

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder that affects 1 in 3,500 males and is caused by mutations in the dystrophin gene. In this paper, we have reported DNA analysis of DMD patients by multiplex polymerase chain reaction (PCR) from various states of northeast India. Of the 69 clinically suspected patients of DMD, deletion was detected by multiplex PCR in 49 (71%) patients. Majority of the deletions (42/49, 85.7%) were located at distal hot spot region that encompasses exons 44-55 and 14.3% of the deletions were located at the proximal hot spot region (exons 2-19). In this study population, the deletion rate was 71% and was more frequent in the distal end exon.
Keywords: Duchenne muscular dystrophy, dystrophin gene, exon deletions, polymerase chain reaction
Introduction
Duchenne muscular dystrophy (DMD) and its less severe allelic form Becker muscular dystrophy (BMD) are X-linked recessive devastating neuromuscular disorder that affects 1 in 3,500 males births and is caused by mutations in the dystrophin gene.[1] The frequency and distribution of deletion in dystrophin gene in DMD population from northeast India is reported in the present work.
Materials and Methods
A total of 69 patients were enrolled in this prospective study with clinically suspected DMD, attending the neurology clinic at Gauhati Medical College Hospital, Guwahati, Assam between October 2009 and September 2011. The diagnosis of DMD was based on the clinical findings of progressive and characteristic pattern of proximal muscle weakness, calf pseudohypertrophy, positive Gower's sign, valley sign, electromyography (EMG), an elevated serum creatine kinase (CK) activity, and the transmission of the pattern as an X-linked trait.
Multiplex polymerase chain reaction for duchenne muscular dystrophy
DNA was isolated from peripheral blood leukocytes using standard phenol/chloroform procedures. Screening of deletions of the dystrophin gene were performed according to Chamberlain et al.[2] and Beggs et al.,[3] using two multiplex PCR assays allowing the amplification of total 17 exons: The Chamberlain reaction using primers for exons 4, 8, 12, 17, 19, 44, 45, 48, 51 and the Beggs reaction performed with primers for the promoter and exons 1, 3, 6, 13, 43, 47, 50, and 52. The PCR products were separated on 2% nusieve + 1% agarose minigels and the bands were visualized by staining with ethidium bromide. Multiplex PCR was performed at Reliance Life Sciences Pvt. Ltd, Mumbai and Super Religare Laboratories Ltd, Mumbai. Written informed consent was obtained from all participants before drawing their blood. The study was approved by the ethics committee of the institution.
Statistical analysis
The statistical analysis was performed with SPSS version 17.0 (SPSS Inc., Chicago, IL). Baseline patient characteristics were reported using the median or mean ± SD for continuous variables according to their distribution. The families were divided in two groups according to the propositus’ Multipex PCR analysis (13 deleted families and 6 non-deleted). A comparative analysis was made using the Kruskal–Wallis test for non-Gaussian distributed continuous variables and Chi-square analysis for categorical variables. Statistical significance was considered at P < 0.05.
Results and Discussion
Out of 69 patients of DMD, 40 (58%) were from the Hindu community and 29 (42%) from the Muslim community, with age ranging from 2-9 years. Family history was present in 19 patients (8 Hindu, 11 Muslim). The mean age of onset was 1.72 + 0.69 years (range 0.8-3 years) and the mean age at presentation was 6.24 + 176 (range 2-9 years). All boys presented with progressive proximal muscle weakness particularly of the lower limbs and the majority (75.3%) complained of calf muscle pseudohypertrophy. The mean creatine kinase value was 8499.39 + 5325 U/L (range 970–19200 U). DNA samples from 69 patients were analyzed for deletions using multiplex PCR. A total of 49 (71%) cases showed intragenic deletions, the localization of these deletions are shown in Figure 1. Majority of the deletions (42/49, 85.7%) were located at distal hot spot region that encompasses exons 44-55 and 14.3% of the deletions were located at the proximal hot spot region (exons 2-19).
Figure 1.
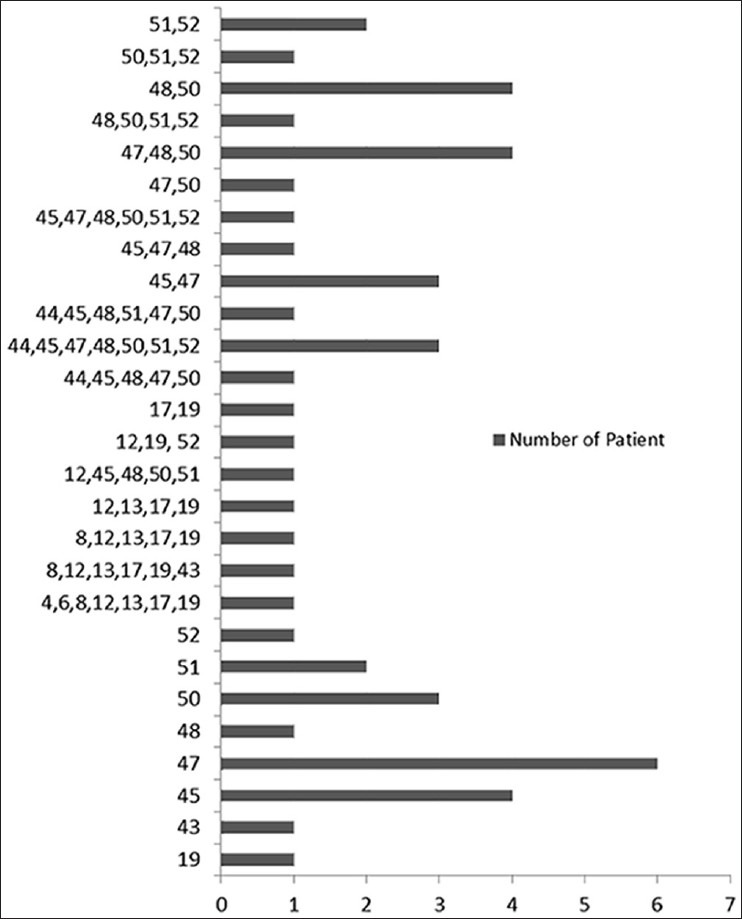
Representing the gene, the localization, and the frequencies of deletions in 49 patients
A total of 130 exons (108 in isolated cases, 22 in familial cases) were deleted in 49 patients, and the average number of exons deleted per patients was 2.9. Single exon was deleted in 19 patients (38.7%), and in 30 patients (61.3%) multiple exonic deletions were seen. Most frequent single exon deleted were 45 and 47, the most common exons involved in multiple deletions were 48 (36.7%), 47 (40.8%), and 50 (42.8%). The most frequent exon deletion in overall deletions were 50 (14.38%), followed by 48 (13.6%), 49 (12%), and 47 (10%). Largest deletions were present in 6 patients extending from exon 44 to 52 in 5 patients and from exon 8 to 43 in 1 patient.
On comparison between the familial and isolated cases of DMD, deletions were found in 72% of isolated cases and 68.4% in familial cases. Out of 19 cases with family history, deletions were detected in 13 patients. Single exon deletion was seen in 7 and multiple exon deletion (upto 3) in 6 cases. A previous study[4] reported approximately 3% higher detection rate in familial cases as compared to sporadic cases. Contrary to this, our study have shown approximately 4% reduction in deletion rate in familial cases (P = 0.0084). It is difficult to draw a definite conclusion as our series had a small sample size.
Multiplex PCR methods allow detection of approximately 98% of deletions, which accounts for 65% of all mutations.[2] Locations of deletions in the dystrophin gene are apparently nonrandom with a preponderance found in two “‘hot spot” regions[5,6] at the 5’ terminus and in the distal half of the central rod domain around exons 44-53.
The proportion of deletions in different populations of India shows a wide variation in the distribution. The reported frequency of deletion in various parts of India range from 62.1% to 74% using multiplex PCR,[7] with which our data conforms. No particular reasons have been persuasively attributed to explain population-based differences in mutations of the dystrophin gene, it is presumed to be due to accumulation of differences in intronic sequences and their distribution over a time period as a consequence of genetic drift. Such sequences unique to the population may favour intragenic deletions due to mismatch at the locus.[8]
In our study, 71% of patients showed deletions. In these patients, detection of DMD was done by deletion analysis using multplex PCR, hence eliminating the need to do muscle biopsy for diagnosis.
As this disease has no effective therapy, early diagnosis of patient can be helpful for carrier detection and genetic counseling. With genetic counseling, further birth of offspring with DMD can be prevented.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Emery AE. Population frequencies of inherited neuromuscular diseases: A world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 2.Chamberlain JS, Gibbs RA, Ranier JE, Caskey CT. A guide to Methods and Applications" New York Academic Press: "PCR Protocols; 1990. "PCR Protocols: A guide to Methods and Applications," New York: Academic Press; 1990; pp. 272–81. [Google Scholar]
- 3.Beggs AH, Kunkel LM. Improved diagnosis of Duchenne/Becker muscular dystrophy. J Clin Invest. 1990;85:613–9. doi: 10.1172/JCI114482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunkel LM, Hejtmancik JF, Caskey CT, Speer A, Monaco AP, Middlesworth W, et al. Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature. 1986;322:73–7. doi: 10.1038/322073a0. [DOI] [PubMed] [Google Scholar]
- 5.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gent in normal and affected individuals. Cell. 1987;50:509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 6.Forrest SM, Cross GS, Flint T, Specr A, Robson KJ, Davies KE. Further studies of gene deletions that cause Duchenne and Bcckcr muscular dystrophies. Genomics. 1988;2:109–14. doi: 10.1016/0888-7543(88)90091-2. [DOI] [PubMed] [Google Scholar]
- 7.Nadkarni JJ, Dastur RS, Viswanathan V, Gaitonde PS, Khadilkar SV. Duchenne and Becker muscular dystrophies: An Indian update on genetics and rehabilitation. Neurol India. 2008;56:248–53. doi: 10.4103/0028-3886.43442. [DOI] [PubMed] [Google Scholar]
- 8.Krawzak M, Cooper DN. Gene deletions causing human genetic diseases: Mechanisms of mutagenesis and their role of local DNA sequences environment. Hum Genet. 1991;86:425–41. doi: 10.1007/BF00194629. [DOI] [PubMed] [Google Scholar]