

Abstract
The metal-reducing δ-proteobacterium Geobacter sulfurreducens produces a large number of c-type cytochromes, many of which have been implicated in the transfer of electrons to insoluble metal oxides. Among these, the dihemic MacA was assigned a central role. Here we have produced G. sulfurreducens MacA by recombinant expression in Escherichia coli and have solved its three-dimensional structure in three different oxidation states. Sequence comparisons group MacA into the family of diheme cytochrome c peroxidases, and the protein indeed showed hydrogen peroxide reductase activity with ABTS2– as an electron donor. The observed KM was 38.5 ± 3.7 μM H2O2 and vmax was 0.78 ± 0.03 μmol H2O2·min–1·mg–1, resulting in a turnover number kcat = 0.46 · s–1. In contrast, no Fe(III) reductase activity was observed. MacA was found to display similar electrochemical properties to other bacterial diheme peroxidases, in additional to the ability to electrochemically mediate electron transfer to the soluble cytochrome PpcA. Differences in activity between CcpA and MacA can be rationalized with structural variations in one of the three loop regions, loop 2, that undergo conformational changes during reductive activation of the enzyme. This loop is adjacent to the active site heme and forms an open loop structure rather than a more rigid helix as in CcpA. For the activation of the protein the loop has to displace the distal ligand to the active site heme, H93, in loop 1. A H93G variant showed an unexpected formation of a helix in loop 2 and disorder in loop 1, while a M297H variant that altered the properties of the electron transfer heme abolished reductive activation.
Keywords: Bacterial cytochrome c peroxidases, Multiheme cytochromes, Mixed-valence state, Active-site loop, Conformational rearrangement, Protein crystallography
The δ-proteobacterial genus Geobacter has become a paradigm system for studying dissimilatory metal-reduction. The species G. sulfurreducens and G. metallireducens are able to use insoluble minerals containing various metal cations such as Fe(III), Mn(IV) or U(VI) as terminal electron acceptors in a respiratory pathway that spans both membranes of the Gram-negative microorganisms (1, 2). In the absence of dioxygen and nitrate in subsurface environments or sediments, the positive midpoint redox potential of the Fe2+/Fe3+ couple makes it a highly suitable electron acceptor. However, as ferric iron hydroxides and hydroperoxides are practically insoluble in water, this requires electron transfer to the outside of the bacterial outer membrane and thus a chain of redox cofactors that connects the menaquinone pool of the inner membrane with the reductases on the outside of the cell. The entire electron transfer pathway of dissimilatory metal reduction in G. sulfurreducens has not been elucidated, although a series of proposals were brought forth and several outer membrane multiheme c-type cytochromes were proposed to act as terminal iron reductases (3-5). In the related γ-proteobacterium Shewanella oneidensis, another widely used model for metal dissimilation, a complex was identified that consists of a porin-like outer membrane protein and two decaheme cytochromes c and that likely is responsible for electron shuttling across the outer membrane. Most recently, the three-dimensional structure of one of the decaheme proteins, MtrF, was solved, showing an unprecedented packing arrangement of heme groups and suggesting a mode of insertion into the transmembrane component of the complex (6).
G. sulfurreducens produces a large number of c-type cytochromes and several of these were proposed to be involved in electron transfer during dissimilatory metal reduction (7). Prominent among them was the ‘metal-reduction-associated cytochrome’ MacA (8), encoded by the gene locus gsu0466. This 35 kDa diheme c-type cytochrome was initially described to function in electron transfer between the inner membrane and the soluble cytochromes of the periplasmic space (7). However, sequence alignments within the genome showed clear homology to the diheme cytochrome c peroxidase CcpA (GSU2813). Peroxidases of this class catalyze the reduction of hydrogen peroxide to water, and their presence indicates the occurrence of reactive oxygen species (ROS) through metabolic activity or in microoxic environments (9, 10). G. sulfurreducens was originally classified as an obligate anaerobe (11), but the analysis of its genome revealed the presence of genes encoding typical enzymes for oxygen detoxification, such as catalase, cytochrome c oxidase or superoxide dismutase. The organism tolerated limited exposure to dioxygen and was even able to utilize it as terminal electron acceptor (12). However, while no direct evidence links MacA to electron transfer in iron dissimilation, a ΔmacA strain also did not show increased sensitivity towards dioxygen (8), so that the physiological role of the protein remains enigmatic.
Proteins of the CcpA family are diheme c-type cytochromes consisting of two distinct domains with a modified globin fold, each with a heme group covalently attached via thioether bonds to a canonical binding motif C-X1-X2-C-H (10). Various structures of CcpA proteins are available that in summary outline a unique activation and reduction mechanism involving major conformational rearrangements (13-18). The two heme groups differ in their axial ligands and, consequently, in their midpoint redox potential. The His/Met-coordinated heme in the C-terminal domain has a positive redox potential ranging from +320 mV in P. aeruginosa (19) to +450 mV in N. europaea (20). It is designated the high-potential (HP) heme and is reduced by the physiological electron donor of CcpA, a small monoheme c-type cytochrome or a cupredoxin (21-23). The second heme group is the site of H2O2 reduction and has a significantly lower midpoint potential (LP), with reported values ranging from –260 mV in N. europaea (20) to –330 mV in P. aeruginosa (19). The mechanistic consequence of this arrangement is that electron transfer from the reduced HP heme to the LP heme does not occur. Instead, the reduction of the HP heme triggers a concerted conformational rearrangement of three loop regions of the protein that opens up a free coordination site at the distal axial position of the LP heme group. The side chains of a conserved Glu and a Gln residue, Q126 and E136 in G. sulfurreducens MacA, swing into the active site, where they act in binding the substrate and in acid-base catalysis (18). The substrate binds to the LP heme and the O–O bond is reductively cleaved using one electron from each heme group. This returns the HP heme to the Fe(III) state and leaves the LP heme as an oxoferryl form (Fe(IV)=O), while one of the hydrogen peroxide oxygens is released as H2O. In this form, the midpoint potential of the oxidized LP heme is drastically increased, so that after the next electron transfer to the HP heme group the electron will swiftly reduce the LP heme to the Fe(III) state, closing the reaction cycle.
To clarify whether MacA plays a role in dissimilatory metal reduction or functions as a peroxidase similar to the closely related CcpA we have produced and analyzed the protein for activity, spectroscopic properties and structure in the three relevant redox states. As a means of probing the redox properties of MacA we have created the variants H93G and M297H to alter the distal ligands to both heme groups and study the effects on activation and reactivity.
EXPERIMENTAL PROCEDURES
Design of Expression Constructs
The macA gene (gsu0466) was amplified from genomic DNA of G. sulfurreducens (DSM 12127) using Phusion polymerase (Finnzymes). The oligonucleotide primers MacA_f_EcoRI (5′-CCCGAATTCA AAAGAGGATGTCATGAAACG-3′) and MacA_r_XhoI (5′-AAACTCGAGTCAGTTGCTGACCGG CCTG-3′) were designed to introduce restriction sites for EcoRI and XhoI, respectively, and the resulting PCR product was purified and cloned into the expression vector pETSN22 (17), a modification of pET22b(+) (Novagen) that introduces an N-terminal StrepTag(II). The correctness of the plasmid was confirmed by sequencing (GATC Biotech).
Site-directed Mutagenesis
In order to create the substitutions H93G and M297H, site-directed mutagenesis was carried out following the QuickChange protocol (Stratagene). In a 50 μl reaction volume containing Pfu polymerase buffer, 50 mM of deoxynucleoside triphosphates mix, 2.5 U of Pfu turbo polymerase, 50 ng of template DNA (pETSN22::macA) and the mutagenesis primers H93G_f (5′-GGAGACATCCATCGGTGGGGGCTGGCAAAAGCCGCC-3′), H93G_r (5′-GGCGGCTTTTGCCAGCCCCCACCGATGGATGTCT CC-3′), M297H_f (5′-AAGGACGCCGTCAAGATCCACGGGAGC GCTCAACTCGGC-3′) and M297H_r (5′-GCCGAGTTGAGCGCTCCCGTGGATCTTGACGGCGTC CTT-3′) were used (substituted triplets underlined). A PCR reaction was carried out with an initial denaturation step at 95 °C for 30 s, followed by 16 cycles of 30 s at 95 °C, 1 min at 55 °C and 7 min at 68 °C. The product was digested with 10 U of DpnI for 1 h at 37 °C and the restriction enzyme was then heat-inactivated for 10 min at 80 °C, before 10 μl of this mixture were transformed into E. coli XL 10 gold cells (Stratagene) that were then incubated at 37 °C in LB medium containing 100 μg·ml–1 of ampicillin and 50 μg·ml–1 of kanamycin. The correctness of the substitutions was confirmed by sequencing (GATC Biotech).
Protein Production
The heterologous production of MacA was carried out using the cytochrome c production strain E. coli BL21(DE3)::pEC86, where the pEC86 plasmid contains the c-type cytochrome maturation genes ccmABCDEFGH (24) that are required for expression of c-type cytochromes during aerobic growth. The expression cultures were grown overnight at 30 °C at 200 rpm without induction with isopropyl thiogalactoside (IPTG), as we commonly observe that the T7 promoter of pETSN22 is sufficiently leaky to allow for good yields of cytochromes without overloading the capacities of the Ccm system produced from pEC86 (17, 25, 26). Ampicillin was added to the growth media to 100 μg·ml–1 and chloramphenicol to 20 μg·ml–1.
Protein Purification
Cells were harvested by centrifugation and resuspended in 2 ml per g cells (wet weight) of running buffer containing 20 mM Tris/HCl buffer (pH 8.0) and 250 mM NaCl. The resuspended cells were then disrupted by three cycles in a microfluidizer (Microfluidics) at 1100 bar. After two centrifugation steps at 20,000·g (20 min, 4 °C) and 100,000·g (1 h, 4 °C) the supernatant, implying the soluble proteins, was loaded with a flow rate of 1 ml·min–1 onto a streptactin superflow column (IBA) and washed with running buffer. Bound protein was eluted in a single step with 2.5 mM D-desthiobiotin in running buffer. The eluate was then loaded onto a size exclusion column (Superdex 200, 16/60, GE Healthcare) at a flow rate of 1 mlJmin–1 using a buffer containing 20 mM Tris/HCl (pH 8.0) and 100 mM NaCl. All chromatographic steps were performed at 25 °C using an ÄKTA prime plus system (GE Healthcare). The eluted fractions were analyzed by SDS-PAGE and the cytochrome-containing fractions were concentrated by ultrafiltration (30,000 Da molecular weight cut-off, vivaspin, Sartorius). Protein was determined by the bicinchoninic acid method (27) using bovine serum albumin as a standard.
Crystallization and Data Collection
MacA was crystallized by the sitting drop vapor diffusion method. 1 μl of protein solution (7.5 mg·ml–1) was added to 1 μl of reservoir solution and equilibrated against that reservoir solution. The wild type protein crystallized in 0.1 M ammonium acetate buffer at pH 5.5, 1.3 M Na/K phosphate and 6 % (v/v) of ethanol. For the characterization of other redox states, the protein was reduced prior to the crystallization experiment and all following steps were carried out in an anoxic glove box (Coy Laboratories) at < 1 ppm O2. The protein reduced with ascorbate solution at pH 7.5 crystallized in 0.1 M HEPES/NaOH buffer at pH 7.5, 0.2 M ammonium acetate and 25 % (w/v) polyethylene glycol 3350. Protein reduced with sodium dithionite at pH 7.5 yielded crystals in 0.1 M sodium citrate buffer at pH 5.6 and 1 M ammonium phosphate. MacA_H93G crystals were obtained in 1 M ammonium sulfate at pH 5.4. In order to avoid the formation of ice, the crystals were transferred to a cryoprotective buffer containing 2.5 M (wild type) or 2.4 M (variants) of Li2SO4. For the reduced crystals 10 % (v/v) of 2R,3R-butane diol served as cryoprotectant. The crystals were mounted in a nylon loop, transferred into the cryoprotectant and flash frozen in liquid nitrogen. Datasets were collected at beam lines X13 (EMBL/DESY, Hamburg, Germany) for the wild type enzyme and X06DA (SLS, Villigen, Switzerland) for the variants, the semi-reduced and the fully reduced form. Indexing, integration and scaling were done with the HKL suite (28). For data collection and refinement statistics see Table 1.
Table 1.
Data Collection and refinement statistics. Numbers in parentheses refer to values within the highest resolution shell.
| wild type (oxidized) | wild type (semi-reduced) | wild type (reduced) | H93G | |
|---|---|---|---|---|
| PDB Accession Number | 4AAL | 4AAM | 4AAN | 4AAO |
| Data collection | ||||
| Space group | P6522 | C2 | C2 | P6522 |
| Cell dimensions | ||||
| a, b, c [Å] | 118.0, 118.0, 242.0 | 95.1, 47.5, 77.9 | 95.6, 46.9, 77.9 | 118.7, 118.7, 246.0 |
| α, β, γ [°] | 90.0, 90.0, 120.0 | 90.0, 91.5, 90.0 | 90.0, 90.9, 90.0 | 90.0, 90.0, 120.0 |
| Monomers per a.u. | 2 | 1 | 1 | 2 |
| Wavelength [Å] | 0.800 | 1.000 | 1.000 | 1.000 |
| Resolution [Å] | 103.0 – 1.95 (2.1 – 1.95) | 40.1 – 2.2 (2.3 – 2.2) | 19.5 – 1.2 (1.3 – 1.2) | 35.5 – 2.3 (2.4 – 2.3) |
| R merge | 0.090 (0.285) | 0.119 (0.229) | 0.086 (0.627) | 0.093 (0.637) |
| Rp.i.m.(42) | 0.025 (0.184) | 0.074 (0.144) | 0.050 (0.374) | 0.023 (0.165) |
| I/σ(I) | 7.9 (3.0) | 6.4 (4.0) | 9.4 (2.0) | 6.3 (1.3) |
| Completeness [%] | 96.4 (97.7) | 99.9 (99.9) | 100.0 (99.9) | 93.5 (100.0) |
| Multiplicity | 13.5 (8.4) | 3.5 (3.5) | 3.9 (3.8) | 16.0 (13.6) |
| Refinement | ||||
| Resolution [Å] | 50.0 – 1.95 | 40.1 – 2.2 | 19.5 – 1.22 | 35.5 – 2.3 |
| No. reflections | 69,037 | 17,665 | 102,772 | 40,924 |
| Rwork/Rfree | 0.206 / 0.256 | 0.164 / 0.227 | 0.149 / 0.164 | 0.160 / 0.184 |
| No. atoms | ||||
| Protein | 4903 | 2456 | 2587 | 4547 |
| Ligand | 316 | 97 | 111 | 229 |
| Water | 599 | 321 | 363 | 310 |
| B-factors [Å2] | ||||
| Protein | 32.7 | 20.5 | 12.4 | 15.5 |
| Ligand | 48.1 | 18.4 | 16.8 | 18.7 |
| Water | 39.9 | 33.8 | 30.9 | 36.5 |
| R.m.s. deviations | ||||
| Bond lengths [Å] | 0.014 | 0.009 | 0.010 | 0.008 |
| Bond angles [°] | 1.570 | 1.174 | 1.070 | 1.124 |
Structure Determination and Refinement
The three-dimensional structure of MacA was solved by molecular replacement using MOLREP (29) from the CCP4 suite (30). G. sulfurreducens CcpA (PDB accession number 3HQ6), with 65 % sequence identity, was used as a search model (17). For structure determination of the reduced forms and variants of MacA the refined structure of fully oxidized MacA was used as a search model. Model building was carried out in COOT (31) and REFMAC5 (32) was used for restrained positional refinement with TLS contributions. The structures of the wild type proteins show the polypeptide chain from residue E23 to the C-terminus at N346. In contrast, the structure of the H93G variant has two gaps in the modeled sequence, where the polypeptide chain is disordered. The correctness of the structures was validated with PROCHECK (33) and figures were generated in PyMOL (34).
UV/vis Spectroscopy
Electron excitation spectra were recorded on a Lambda 40 spectrometer (Perkin Elmer) using Suprasil screw cap cuvettes with butyl rubber septa (Hellma). All experiments were carried out using a buffer containing 20 mM Tris/HCl, pH 7.5, and 100 mM NaCl, and a protein concentration of 10 μM. To obtain the half and fully reduced states the samples were handled in an anoxic glove box under a N2/H2 atmosphere. For these experiments all buffers and protein solutions were alternately degassed and flushed with N2 using modified Schlenk techniques. The protein was reduced by adding either 100 μM sodium ascorbate, pH 7.5, or sodium dithionite, pH 7.5. In each case 10 μM diaminodurene (2,3,5,6-tetramethyl-p-phenylendiamine) were added as a redox mediator (35).
EPR Spectroscopy
EPR spectra were recorded on a Bruker Elexsys 500 continuous-wave spectrometer at a microwave frequency of 9.343 GHz. All samples were measured at 11 K with a microwave power of 5 mW, a modulation frequency of 100 kHz and a modulation amplitude of 12 G. The protein concentration was 22 mg·ml–1 (3 mM). The semi-reduced form was obtained by incubation with excess sodium ascorbate and subsequent removal of the reductant on a NAP 5 gravity-flow desalting column (Sephadex G25, GE Healthcare), followed by concentration of the sample to the original value via ultrafiltration.
Peroxidase Assays
Cytochrome c Peroxidase activity was analyzed and detected by using the artificial electron donor 2,2′-azino-bis(3-ethylenbenzthiazoline-6-sulfonic acid) (ABTS2–) (36). Because no activity was detected under oxic conditions the experiments were set up in an inert gas glove box and all solutions were degassed prior to the measurement. The protein was first reduced with an excess of sodium ascorbate to obtain the semi-reduced state. Excess reductant was then removed via a 5 ml NAP column (GE Healthcare). In a microcuvette (Sarstedt), 300 nM MacA, 10 mM potassium phosphate buffer with different pH values and 2.925 mM ABTS2– were provided. The increase of absorbance was followed with the Biovave II photometer (Biorad) at 420 nm (ε420 = 36 mM–1·cm–1) after adding H2O2 in concentrations between 2.5 and 70 μM. The H2O2 concentration was confirmed photometrically by its absorption at 240 nm (ε420 = 39.4 M–1·cm–1) (37).
Iron Reduction Assays
For ferric reductase activity measurements, MacA was reduced with sodium dithionite and excess reductant was removed on a NAP 5 gravity-flow desalting column (Sephadex G25, GE Healthcare). 10 μM of protein in 20 mM of HEPES/NaOH buffer at pH 7.0 were then mixed with solutions containing different concentrations of FeCl3, Fe2(SO4)2–, or Fe(III) citrate. Possible electron transfer from MacA to Fe(III) was monitored by following the decrease of the α-band at 554 nm during 10 min.
Electrochemistry
Protein film voltammetry experiments were preformed on a PGSTAT 12 AutoLab (Ecochemie) potentiostat, equipped with FRA and EDC modules. A three-electrode configuration was used in a water jacketed glass cell. A platinum wire is used as the counter electrode and a saturated calomel reference electrode is used. Potentials are reported here versus a standard hydrogen electrode (SHE). Calomel potentials are corrected +242 mV. All experiments were done at 0°C unless otherwise noted. A cell solution of 10 mM CHES, HEPES, MES, and TAPS with 100 mM NaCl allowed for a broad range of pH values to be investigated. When necessary the rotating electrode is rotated using an EG&G rotator. Protein films are generated on pyrolytic edge plane graphite electrodes (PGE) by directly depositing 1 μL of a 300 μM stock protein solution directly onto the electrode surface and incubating for five minutes. Excess protein is rinsed from the electrode surface with cell solution buffer.
Electrochemical analyses in the absence of substrates were collected at the bench-top with the electrochemical cell surrounded by a Faraday cage to eliminate electrochemical noise from the system, and under an argon purge. Catalytic electrochemical experiments were preformed in an MBraun Labmaster glovebox in an anaerobic environment. Data were collected with the GPES software package (Ecochemie). Nonturnover signals were analyzed by subtraction of the graphite baseline electrochemical response from the raw data using the SOAS package (38). A linear baseline is subtracted from the cathodic scan of the raw catalytic data to extract kinetic parameters and analyze the catalytic data.
RESULTS AND DISCUSSION
Production of Recombinant MacA
The maturation of c-type cytochromes requires a complex machinery that in Escherichia coli is produced exclusively during anaerobic growth. In order to obtain recombinant MacA from aerobically grown cultures of E. coli, the accessory plasmid pEC86 was used that constitutively expresses the eight ccm genes that encode the cytochrome c maturation machinery of this γ-proteobacterium (24). Under these conditions the cell pellet after centrifugation had a dark red color and the yields of MacA amounted on average to 1.5 mg per g of cell pellet (wet weight). After cell lysis, MacA was isolated from the soluble fraction via StrepTag(II) affinity chromatography and subsequently subjected to size exclusion chromatography, from where it eluted as a dimer.
Catalytic Properties of MacA
MacA was first assayed for iron reductase activity using Fe(III) ions as a substrate. No activity was observed, although a variety of c-type cytochromes that physiologically function exclusively in electron transfer were shown to be able to reduce the excellent electron acceptor Fe(III) (2). In order to compare MacA to other members of the CcpA family, peroxidase activity was subsequently assayed using ABTS2– as an artificial electron donor (36). In the oxidized state the protein did not show peroxidase activity, but it did so following reductive activation with sodium ascorbate. MacA thus is a second diheme cytochrome c peroxidase in G. sulfurreducens. The activity was pH-dependent and peaked at pH 5.5, with KM = 38.5 ± 3.7 IM H2O2 and vmax = 0.78 ± 0.03 μmol H2O2·min–1·mg–1, resulting in a turnover number kcat = 0.46 · s–1. This affinity is substantially lower than the one determined for CcpA from the same organism (17), where the same assay yielded KM = 6.2 μM H2O2 and vmax = 26.4 ± 0.5 μmol H2O2·min–1·mg–1, with a turnover number kcat = 15.5 s–1.
Three-dimensional Structures
Using X-ray diffraction methods the structure of MacA (Figure 1) was solved in the oxidized, ascorbate-reduced and dithionite-reduced states, providing the full set of redox states for a single CcpA family protein for the first time (Figure 2). MacA consists of two globular domains that hold one of the heme groups each. Residues P40 to T184 form the N-terminal domain that harbors the LP heme group covalently bound to C73 and C76 via thioether bonds. The proximal ligand to the LP heme is H77 with a Fe–Nε bond distance of 2.0 Å. H93 is the corresponding distal axial ligand and in oxidized MacA its Nε atom is coordinated to the heme iron at 2.1 Å. The C-terminal domain encompasses D189 through A324, and here the HP heme group is bound to C219 and C222. The HP heme iron is coordinated on the proximal side by the Nε nitrogen of H223 at 2.1 Å distance, and by the Sγ atom of M297 at a longer distance of 2.3 Å. The conserved W116 is located between the heme groups and the commonly observed Ca2+ ion was present in all structures (Figure 1). The structure of semi-reduced MacA was refined to 2.2 Å (Figure 2B) resolution and that of the fully reduced form to 1.2 Å (Figure 2C), the highest resolution available for a CcpA family protein to date. As expected, the structures emphasize that reduction of the HP heme group alone is sufficient to trigger a major rearrangement of three loop regions of the protein, eventually granting access to the active site at the distal axial position of the LP heme group. In MacA, the loop regions are E88-N101 (loop 1), I112-N139 (loop 2) and Y233-F267 (loop 3).
Figure 1.

Three-dimensional structure of G. sulfurreducens MacA. Cartoon representation of the monomer of MacA in the oxidized (FeIII...FeIII) form in stereo. The peptide chain is colored from blue at the N-terminus to red at the C-terminus and the high- (HP) and low-potential (LP) heme groups as well as the Ca2+ ion located between the two domains of the protein are indicated.
Figure 2.

Structural rearrangements of G. sulfurreducens MacA in different redox states. While the main part of the enzyme remains structurally unaltered, three loops regions change conformation upon reduction. A) In the oxidized (Fe(III) – Fe(III)) state, loop regions 1, 2 and 3 are oriented such that H93 in loop1 coordinates the active site (LP) heme iron and prevents the binding of substrate. B) Sodium ascorbate only reduces the HP heme, leading to the (Fe(III) – Fe(II)) state and a consecutive rearrangements of loops 3-2-1 to open up the active site. The catalytically relevant Q126 and E136 reorient towards the LP heme iron. C) Complete reduction to the (Fe(II) – Fe(II)) state with sodium dithionite does not trigger further structural changes.
The structure of oxidized MacA is highly similar to that of CcpA from the same organism (17), with a root-mean-squared deviation of 0.64 Å for all atoms of the two structures in their fully oxidized states (Figure 3A). However, a prominent difference was observed that possibly explains the functional differences of the two proteins. In the region between residues G129 and N139, the structure of CcpA consists of an α-helical segment, while MacA attains a more extended and consequently less well ordered loop structure. The positions and conformations of residues E136 that are involved in catalysis differ drastically (Figure 3B). This flexible loop structure in MacA possibly also explains the movement of residue S134 in the H93G variant towards the active site, where it becomes the distal axial ligand to the LP heme group as detailed below. In its semi-reduced and reduced forms, MacA also shows high similarities to the constantly open form of the N. europaea enzyme. One single α-helix near the HP heme is absent in the MacA structure and the loop between residues R254 and D263 shows a relatively unstructured segment in MacA, but an ordered α-helix in the N. europaea enzyme (20).
Figure 3.

Comparison of the two diheme peroxidases of G. sulfurreducens, CcpA and MacA. A) Structural alignment of CcpA (black) and MacA (red). The overall structure of the proteins is highly conserved, and all known functional features of CcpA proteins are present in the structure of MacA. B) Superposition of the LP heme environment of CcpA (white) and MacA (green) of G. sulfurreducens. In spite of a high overall similarity of both proteins, the loop 2 region encompassing residues 120–140 shows significant differences. This region undergoes conformational changes during reductive activation of the peroxidases and the observed differences are presumed to be responsible for the observed differences in activity.
In summary, MacA shows all features of a cytochrome c peroxidase of the CcpA type. It reduces hydrogen peroxide with ABTS2– as an artificial electron donor, but does not show reducing activity towards ferric iron. MacA is structurally similar to CcpA form the same organism, although its enzymatic activity is significantly lower. MacA displays highly similar reactivity to other bacterial cytochrome c peroxidases (37, 38), though it also reveals an ability to channel electrons to the soluble tri-heme cytochrome PpcA. Thus, G. sulfurreducens possesses two hydrogen peroxide reductases required for oxygen detoxification, in agreement with its reclassification as being microaerotolerant (12), while MacA may have two functions. So far no data is available concerning the regulation and timing of the expression of the ccpA and macA genes. Although both CcpA and MacA were shown to be among the proteins whose levels increase under oxidative stress, a ΔmacA strain did not show a phenotype with increased dioxygen sensitivity (8). The surface of MacA shows a positive electrostatic potential, which is in agreement with CcpA, but distinguishes both from all other CcpA family members described previously (17). For the G. sulfurreducens proteins this might indicate a closer association with the negatively charged surface of the cytoplasmic membrane, which would facilitate electron transfer to the outer membrane via PpcA.
Spectroscopic Properties
Electron excitation spectra of isolated MacA show the typical features of c-type cytochromes that are dominated by the characteristic Soret band (Figure 4). In the oxidized state this band is visible at 406.5 nm, with an additional, broad maximum at 525 nm. A shoulder at 620 nm is indicative of high spin Fe(III) and is ascribed to a high spin/low spin equilibrium of the HP heme due to the long Fe–S bond to the distal methionine ligand. Upon reduction with sodium ascorbate only the HP heme is reduced to low spin Fe(II), its Soret band is shifted to 419 nm and the α- and β-bands emerge with maxima at 554 nm and 524 nm, respectively. In CcpA peroxidases, this reduction triggers a major conformational shift that leads to dissociation of the distal histidine ligand of the LP heme and the exposure of the Fe ion as the active site for H2O2 reduction. The now five-coordinate iron of the LP heme is in the high spin state so that the feature at 620 nm in the UV/vis spectrum is retained. Sodium dithionite can be used to reduce the LP heme as well, resulting in a purely reduced spectrum and the disappearance of the high spin feature at 620 nm. In all aspects, MacA behaved very similar to G. sulfurreducens CcpA (17), underlining its likely role in oxygen detoxification. In the H93G variant the spectroscopic properties were largely unchanged, and the high spin signal was also detectable in both the oxidized and ascorbate-reduced states. Due to the point mutation the LP heme was expected to be in the high spin state in the oxidized form, and accordingly the shape of the 620 nm feature was sharpened (Figure 6). This high spin signature disappeared entirely in the M297H variant that had the distal axial ligand of the HP heme exchanged, leading to a significant drop in the midpoint redox potential of this group. In consequence, treatment with sodium ascorbate does not generate a mixed Fe(II)-Fe(III) form of MacA M297H as it did for the wild type protein. In the oxidized state, the bands were shifted slightly to 405.5 nm and 534 nm, respectively, and in the dithionite-reduced state the α-, β- and γ-bands appeared at 552 nm, 522 nm and 418 nm (Figure 4).
Figure 4.

Electron excitation spectra of MacA. The enzyme shows the typical Soret band at 408 nm in the oxidized state (red), with additional shoulders at 535 nm and at 630 nm. The latter originates from high spin Fe(III) that is partially present at the HP heme group. Upon inclubation with sodium ascorbate (blue), the HP heme group is reduced, while the active site HP heme group remains oxidized and the spectrum shows a mixed-valent state of MacA. Sodium dithionite reduces both heme groups (black), resulting in a spectrum with α-, β-, and γ/Soret-bands at 554 nm, 524 nm and 419 nm, respectively.
Figure 6.
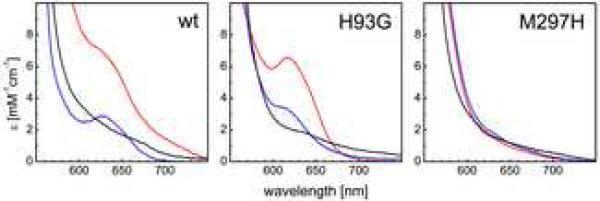
High spin heme signatures in electron excitation spectra of wild type MacA and variants. A) In the wild type, the oxidized state (red) shows a high spin feature originating from an equilibrium at the stretched Fe–S bond at the HP heme. Upon reduction with sodium ascorbate (blue), the HP heme becomes Fe(II) low spin, but a new high spin feature arises from the still oxidized LP heme that is five-coordinate after activation. In the fully reduced state (black) both hemes are low spin. B) In the H93G variant the expectation that the LP heme would always be 5-coordinate was not fulfilled. The high spin signature changes only slightly, and this was explained by the crystal structure where S134 became a distal ligand to the LP heme. C) The M297H variant shifts the potential of the HP heme, resulting in a low spin configuration with His/His coordination in the oxidized state and no change upon addition of ascorbate.
Electron paramagnetic resonance (EPR) spectroscopy confirmed the results from UV/vis spectroscopy, but revealed additional details concerning the states of the heme groups. In the oxidized state of MacA the X-band spectrum showed a dominant axial high spin signal with g⊥=1.92 and g∥=5.58 (Figure 5A). In contrast to a high spin / low spin equilibrium as reported for other CcpA proteins, the HP heme group of MacA seems to be almost completely in the high spin state, while the LP heme shows a low spin signature with gy=2.35 and gz=2.95. Treatment with ascorbate reduces the HP heme to an EPR silent Fe(II) state and triggers the conformational changes that open the LP heme group. Interestingly, the EPR spectra become more complex upon reduction (Figure 5B). Three signals are visible in this state, a high spin signal at g=5.58, similar to the oxidized state but with lower intensity. This signal lacks the prominent g⊥=1.92 feature and is assigned to the five-coordinate high spin form of the LP heme. In addition, however, two distinct low spin signals are visible that share gy=2.36, but differ in gx and gy (1.51/1.60 and 2.78/2.89, respectively). Both likely represent populations of the LP heme with a distal axial ligand. Such ligands may be water and/or dioxygen, but this remains speculative as electron density maps of the ascorbate-reduced protein are ambiguous, possibly due to a mixture of liganded states. As expected, further reduction of the LP heme group with dithionite leaves the enzyme in an EPR-silent state (Figure 5C).
Figure 5.

EPR spectra of G. sulfurreducens MacA recorded at 11 K. A) The oxidized state is dominated by a high spin Fe(III) signal at g=1.92, 5.58, originating from the His/Met-coordinated HP heme. The LP heme is in a low spin configuration with g values of 2.35, 2.95. B) Upon reduction with ascorbate, the contribution of the HP heme disappears, but three signals remain that represent different states of the HP heme. A high spin signal remains at g∥=5.58, but lacks the prominent g⊥=1.92 signal of the HP heme. Two different low spin signals are centered around gy=2.36, but differ slightly in their gx and gy values. C) After reduction of the LP heme with sodium dithionite the sample is EPR silent as expected.
The structural and spectroscopic data for MacA confirms earlier mechanistic proposals for CcpA peroxidases (10), and the availability of high-resolution structures for three redox states of MacA from the same organism allows for a detailed comparison. The complex rearrangement of three loop regions in essence is a way of conveying the information that the HP heme group has obtained an electron and the system thus is ready to react with substrate to the active site at the LP heme. The reduction of the HP heme leads to a switch of the conformation of loop region 3 by a yet undisclosed mechanism. Loop 3 is better defined in the reduced structures, so that it can be assumed that the reduced conformation is the more stable one. This is of importance, as loop 3 has to trigger the conformational change of loop 2 that in turn induces loop 1 to switch to its open conformation and permit substrate access to the active site.
Protein Electrochemistry
MacA was investigated using protein film voltammetry at PGE electrodes to assess the redox properties of the LP heme. MacA revealed stable voltammograms at pH 7.5 corresponding to the LP heme, as shown in Figure 7A (left). Subtraction of the non-farradaic component of the voltammograms reveals two electrochemical features with midpoint potentials (Em) of -237 mV and -138 mV (vs. hydrogen), in a fashion similar to those reported for the Pseudomonas aeruginosa enzyme (39), where the two features have been associated with the His-bound and –free conformations of the LP heme active site. When the HP heme is reduced with ascorbate prior to adsorption (Figure 7A, right), the higher potential feature is removed, revealing a single redox couple at -241 mV vs SHE, corresponding to the LP heme free from H93 coordination. As with the P. aeruginosa and Geobacter CCP enzymes (39, 40), exposure of such protein films to H2O2 (Figure 7B) result in reducing sigmoidal “waves”, centered at potential of LP heme, and which can be analyzed to determine Km (25 μM at pH 7.5). To probe the possible role of MacA in metal reduction chemistry, electrochemically-detected protein:protein interactions (41) were investigated to test the hypothesis that in metal reduction chemistry, MacA passes electrons on to the small triheme Geobacter cytochrome PpcA (7). When MacA is adsorbed to PGE, and PpcA is introduced into the cell solution (Figure 7C) electrocatalysis is observed. Notably, these reductive catalytic currents require MacA to be adsorbed initially, and they have a magnitude that scales with the concentration of PpcA. The catalytic currents do not saturate at concentrations that can be achieved, suggesting that the MacA:PpcA interaction must be labile. In the converse experiment, when PpcA is adsorbed onto PGE electrodes, a broad envelop of current is observed due to the three hemes of similar potential (dotted line, Figure 7D), yet inclusion of micromolar concentrations of MacA into the cell solution does not result in noticeable electrocatalysis.
Figure 7.

(A) Electrochemical characterization of MacA in the fully oxidized (left) and ascorbatereduced (right) state as raw (black) and baseline-subtracted (colored) voltammograms. (B) Electrocatlytic reduction of H2O2 by MacA adsorbed at PGE electrodes, pH 7.5, v = 20 mV/s, rotation rate = 1500 rpm. (C) Electrocatlytic reduction of PpcA free in solution by MacA adsorbed at PGE, from a concentration of 1 to 10 μM PpcA. (D) PpcA adsorbed at PGE (dotted line) and then exposed to excess MacA in solution (solid red).
Site-directed Mutagenesis of Heme Ligands
In order to elucidate whether H93, the distal ligand to the LP heme, is a determining factor for the overall conformation of the enzyme we created a H93G variant of MacA, studied its activity and solved its structure by X-ray crystallography to a resolution of 2.3 Å. In the variant, the C-terminal domain remains largely unchanged, but structural rearrangements occur in the N-terminal domain with the active site LP heme. Due to the substitution of H93 for G, the protein now recruits the side chain of nearby S134 as a distal heme ligand, retaining the six-fold coordination and its low spin state. In consequence the α-helix containing L123-V135 approaches the active site and displaces the active-site loop 1 (E88-N101) (Figure 8). This flexibility is likely due to the decreased rigidity of the loop 2 region. Functionally, a more flexible loop 2 may be less well suited to displace loop 1 upon activation, resulting eventually in a decrease in specific activity of MacA with respect to CcpA. With S134 in place, the H93G variant of MacA is inactive in the oxidized state and requires reductive activation, as does the wild type. The second variant studied spectroscopically, MacA M297H, could not be crystallized. As the protein shows a decrease in redox potential of the HP heme by at least 250 mV and no additional high spin signal in the spectrum we assume that H297 does ligate the heme iron. However, this may lead to structural disorder in the N-terminal domain that then counteracts the formation of well-diffracting crystals. In MacA M297H, reductive activation using sodium ascorbate was no longer possible. Consequently this variant did not show hydrogen peroxide reductase activity, confirming the sequence of events required for activation as outlined above.
Figure 8.

Structural details of MacA. A) The LP heme environment of the H93G variant of MacA (stereo representation). In the oxidized state of the protein the removal of the distal ligand H93 to the LP heme led to structural disorder in loop region 1 (red). Loop region 2 (blue) that shows less secondary structure in the wild type, now attains a helical conformation, and the conserved S134 coordinates the LP heme as a distal ligand. The LP heme thus retains its low spin state, as seen by UV/vis spectroscopy. B) Electron Density for the reduced form of MacA at 1.2 Å resolution. The stereo image shows the LP heme group in the open form, with a 2Fo–Fc electron density map contoured at the 1σ level.
With the present study, the available data on CcpA enzymes converges towards an increasingly comprehensive picture of the process of reductive activation that is key to avoiding unspecific reduction reactions to occur through these reactive catalysts. The molecular details of H2O2 reduction at the LP heme itself are less well understood, but the outstanding diffraction quality of MacA crystals, in particular in the reduced state, may be a crucial prerequisite to characterize substrate binding and conversion at atomic resolution in further studies.
ACKNOWLEDGMENT
We thank Linda Thöny-Meyer for kindly providing the pEC86 plasmid and Johannes Gescher for assistance with iron reduction assays. Diffraction data were collected at beam lines X13 (EMBL/DESY, Hamburg, Germany) and X06DA (SLS, Villigen, Switzerland).
Funding This research was supported by Deutsche Forschungsgemeinschaft (grant Ei 520/1; Ei 520/5 and IRTG 1478), and the National Institutes of Health (project grant R01-GM072663).
ABBREVIATIONS
- CcpA
bacterial diheme cytochrome c peroxidase
- EPR
electron paramagnetic resonance
- ABTS
2,2′-azino-bis(3-ethylenbenzthiazoline-6-sulfonic acid)
- PGE
pyrolytic graphite edge
Footnotes
Accession Codes Coordinates and structure factors have been deposited with the Protein Data Bank as entries 4AAL (oxidized), 4AAM (semi-reduced), 4AAN (reduced) and 4AAO (H93G).
REFERENCES
- 1.Lovley DR. Dissimilatory metal reduction. Annu. Rev. Microbiol. 1993;47:263–290. doi: 10.1146/annurev.mi.47.100193.001403. [DOI] [PubMed] [Google Scholar]
- 2.Lovley DR, Holmes DE, Nevin KP. Dissimilatory Fe(III) and Mn(IV) reduction. Adv. Microb. Physiol. 2004;49:219–286. doi: 10.1016/S0065-2911(04)49005-5. [DOI] [PubMed] [Google Scholar]
- 3.Leang C, Coppi MV, Lovley DR. OmcB, a c-type polyheme cytochrome, involved in Fe(III) reduction in Geobacter sulfurreducens. J. Bacteriol. 2003;185:2096–2103. doi: 10.1128/JB.185.7.2096-2103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta T, Coppi MV, Childers SE, Lovley DR. Outer membrane c-type cytochromes required for Fe(III) and Mn(IV) oxide reduction in Geobacter sulfurreducens. Appl. Environ. Microbiol. 2005;71:8634–8641. doi: 10.1128/AEM.71.12.8634-8641.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber KA, Achenbach LA, Coates JD. Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction. Nat. Rev. Microbiol. 2006;4:752–764. doi: 10.1038/nrmicro1490. [DOI] [PubMed] [Google Scholar]
- 6.Clarke TA, Edwards MJ, Gates AJ, Hall A, White GF, Bradley J, Reardon CL, Shi L, Beliaev AS, Marshall MJ, Wang ZM, Watmough NJ, Fredrickson JK, Zachara JM, Butt JN, Richardson DJ. Structure of a bacterial cell surface decaheme electron conduit. Proc. Natl. Acad. Sci. U. S. A. 2011;108:9384–9389. doi: 10.1073/pnas.1017200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Methé BA, Nelson KE, Eisen JA, Paulsen IT, Nelson W, Heidelberg JF, Wu D, Wu M, Ward N, Beanan MJ, Dodson RJ, Madupu R, Brinkac LM, Daugherty SC, DeBoy RT, Durkin AS, Gwinn M, Kolonay JF, Sullivan SA, Haft DH, Selengut J, Davidsen TM, Zafar N, White O, Tran B, Romero C, Forberger HA, Weidman J, Khouri H, Feldblyum TV, Utterback TR, Van Aken SE, Lovley DR, Fraser CM. Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science. 2003;302:1967–1969. doi: 10.1126/science.1088727. [DOI] [PubMed] [Google Scholar]
- 8.Butler JE, Kaufmann F, Coppi MV, Nunez C, Lovley DR. MacA, a diheme c-type cytochrome involved in Fe(III) reduction by Geobacter sulfurreducens. J. Bacteriol. 2004;186:4042–4045. doi: 10.1128/JB.186.12.4042-4045.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atack JM, Kelly DJ. Structure, mechanism and physiological roles of bacterial cytochrome c peroxidases. Adv. Microb. Physiol. 2007;52:73–106. doi: 10.1016/S0065-2911(06)52002-8. [DOI] [PubMed] [Google Scholar]
- 10.Pettigrew GW, Echalier A, Pauleta SR. Structure and mechanism in the bacterial dihaem cytochrome c peroxidases. J. Inorg. Biochem. 2006;100:551–567. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Caccavo F, Jr., Lonergan DJ, Lovley DR, Davis M, Stolz JF, McInerney MJ. Geobacter sulfurreducens sp. nov., a hydrogen- and acetate-oxidizing dissimilatory metal-reducing microorganism. Appl. Environ. Microbiol. 1994;60:3752–3759. doi: 10.1128/aem.60.10.3752-3759.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin WC, Coppi MV, Lovley DR. Geobacter sulfurreducens can grow with oxygen as a terminal electron acceptor. Appl. Environ. Microbiol. 2004;70:2525–2528. doi: 10.1128/AEM.70.4.2525-2528.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Smet L, Savvides SN, Van Horen E, Pettigrew G, Van Beeumen JJ. Structural and mutagenesis studies on the cytochrome c peroxidase from Rhodobacter capsulatus provide new insights into structure-function relationships of bacterial di-heme peroxidases. J. Biol. Chem. 2006;281:4371–4379. doi: 10.1074/jbc.M509582200. [DOI] [PubMed] [Google Scholar]
- 14.Dias JM, Alves T, Bonifacio C, Pereira AS, Trincão J, Bourgeois D, Moura I, Romão MJ. Structural basis for the mechanism of Ca2+ activation of the di-heme cytochrome c peroxidase from Pseudomonas nautica 617. Structure. 2004;12:961–973. doi: 10.1016/j.str.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 15.Echalier A, Goodhew CF, Pettigrew GW, Fülöp V. Activation and catalysis of the di-heme cytochrome c peroxidase from Paracoccus pantotrophus. Structure. 2006;14:107–117. doi: 10.1016/j.str.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Fülöp V, Ridout CJ, Greenwood C, Hajdu J. Crystal structure of the di-haem cytochrome c peroxidase from Pseudomonas aeruginosa. Structure. 1995;3:1225–1233. doi: 10.1016/s0969-2126(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann M, Seidel J, Einsle O. CcpA from Geobacter sulfurreducens Is a Basic Di-Heme Cytochrome c Peroxidase. J. Mol. Biol. 2009;393:951–965. doi: 10.1016/j.jmb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu H, Schuller DJ, Lanzilotta WN, Sundaramoorthy M, Arciero DM, Hooper AB, Poulos TL. Crystal structure of Nitrosomonas europaea cytochrome c peroxidase and the structural basis for ligand switching in bacterial di-heme peroxidases. Biochemistry. 2001;40:13483–13490. doi: 10.1021/bi011481h. [DOI] [PubMed] [Google Scholar]
- 19.Ellfolk N, Ronnberg M, Aasa R, Andreasson LE, Vanngard T. Properties and function of the two hemes in Pseudomonas cytochrome c peroxidase. Biochim. Biophys. Acta. 1983;743:23–30. doi: 10.1016/0167-4838(83)90413-2. [DOI] [PubMed] [Google Scholar]
- 20.Arciero DM, Hooper AB. A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J. Biol. Chem. 1994;269:11878–11886. [PubMed] [Google Scholar]
- 21.Foote N, Peterson J, Gadsby PM, Greenwood C, Thomson AJ. A study of the oxidized form of Pseudomonas aeruginosa cytochrome c551 peroxidase with the use of magnetic circular dichroism. Biochem. J. 1984;223:369–378. doi: 10.1042/bj2230369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pauleta SR, Cooper A, Nutley M, Errington N, Harding S, Guerlesquin F, Goodhew CF, Moura I, Moura JJ, Pettigrew GW. A copper protein and a cytochrome bind at the same site on bacterial cytochrome c peroxidase. Biochemistry. 2004;43:14566–14576. doi: 10.1021/bi0485833. [DOI] [PubMed] [Google Scholar]
- 23.Pauleta SR, Guerlesquin F, Goodhew CF, Devreese B, Van Beeumen J, Pereira AS, Moura I, Pettigrew GW. Paracoccus pantotrophus pseudoazurin is an electron donor to cytochrome c peroxidase. Biochemistry. 2004b;43:11214–11225. doi: 10.1021/bi0491144. [DOI] [PubMed] [Google Scholar]
- 24.Arslan E, Schulz H, Zufferey R, Kunzler P, Thöny-Meyer L. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 1998;251:744–747. doi: 10.1006/bbrc.1998.9549. [DOI] [PubMed] [Google Scholar]
- 25.Heitmann D, Einsle O. Structural and biochemical characterization of DHC2, a novel diheme cytochrome c from Geobacter sulfurreducens. Biochemistry. 2005;44:12411–12419. doi: 10.1021/bi0509999. [DOI] [PubMed] [Google Scholar]
- 26.Lukat P, Hoffmann M, Einsle O. Crystal packing of the c6-type cytochrome OmcF from Geobacter sulfurreducens is mediated by an N-terminal Strep-tag II. Acta Crystallogr D. 2008;64:919–926. doi: 10.1107/S0907444908021306. [DOI] [PubMed] [Google Scholar]
- 27.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 30.Collaborative Computational Project No. 4 The CCP4 Suite: Programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.DeLano WL. The PyMOL Molecular Graphic System. DeLano Scientific; San Carlos: 2002. [Google Scholar]
- 35.Hill BC, Nicholls P. Reduction and Activity of Cytochrome c in the Cytochrome-c-Cytochrome aa3 Complex. Biochem. J. 1980;187:809–818. doi: 10.1042/bj1870809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Childs RE, Bardsley WG. The steady-state kinetics of peroxidase with 2,2' azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem. J. 1975;145:93–103. doi: 10.1042/bj1450093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson DP, Kiesow LA. Enthalpy of decomposition of hydrogen peroxide by catalase at 25° C (with molar extinction coefficients of H2O2 solutions in the UV) Anal. Biochem. 1972;49:474–478. doi: 10.1016/0003-2697(72)90451-4. [DOI] [PubMed] [Google Scholar]
- 38.Fourmond V, Hoke K, Heering HA, Baffert C, Leroux F, Bertrand P, Léger C. SOAS: A free program to analyze electrochemical data and other one-dimensional signals. Bioelectrochemistry. 2009;76:141–147. doi: 10.1016/j.bioelechem.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Becker CF, Watmough NJ, Elliott SJ. Electrochemical Evidence for Multiple Peroxidatic Heme States of the Diheme Cytochrome c Peroxidase of Pseudomonas aeruginosa. Biochemistry. 2009;48:87–95. doi: 10.1021/bi801699m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellis KE, Seidel J, Einsle O, Elliott SJ. Geobacter sulfurreducens Cytochrome c Peroxidases: Electrochemical Classification of Catalytic Mechanisms. Biochemistry. 2011;50:4513–4520. doi: 10.1021/bi200399h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Firer-Sherwood MA, Bewley KD, Mock JY, Elliott SJ. Tools for resolving complexity in the electron transfer networks of multiheme cytochromes c. Metallomics. 2011;3:344–348. doi: 10.1039/c0mt00097c. [DOI] [PubMed] [Google Scholar]
- 42.Weiss M, Hilgenfeld R. On the use of the merging R factor as a quality indicator for X-ray data. J. Appl. Cryst. 1997;30:203–205. [Google Scholar]