRE4230035 (ZCB) and HL092245 (JDT).
Contributor Information
Zachary C. Berwick, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, IN 46202, USA
Steven P. Moberly, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, IN 46202, USA
Meredith C. Kohr, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, IN 46202, USA
Ethan B. Morrical, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, IN 46202, USA
Michelle M. Kurian, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, IN 46202, USA
Gregory M. Dick, Department of Exercise Physiology, Center for Cardiovascular and Respiratory Sciences, West Virginia University School of Medicine, Morgantown, USA
Johnathan D. Tune, Email: jtune@iupui.edu, Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, IN 46202, USA
References
- 1.Ahmed A, Waters CM, Leffler CW, Jaggar JH. Ionic mechanisms mediating the myogenic response in newborn porcine cerebral arteries. Am J Physiol Heart Circ Physiol. 2004;287:H2061–H2069. doi: 10.1152/ajpheart.00660.2004. [DOI] [PubMed] [Google Scholar]
- 2.Bai XJ, Iwamoto T, Williams AG, Jr, Fan WL, Downey HF. Coronary pressure-flow autoregulation protects myocardium from pressure-induced changes in oxygen consumption. Am J Physiol. 1994;266:H2359–H2368. doi: 10.1152/ajpheart.1994.266.6.H2359. [DOI] [PubMed] [Google Scholar]
- 3.Bassenge E, Heusch G. Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol. 1990;116:77–165. doi: 10.1007/3540528806_4. [DOI] [PubMed] [Google Scholar]
- 4.Berwick ZC, Dick GM, Moberly SP, Kohr MC, Sturek M, Tune JD. Contribution of voltage-dependent K(+) channels to metabolic control of coronary blood flow. J Mol Cell Cardiol. 2011 doi: 10.1016/j.yjmcc.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berwick ZC, Payne GA, Lynch B, Dick GM, Sturek M, Tune JD. Contribution of adenosine A(2A) and A(2B) receptors to ischemic coronary dilation: role of K(V) and K(ATP) channels. Microcirculation. 2010;17:600–607. doi: 10.1111/j.1549-8719.2010.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borbouse L, Dick GM, Payne GA, Berwick ZC, Neeb ZP, Alloosh M, Bratz IN, Sturek M, Tune JD. Metabolic syndrome reduces the contribution of K + channels to ischemic coronary vasodilation. Am J Physiol Heart Circ Physiol. 2010;298:H1182–H1189. doi: 10.1152/ajpheart.00888.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brading AF, Burdyga TV, Scripnyuk ZD. The effects of papaverine on the electrical and mechanical activity of the guinea-pig ureter. J Physiol. 1983;334:79–89. doi: 10.1113/jphysiol.1983.sp014481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bratz IN, Swafford AN, Jr, Kanagy NL, Dick GM. Reduced functional expression of K(+) channels in vascular smooth muscle cells from rats made hypertensive with N{omega}-nitro-L-arginine. Am J Physiol Heart Circ Physiol. 2005;289:H1284–H1290. doi: 10.1152/ajpheart.01053.2004. [DOI] [PubMed] [Google Scholar]
- 9.Broten TP, Feigl EO. Role of myocardial oxygen and carbon dioxide in coronary autoregulation. Am J Physiol. 1992;262:H1231–H1237. doi: 10.1152/ajpheart.1992.262.4.H1231. [DOI] [PubMed] [Google Scholar]
- 10.Canty JM, Jr, Smith TP., Jr Modulation of coronary autoregulatory responses by endothelium-derived nitric oxide. Int J Cardiol. 1995;50:207–215. doi: 10.1016/0167-5273(95)02379-B. [DOI] [PubMed] [Google Scholar]
- 11.Canty JM, Jr, Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J Mol Cell Cardiol. 2011 doi: 10.1016/j.yjmcc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 13.Davis MJ, Meininger GA, Zawieja DC. Stretch-induced increases in intracellular calcium of isolated vascular smooth muscle cells. Am J Physiol. 1992;263:H1292–H1299. doi: 10.1152/ajpheart.1992.263.4.H1292. [DOI] [PubMed] [Google Scholar]
- 14.DeFily DV, Chilian WM. Coronary microcirculation: autoregulation and metabolic control. Basic Res Cardiol. 1995;90:112–118. doi: 10.1007/BF00789441. [DOI] [PubMed] [Google Scholar]
- 15.Dick GM, Bratz IN, Borbouse L, Payne GA, Dincer UD, Knudson JD, Rogers PA, Tune JD. Voltage-dependent K + channels regulate the duration of reactive hyperemia in the canine coronary circulation. Am J Physiol Heart Circ Physiol. 2008;294:H2371–H2381. doi: 10.1152/ajpheart.01279.2007. [DOI] [PubMed] [Google Scholar]
- 16.Dole WP. Autoregulation of the coronary circulation. Prog Cardiovasc Dis. 1987;29:293–323. doi: 10.1016/S0033-0620(87)80005-1. [DOI] [PubMed] [Google Scholar]
- 17.Dole WP, Nuno DW. Myocardial oxygen tension determines the degree and pressure range of coronary autoregulation. Circ Res. 1986;59:202–215. doi: 10.1161/01.RES.59.2.202. [DOI] [PubMed] [Google Scholar]
- 18.Driscol TE, Moir TW, Eckstein RW. Autoregulation of coronary blood flow: effect of interarterial pressure gradients. Circ Res. 1964;15:103–111. [PubMed] [Google Scholar]
- 19.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009–1086. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 20.Ebeigbe AB, Aloamaka CP. Mechanism of hydralazine-induced relaxation of arterial smooth muscle. Cardiovasc Res. 1985;19:400–405. doi: 10.1093/cvr/19.7.400. [DOI] [PubMed] [Google Scholar]
- 21.Feigl EO. Coronary physiology. Physiol Rev. 1983;63:1–205. doi: 10.1152/physrev.1983.63.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Feigl EO. Coronary autoregulation. J Hypertens Suppl. 1989;7:S55–S58. [PubMed] [Google Scholar]
- 23.Feigl EO, Neat GW, Huang AH. Interrelations between coronary artery pressure, myocardial metabolism and coronary blood flow. J Mol Cell Cardiol. 1990;22:375–390. doi: 10.1016/0022-2828(90)91474-L. [DOI] [PubMed] [Google Scholar]
- 24.Gregg DE. Effect of coronary perfusion pressure or coronary flow on oxygen usage of the myocardium. Circ Res. 1963;13:497–500. doi: 10.1161/01.RES.13.6.497. [DOI] [PubMed] [Google Scholar]
- 25.Heaps CL, Jeffery EC, Laine GA, Price EM, Bowles DK. Effects of exercise training and hypercholesterolemia on adenosine activation of voltage-dependent K + channels in coronary arterioles. J Appl Physiol. 2008;105:1761–1771. doi: 10.1152/japplphysiol.90958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heaps CL, Tharp DL, Bowles DK. Hypercholesterolemia abolishes voltage-dependent K + channel contribution to adenosine-mediated relaxation in porcine coronary arterioles. Am J Physiol Heart Circ Physiol. 2005;288:H568–H576. doi: 10.1152/ajpheart.00157.2004. [DOI] [PubMed] [Google Scholar]
- 27.Herlihy JT, Bockman EL, Berne RM, Rubio R. Adenosine relaxation of isolated vascular smooth muscle. Am J Physiol. 1976;230:1239–1243. doi: 10.1152/ajplegacy.1976.230.5.1239. [DOI] [PubMed] [Google Scholar]
- 28.Heusch G. Adenosine and maximum coronary vasodilation in humans: myth and misconceptions in the assessment of coronary reserve. Basic Res Cardiol. 2010;105:1–5. doi: 10.1007/s00395-009-0074-7. [DOI] [PubMed] [Google Scholar]
- 29.Hill MA, Davis MJ, Meininger GA, Potocnik SJ, Murphy TV. Arteriolar myogenic signalling mechanisms: implications for local vascular function. Clin Hemorheol Microcirc. 2006;34:67–79. [PubMed] [Google Scholar]
- 30.Hinshaw LB. Mechanism of renal autoregulation: role of tissue pressure and description of a multifactor hypothesis. Circ Res. 1964;(suppl 15):120–131. [PubMed] [Google Scholar]
- 31.Ishikawa T, Hume JR, Keef KD. Regulation of Ca2 + channels by cAMP and cGMP in vascular smooth muscle cells. Circ Res. 1993;73:1128–1137. doi: 10.1161/01.RES.73.6.1128. [DOI] [PubMed] [Google Scholar]
- 32.Johnson PC. Review of previous studies and current theories of autoregulation. Circ Res. 1964;(suppl 15):2–9. [PubMed] [Google Scholar]
- 33.Johnson PC, Waugh WH, Hinshaw LB. Autoregulation of Blood Flow. Science. 1963;140:203–207. doi: 10.1126/science.140.3563.203. [DOI] [PubMed] [Google Scholar]
- 34.Jones CJ, Kuo L, Davis MJ, Chilian WM. Myogenic and flow-dependent control mechanisms in the coronary microcirculation. Basic Res Cardiol. 1993;88:2–10. doi: 10.1007/BF00788525. [DOI] [PubMed] [Google Scholar]
- 35.Kauser K, Stekiel WJ, Rubanyi G, Harder DR. Mechanism of action of EDRF on pressurized arteries: effect on K + conductance. Circ Res. 1989;65:199–204. doi: 10.1161/01.RES.65.1.199. [DOI] [PubMed] [Google Scholar]
- 36.Knudson JD, Dincer UD, Bratz IN, Sturek M, Dick GM, Tune JD. Mechanisms of coronary dysfunction in obesity and insulin resistance. Microcirculation. 2007;14:317–338. doi: 10.1080/10739680701282887. [DOI] [PubMed] [Google Scholar]
- 37.Komaru T, Lamping KG, Dellsperger KC. Role of adenosine in vasodilation of epimyocardial coronary microvessels during reduction in perfusion pressure. J Cardiovasc Pharmacol. 1994;24:434–442. doi: 10.1097/00005344-199409000-00012. [DOI] [PubMed] [Google Scholar]
- 38.Krajcar M, Heusch G. Local and neurohumoral control of coronary blood flow. Basic Res Cardiol. 1993;88(Suppl 1):25–42. doi: 10.1007/978-3-642-72497-8_3. [DOI] [PubMed] [Google Scholar]
- 39.Kuo L, Chilian WM, Davis MJ. Coronary arteriolar myogenic response is independent of endothelium. Circ Res. 1990;66:860–866. doi: 10.1161/01.RES.66.3.860. [DOI] [PubMed] [Google Scholar]
- 40.Kuo L, Chilian WM, Davis MJ. Interaction of pressure-and flow-induced responses in porcine coronary resistance vessels. Am J Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- 41.Kuo L, Davis MJ, Chilian WM. Myogenic activity in isolated subepicardial and subendocardial coronary arterioles. Am J Physiol. 1988;255:H1558–H1562. doi: 10.1152/ajpheart.1988.255.6.H1558. [DOI] [PubMed] [Google Scholar]
- 42.Laird JD, Breuls PN, van der MP, Spaan JA. Can a single vasodilator be responsible for both coronary autoregulation and metabolic vasodilation? Basic Res Cardiol. 1981;76:354–358. doi: 10.1007/BF01908321. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Chai Q, Gutterman DD, Liu Y. Elevated glucose impairs cAMP-mediated dilation by reducing Kv channel activity in rat small coronary smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H1213–H1219. doi: 10.1152/ajpheart.00226.2003. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Gutterman DD. Vascular control in humans: focus on the coronary microcirculation. Basic Res Cardiol. 2009;104:211–227. doi: 10.1007/s00395-009-0775-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Gutterman DD. The coronary circulation in diabetes: influence of reactive oxygen species on K + channel-mediated vasodilation. Vascul Pharmacol. 2002;38:43–49. doi: 10.1016/S1537-1891(02)00125-8. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Terata K, Rusch NJ, Gutterman DD. High glucose impairs voltage-gated K(+) channel current in rat small coronary arteries. Circ Res. 2001;89:146–152. doi: 10.1161/hh1401.093294. [DOI] [PubMed] [Google Scholar]
- 48.McHale PA, Dube GP, Greenfield JC., Jr Evidence for myogenic vasomotor activity in the coronary circulation. Prog Cardiovasc Dis. 1987;30:139–146. doi: 10.1016/0033-0620(87)90006-5. [DOI] [PubMed] [Google Scholar]
- 49.Miller FJ, Jr, Dellsperger KC, Gutterman DD. Myogenic constriction of human coronary arterioles. Am J Physiol. 1997;273:H257–H264. doi: 10.1152/ajpheart.1997.273.1.H257. [DOI] [PubMed] [Google Scholar]
- 50.Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 51.Osher WJ. Pressure-flow relationship of the coronary system. Am J Physiol. 1953;172:403–416. doi: 10.1152/ajplegacy.1953.172.2.403. [DOI] [PubMed] [Google Scholar]
- 52.Pesic A, Madden JA, Pesic M, Rusch NJ. High blood pressure upregulates arterial L-type Ca2 + channels: is membrane depolarization the signal? Circ Res. 2004;94:e97–e104. doi: 10.1161/01.RES.0000131495.93500.3c. [DOI] [PubMed] [Google Scholar]
- 53.Pratt PF, Bonnet S, Ludwig LM, Bonnet P, Rusch NJ. Upregulation of L-type Ca2 + channels in mesenteric and skeletal arteries of SHR. Hypertension. 2002;40:214–219. doi: 10.1161/01.HYP.0000025877.23309.36. [DOI] [PubMed] [Google Scholar]
- 54.Rogers PA, Chilian WM, Bratz IN, Bryan RM, Jr, Dick GM. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292:H1404–H1411. doi: 10.1152/ajpheart.00696.2006. [DOI] [PubMed] [Google Scholar]
- 55.Rogers PA, Dick GM, Knudson JD, Focardi M, Bratz IN, Swafford AN, Jr, Saitoh S, Tune JD, Chilian WM. H2O2-induced redox-sensitive coronary vasodilation is mediated by 4-aminopyridine-sensitive K + channels. Am J Physiol Heart Circ Physiol. 2006;291:H2473–H2482. doi: 10.1152/ajpheart.00172.2006. [DOI] [PubMed] [Google Scholar]
- 56.Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, Knudson JD, Dick GM, Swafford A, Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol. 2006;26:2614–2621. doi: 10.1161/01.ATV.0000249408.55796.da. [DOI] [PubMed] [Google Scholar]
- 57.Schrader J, Haddy FJ, Gerlach E. Release of adenosine, inosine and hypoxanthine from the isolated guinea pig heart during hypoxia, flow-autoregulation and reactive hyperemia. Pflugers Arch. 1977;369:1–6. doi: 10.1007/BF00580802. [DOI] [PubMed] [Google Scholar]
- 58.Schulz R, Guth BD, Heusch G. No effect of coronary perfusion on regional myocardial function within the autoregulatory range in pigs. Evidence against the Gregg phenomenon. Circulation. 1991;83:1390–1403. doi: 10.1161/01.CIR.83.4.1390. [DOI] [PubMed] [Google Scholar]
- 59.Setty S, Sun W, Tune JD. Coronary blood flow regulation in the prediabetic metabolic syndrome. Basic Res Cardiol. 2003;98:416–423. doi: 10.1007/s00395-003-0418-7. [DOI] [PubMed] [Google Scholar]
- 60.Smith TP, Jr, Canty JM., Jr Modulation of coronary autoregulatory responses by nitric oxide. Evidence for flow-dependent resistance adjustments in conscious dogs. Circ Res. 1993;73:232–240. doi: 10.1161/01.RES.73.2.232. [DOI] [PubMed] [Google Scholar]
- 61.Stepp DW, Kroll K, Feigl EO. K + ATP channels and adenosine are not necessary for coronary autoregulation. Am J Physiol. 1997;273:H1299–H1308. doi: 10.1152/ajpheart.1997.273.3.H1299. [DOI] [PubMed] [Google Scholar]
- 62.Tare M, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- 63.Thorneloe KS, Nelson MT. Ion channels in smooth muscle: regulators of intracellular calcium and contractility. Can J Physiol Pharmacol. 2005;83:215–242. doi: 10.1139/Y05-016. [DOI] [PubMed] [Google Scholar]
- 64.Warltier DC, Hardman HF, Gross GJ. Transmural perfusion gradients distal to various degrees of coronary artery stenosis during resting flow or at maximal vasodilation. Basic Res Cardiol. 1979;74:494–508. doi: 10.1007/BF01907643. [DOI] [PubMed] [Google Scholar]
- 65.Weisfeldt ML, Shock NW. Effect of perfusion pressure on coronary flow and oxygen usage of nonworking heart. Am J Physiol. 1970;218:95–101. doi: 10.1152/ajplegacy.1970.218.1.95. [DOI] [PubMed] [Google Scholar]
- 66.Wong AY, Klassen GA. Vasomotor coronary oscillations: a model to evaluate autoregulation. Basic Res Cardiol. 1991;86:461–475. doi: 10.1007/BF02190714. [DOI] [PubMed] [Google Scholar]
- 67.Wu GB, Zhou EX, Qing DX, Li J. Role of potassium channels in regulation of rat coronary arteriole tone. Eur J Pharmacol. 2009;620:57–62. doi: 10.1016/j.ejphar.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 68.Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, Ogasawara Y, Kajiya F. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation. 2003;107:1040–1045. doi: 10.1161/01.CIR.0000050145.25589.65. [DOI] [PubMed] [Google Scholar]
- 69.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-Induced Dilation in Human Coronary Arterioles: Role of Protein Kinase G Dimerization and Large-Conductance Ca2 +-Activated K + Channel Activation. Circ Res. 2012;110:471–480. doi: 10.1161/CIRCRESAHA.111.258871. [DOI] [PMC free article] [PubMed] [Google Scholar]