

Abstract
Imaging multiple fluorescent proteins (FPs) by two-photon microscopy has numerous applications for studying biological processes in thick and live samples. Here we demonstrate a setup utilizing a single broadband laser and a phase-only pulse-shaper to achieve imaging of three FPs (mAmetrine, TagRFPt, and mKate2) in live mammalian cells. Phase-shaping to achieve selective excitation of the FPs in combination with post-imaging linear unmixing enables clean separation of the fluorescence signal of each FP. This setup also benefits from low overall cost and simple optical alignment, enabling easy adaptation in a regular biomedical research laboratory.
OCIS codes: (320.5540) Pulse shaping, (180.2520) Fluorescence microscopy, (190.4180) Multiphoton processes, (180.4315) Nonlinear microscopy
1. Introduction
Fluorescent proteins (FP) are widely used as genetically encoded fluorescent reporters in biomedical research [1]. Over the last few decades, a broad range of FP variants have been developed, with fluorescence emission from blue to far red [2]. Combinations of FP spectral variants allow multi-color labeling in the same biological sample, a technique which has been used in a wide range of biomedical applications, such as multi-color flow cytometry [3], simultaneous tracking of multiple organelles [4] and/or single proteins [5], super-resolution structural studies [6], monitoring of protein conformational changes [7, 8], as well as protein-protein colocalization [9] and interactions [10]. Multi-color FP studies have also enabled the tracing of neuronal networks [11] and cell colonial expansions [12, 13]. Because they are strongly expressed in selective organs or cells, FPs provide the most convenient labeling for live animal imaging [14, 15].
Two-photon fluorescence microscopy (2PFM) offers significant advantages over one-photon fluorescence microscopy for deep tissue imaging due to its increased penetration depth and decreased photobleaching [16, 17]. Although it is widely used for live or thick samples, 2PFM has primarily been applied for imaging one or two FPs at a time [18–20]. In order to image more colors, multiple wavelengths are needed to excite FPs with different two-photon absorption peaks. Conventionally, multi-color 2PFM is realized by tuning a femtosecond laser sequentially to excite each fluorophore. However, the slow tuning process (in seconds to minutes) restricts its application to time insensitive experiments [21]. Using one or two femtosecond oscillators and a coupled optical parametric oscillator (OPO), two or three distinct excitation wavelengths can be generated [22, 23]. Alternatively, three-color pulses can also be obtained by second harmonic generation (SHG) of the 1550 nm pump laser and soliton self-frequency shifts at 1728 nm and 1900 nm [24]. OPO systems are costly, while fiber-broadened sources can suffer from instability and difficulty with alignment, which complicates their use in normal biomedical laboratories. Furthermore, the multiple wavelengths generated in these systems are correlated, which limits flexibility in FP choice. Another solution is to use oscillators with broad spectral bandwidth, spanning ~one hundred nanometers or greater [25–29]. The transform-limited pulse of such a broadband laser normally covers multiple FPs’ excitation spectra [30]. While the use of transform-limited pulses enables simultaneous excitation of multiple FPs, it does not provide selectivity and excites them at fixed efficiency ratios. This is undesirable as adjusting relative FP brightness becomes impossible, and undesirable signals such as autofluorescence can be enhanced. In order to solve these problems, pulse shaping methods have been developed to tailor the ultrafast pulses for selective excitation of fluorescent proteins over autofluorescence [25, 27] and for the selective enhancement of fluorescence from particular fluorophores [31, 32]. Phase-shaping takes advantage of the broad spectral range of ultrashort pulses, controlling the relative phase between different frequency components to enhance or suppress two-photon excitation [25, 33, 34]. To illustrate the role of spectral phase in this process, consider the fluorescence signal S produced after two-photon excitation:
| (1) |
where g(ω) is the two-photon excitation spectrum of the fluorophore and is the second harmonic power spectrum of the laser pulse [25, 27, 35, 36]:
| (2) |
The second harmonic power spectrum shows a clear dependence on the spectral phase and illustrates the different frequency combinations available to produce two-photon excitation. By manipulating the spectral phase, the two-photon excited fluorescence signal itself can be controlled. The spectral phase can be readily adjusted in a 4f pulse-shaper [37]. Here we use a pulse-shaper based on a Spatial Light Modulator (SLM), which permits the creation of phase masks that adjust the relative phase of individual frequency components. Phase masks on the SLM can be changed on a time scale of ~50ms, giving a significant time advantage over tunable laser systems [37].
We report using phase-shaping of ultrafast pulses to selectively excite three FPs expressed in mammalian cells. By combining this with post-imaging linear unmixing, we demonstrate the feasibility of using a single broadband laser and SLM pulse-shaper to achieve three-color 2PFM in live cells. The relatively less expensive price, ease of alignment, and flexibility in choosing FPs make this two-photon modality attractive for use in many biomedical laboratories. This method could be readily extended beyond three-color imaging with even broader bandwidth commercially available Titanium:Sapphire oscillators.
2. Experimental setup
The microscopy setup used in this experiment is shown in Fig. 1 . Pulses from a 75 MHz Titanium:Sapphire (Ti:Sa) oscillator (Femtolasers Synergy), with 80 nm bandwidth centered at 790 nm, were phase-shaped by a femtoJock® box equipped with a single-mask 128-pixel SLM (Biophotonic Solutions lnc., East Lansing, MI). The phase-shaped beam was then sent into the scan head of a Prairie Technologies modified Olympus BX51WI upright microscope for imaging. An Olympus UPlanApo 60x, 1.2NA water-immersion microscope objective was used to focus onto the sample and collect fluorescence signal. Excitation power at the sample was approximately 3.5 mW. The signal was then separated from the excitation light and spectrally split into two PMT channels for multicolor detection. Multiphoton Intrapulse Interference Phase Scan (MIIPS) [38–41] was used to pre-compensate for dispersion caused by the microscope optics and achieve a near transform-limited (TL) pulse prior to application of the desired phase mask.
Fig. 1.
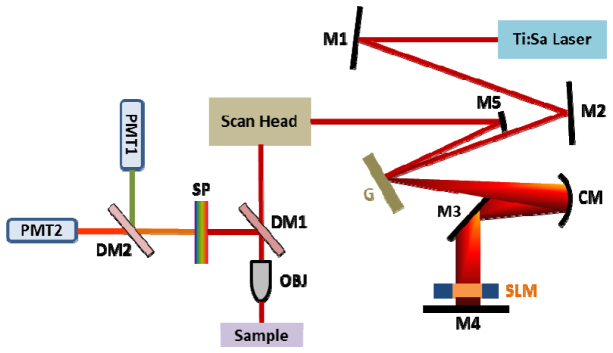
Experimental setup. M1-M5: mirrors, G = grating, CM = curved mirror, SLM = spatial light modulator, DM1 = 660DCXR dichroic mirror, DM2 = 595DCXR dichroic mirror, OBJ = 60x NA1.2 water immersion objective; SP = 650nm short pass filter.
For 2PFM of multiple FPs, all of the FPs must be able to be efficiently excited by the broadband Ti:Sa laser and the emission spectra of the FPs should be distinct enough that they can be separated by filter selection and unmixing processing. To suit our broadband Ti:Sa laser’s spectral range [Fig. 2(a) , long dashed black curve], we chose a yellow FP with a large Stokes shift, mAmetrine [8], and took advantage of TagRFP/TagRFPt [42, 43] and mKate2’s [44] strong transitions to excited states higher than S1 at shorter wavelengths [19, 30, 45] [Fig. 2(a), dotted green, orange and crimson curves, respectively]. By applying binary spectral phase masks to the SLM, we created phase-shaped pulses with second harmonic generation (SHG) spectra centered at 390 nm (the “blue phase-shaped pulse”) and 410 nm (the “red phase-shaped pulse”), respectively [Fig. 2(b)]. The phase masks were made using prime number binary phase shaping, with a flat-phase section of 40 nm centered at twice the desired two-photon excitation wavelength maximum and a semi-random choice of 0 or pi as described by Comstock et al. [33]. The masks were roughly symmetric around the flat region (centered at 780 nm for the blue phase-shaped pulse and 820 nm for the red phase-shaped pulse). Further optimization of the pulse shapes is under active investigation. The red and blue phase-shaped pulses, as well as TL pulses, were applied to multiple cells in each sample. The excited fluorescence was split into green and red spectral regions and directed to separate PMTs. The scanning process was controlled by Prairie Technologies’ PrairieView software. Final images are averages of 16 scans across the region of interest with pixel dwell times of 4 µs. Image processing and analysis was performed in Matlab.
Fig. 2.
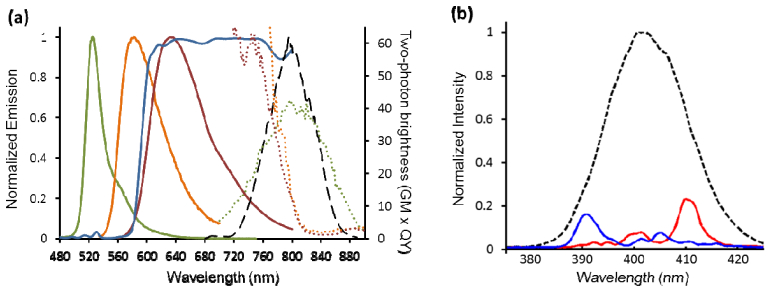
Fluorescent protein spectra and selective excitations. (a) Two-photon brightness (dotted curves) and fluorescence emission spectra (solid curves) of mAmetrine, TagRFPt and mKate2 are plotted in green, orange, and red, respectively (adapted from ref [30]). We note that TagRFPt, which was used in this study, is known to have a very similar spectral response to TagRFP shown here [43, 45]. Normalized spectrum of the broadband Ti:Sa laser is plotted in black (long dashed curve). The 595DCXR dichroic mirror transform function is plotted in blue. (b) SHG signal from a β-BaBO4 crystal for a TL pulse (dashed line) and two selective two-photon excitations with SHG centered at 390 nm (blue phase-shaped pulse, blue solid line) and 410 nm (red phase-shaped pulse, red solid line).
COS-7 cells were plated on three 35 mm tissue culture dishes for 24 hours before being separately transfected with mAmetrine, TagRFPt or mKate2 plasmids. 24 hours later, cells were resuspended and mix-cultured in a 50 mm dish for another 24 hours prior to imaging. A separate set of cells was plated on a 35 mm tissue culture dish, co-transfected with all three FPs after 24 hours, and cultured for an additional 48 hours. Immediately prior to imaging the culture media was replaced with pre-warmed Ringer’s Buffer. The water-immersion objective was submerged directly into the buffer for image acquisition.
3. Live cell imaging
To test whether the TL pulse excites all FPs and whether the two tailored pulses produce differential excitation for mAmetrine, TagRFPt and mKate2, we imaged a mixed population of COS-7 cells, in which each transfected cell expressed only one of the three FPs. We first imaged the sample with TL pulses [Figs. 3(a) and 3(b)]. Taking into account the FP emission spectra and the dichroic mirror used in our setup [Fig. 2(a)], mAmetrine, TagRFPt and mKate2 expressing cells were expected to have different fluorescence distributions into the green or red PMT channels. As expected, the putative mAmetrine cells (solid arrows) have fluorescence predominantly distributed to the green channel, while the putative TagRFPt cells (open arrows) have fluorescence evenly distributed to both channels. The putative mKate2 cells (solid arrow head) have fluorescence predominantly distributed to the red channel. Application of each phase-shaped pulse results in two fluorescence images, one from each PMT [Figs. 3(c)-3(f)]. As expected, the fluorophores were selectively excited by the different pulse shapes. [Fig. 3(c) vs Figs. 3(e) and 3(d) vs 3(f)]. The Ametrine cell, with its broad excitation spectrum, is excited by both red and blue phase-shaped pulses but still appears in the green PMT channel. TagRFPt and mKate2, however, are preferentially excited by the blue phase-shaped pulse.
Fig. 3.

Images of two-photon excitation of a mixed sample, in which each mammalian COS-7 cell only expresses one of either mAmetrine, TagRFPt or mKate2 fluorescent proteins. Cells were excited by the TL (left column), blue phase-shaped (middle column) and red phase-shaped (right column) pulses. Solid arrows, putative mAmetrine-expressing cells; open arrows, putative TagRFPt-expressing cells; solid arrowhead, putative mKate2-expressing cell. The TL pulses create higher image intensities than the shaped pulses do. To avoid saturation, lower PMT voltages have been applied to lower the fluorescence intensity of ~7 fold. Additionally, brightness and contrast of panels (a) and (b) have been scaled down ~10 fold to avoid saturation in display. Scale bar is 20 µm.
4. Linear unmixing
Although phase-shaping enables successful selective excitation of the fluorophores, the problem remains of how to identify them in complex samples containing multiple cells expressing different fluorophores. Generally, a dedicated detection channel is needed for each fluorescent color present [46] – leading to experimental setups that come at significant costs. An additional problem is the possibility of overlapping emission spectra, allowing signal from one fluorophore to leak into the detection channel of another [21, 46, 47]. This is potentially a more severe problem for live tissue imaging setups, in which emission filters are normally taken away in order to acquire maximal fluorescence signal for deeper light penetration [48]. The existing solution to the problem of separating and identifying different fluorophores is linear spectral unmixing, which records the emitted fluorescence spectra taken under different excitation conditions and then treats those spectra as linear combinations of the spectra of component fluorophores [21, 46, 47, 49, 50]. That is, the equation is solved, where S is the measured spectrum, A is a constant quantifying the amount of overlap between component spectra, and R is the reference spectrum of the single fluorophore i [21].
Here we employ a simplified unmixing method [51–53] that avoids the longer image acquisition time and experimental complexity of acquiring fluorescence spectra. We instead use two detectors to record the integrated green and red fluorescence for two different excitation conditions (red and blue phase-shaped pulses) to provide a total of 4 independent fluorescence images. For unmixing we solve the equation , where is the observed image under imaging condition i containing a mix of fluorophores, Nk is the unknown intensity of the single fluorophore k, and Ci is the constant quantifying the fraction of Nk in . The constants C are determined individually for each of the fluorophores used and are based on two parameters, α and β, found from images of reference cells expressing single fluorophores. The α and β parameters are ratios of fluorescence signal intensity recorded with different excitation and emission conditions: and where blue/red denotes the phase-shaped pulse used for excitation and GREEN/RED denotes the short or long wavelength emission detection channel.
These α and β values are then used to calculate the distribution of the total fluorescence intensity into each imaging condition, summarized in Table 1 :
Table 1. Constants quantifying the fractions of fluorophores intensity in observed images.
| GREEN Emission | RED Emission | |
|---|---|---|
| blue phase-shaped pulse
|
|
|
| red phase-shaped pulse |
This allows us to solve the matrix equation
| (3) |
for the unknown image intensity values IA, IR, and IK for mAmetrine, TagRFPt, and mKate2 fluorescence, respectively. In our unmixing process, we only utilize three images to separate three FPs [Figs. 3(c)-3(d)]. We purposely neglect the red channel image taken with the red phase-shaped pulse [Fig. 3(f)] because it has the lowest pixel intensity. This is because the red phase-shaped pulse only preferentially excites mAmetrine, yet the mAmetrine fluorescence is barely distributed to the red channel. To smooth shot noise, a Gaussian filter with a radius of 1 pixel was applied to raw images after processing. After background correction, linear unmixing outputs three images, each of which consists of fluorescent signal from one of the three FPs. The unmixing results corresponding to the images presented in Fig. 3 are shown in Fig. 4(a) , confirming the putative assignments of the FPs expressed in each cell. The merged image generated from the linear unmixing results shows nearly pure colors in all cells as expected [Fig. 4(a), merge]. In addition, we performed the same imaging and unmixing procedures on cells co-expressing all three FPs. The resulting image, shown in Fig. 4(b) shows mixed expression of all three FPs, generating a variety of colors as would be expected for co-expression with varying relative FP concentration.
Fig. 4.
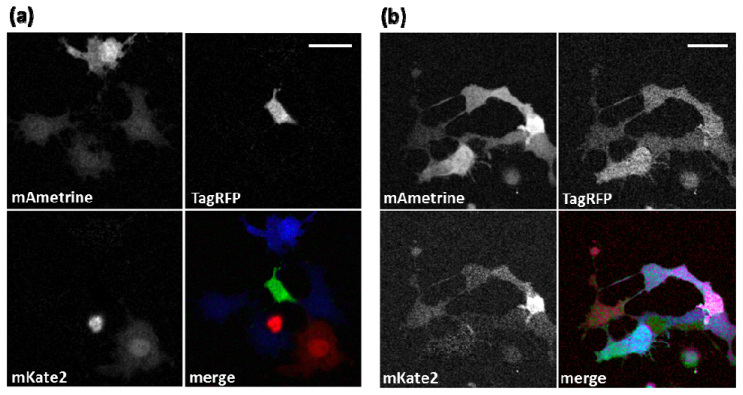
Linear unmixing. (a) Signal of mAmetrine, TagRFPt and mKate2 from Fig. 3 (middle and right columns) is restored in three separate images. (b) Pseudo-colored merge image of a sample co-expressing all three FPs after linear unmixing. In (a) and (b), mAmetrine, TagRFPt and mKate2 are pseudo-colored in blue, green and red in the merge image, respectively. Gamma correction (0.5) was applied to both panels in order to better present the dimly labeled cells. Scale bars are 20 µm.
5. Discussion and Conclusions
Using a broadband femtosecond laser and ultrafast phase-shaping techniques, we have developed a two-photon imaging modality allowing selective excitation of three FPs with distinct fluorescence spectra. By combining this with linear unmixing from two PMT channels, the fluorescence signal of each of the three FPs was restored with high separation. With two phase-shaped pulses, a total of four raw images were taken by two PMTs, which allows unmixing of as many as four FPs with distinct fluorescence spectra. Addition of more PMTs would permit either detection of more FP species or improved separation and reduced noise in the unmixing results [54, 55]. The use of a single broadband laser not only reduces the overall cost of the system, but also avoids the alignment complications of other multi-color 2PFM setups, allowing easy adaptation to most biomedical laboratories. Since phase-shaping provides versatile narrow excitation selectivity within the laser spectrum [33], selective FP excitation is only limited by the laser bandwidth. Using emerging laser sources with ultra-broad bandwidths, such as the VENTEON systems (VENTEON Laser Technologies GmbH), would easily expand our setup to nearly all available FPs [30] with precise selective excitations [56]. Such excitations could also be tailored to ensure equal fluorescence signal for the FPs of interest. In addition, phase-shaping may offer reduced photodamage in some imaging contexts. Depending on sample composition, photodamage may scale linearly (for example in pigment-rich tissues) or nonlinearly with peak intensity [57, 58]. Phase-shaped pulses designed for high 2PF signal from the FPs of interest have lower peak intensities than TL pulses and should provide reduced photodamage in samples where two and three photon absorption processes are the dominant photodamage mechanisms.
Acknowledgments
FP spectra in Fig. 2(a) were adapted from reference [30]. We gratefully acknowledge support from the National Institutes of Health (grant # 1-R21-EB-012686-01-A1). M.H. Brenner was supported by the Michigan Molecular Biophysics Training Grant and a University of Michigan Rackham Merit Fellowship.
References and links
- 1.Tsien R. Y., “The green fluorescent protein,” Annu. Rev. Biochem. 67(1), 509–544 (1998). 10.1146/annurev.biochem.67.1.509 [DOI] [PubMed] [Google Scholar]
- 2.Shaner N. C., Steinbach P. A., Tsien R. Y., “A guide to choosing fluorescent proteins,” Nat. Methods 2(12), 905–909 (2005). 10.1038/nmeth819 [DOI] [PubMed] [Google Scholar]
- 3.Tkaczyk E. R., Tkaczyk A. H., “Multiphoton flow cytometry strategies and applications,” Cytometry A 79(10), 775–788 (2011). 10.1002/cyto.a.21110 [DOI] [PubMed] [Google Scholar]
- 4.Qian M., Cai D., Verhey K. J., Tsai B., “A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection,” PLoS Pathog. 5(6), e1000465 (2009). 10.1371/journal.ppat.1000465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai D., McEwen D. P., Martens J. R., Meyhofer E., Verhey K. J., “Single molecule imaging reveals differences in microtubule track selection between Kinesin motors,” PLoS Biol. 7(10), e1000216 (2009). 10.1371/journal.pbio.1000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lippincott-Schwartz J., Patterson G. H., “Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging,” Trends Cell Biol. 19(11), 555–565 (2009). 10.1016/j.tcb.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai D., Hoppe A. D., Swanson J. A., Verhey K. J., “Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells,” J. Cell Biol. 176(1), 51–63 (2007). 10.1083/jcb.200605097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ai H. W., Hazelwood K. L., Davidson M. W., Campbell R. E., “Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors,” Nat. Methods 5(5), 401–403 (2008). 10.1038/nmeth.1207 [DOI] [PubMed] [Google Scholar]
- 9.Giepmans B. N., Adams S. R., Ellisman M. H., Tsien R. Y., “The fluorescent toolbox for assessing protein location and function,” Science 312(5771), 217–224 (2006). 10.1126/science.1124618 [DOI] [PubMed] [Google Scholar]
- 10.Kogure T., Karasawa S., Araki T., Saito K., Kinjo M., Miyawaki A., “A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy,” Nat. Biotechnol. 24(5), 577–581 (2006). 10.1038/nbt1207 [DOI] [PubMed] [Google Scholar]
- 11.Livet J., Weissman T. A., Kang H., Draft R. W., Lu J., Bennis R. A., Sanes J. R., Lichtman J. W., “Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system,” Nature 450(7166), 56–62 (2007). 10.1038/nature06293 [DOI] [PubMed] [Google Scholar]
- 12.Snippert H. J., van der Flier L. G., Sato T., van Es J. H., van den Born M., Kroon-Veenboer C., Barker N., Klein A. M., van Rheenen J., Simons B. D., Clevers H., “Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells,” Cell 143(1), 134–144 (2010). 10.1016/j.cell.2010.09.016 [DOI] [PubMed] [Google Scholar]
- 13.Red-Horse K., Ueno H., Weissman I. L., Krasnow M. A., “Coronary arteries form by developmental reprogramming of venous cells,” Nature 464(7288), 549–553 (2010). 10.1038/nature08873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng G., Mellor R. H., Bernstein M., Keller-Peck C., Nguyen Q. T., Wallace M., Nerbonne J. M., Lichtman J. W., Sanes J. R., “Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP,” Neuron 28(1), 41–51 (2000). 10.1016/S0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- 15.Misgeld T., Kerschensteiner M., Bareyre F. M., Burgess R. W., Lichtman J. W., “Imaging axonal transport of mitochondria in vivo,” Nat. Methods 4(7), 559–561 (2007). 10.1038/nmeth1055 [DOI] [PubMed] [Google Scholar]
- 16.Denk W., Strickler J. H., Webb W. W., “Two-photon laser scanning fluorescence microscopy,” Science 248(4951), 73–76 (1990). 10.1126/science.2321027 [DOI] [PubMed] [Google Scholar]
- 17.Denk W., “Two-photon excitation in functional biological imaging,” J. Biomed. Opt. 1(3), 296–304 (1996). 10.1117/12.242945 [DOI] [PubMed] [Google Scholar]
- 18.Kawano H., Kogure T., Abe Y., Mizuno H., Miyawaki A., “Two-photon dual-color imaging using fluorescent proteins,” Nat. Methods 5(5), 373–374 (2008). 10.1038/nmeth0508-373 [DOI] [PubMed] [Google Scholar]
- 19.Tillo S. E., Hughes T. E., Makarov N. S., Rebane A., Drobizhev M., “A new approach to dual-color two-photon microscopy with fluorescent proteins,” BMC Biotechnol. 10(1), 6 (2010). 10.1186/1472-6750-10-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahai E., Wyckoff J., Philippar U., Segall J. E., Gertler F., Condeelis J., “Simultaneous imaging of GFP, CFP and collagen in tumors in vivo using multiphoton microscopy,” BMC Biotechnol. 5(1), 14 (2005). 10.1186/1472-6750-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lansford R., Bearman G., Fraser S. E., “Resolution of multiple green fluorescent protein color variants and dyes using two-photon microscopy and imaging spectroscopy,” J. Biomed. Opt. 6(3), 311–318 (2001). 10.1117/1.1383780 [DOI] [PubMed] [Google Scholar]
- 22.Entenberg D., Wyckoff J., Gligorijevic B., Roussos E. T., Verkhusha V. V., Pollard J. W., Condeelis J., “Setup and use of a two-laser multiphoton microscope for multichannel intravital fluorescence imaging,” Nat. Protoc. 6(10), 1500–1520 (2011). 10.1038/nprot.2011.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahou P., Zimmerley M., Loulier K., Matho K. S., Labroille G., Morin X., Supatto W., Livet J., Débarre D., Beaurepaire E., “Multicolor two-photon tissue imaging by wavelength mixing,” Nat. Methods 9(8), 815–818 (2012). 10.1038/nmeth.2098 [DOI] [PubMed] [Google Scholar]
- 24.Wang K., Liu T. M., Wu J., Horton N. G., Lin C. P., Xu C., “Three-color femtosecond source for simultaneous excitation of three fluorescent proteins in two-photon fluorescence microscopy,” Biomed. Opt. Express 3(9), 1972–1977 (2012). 10.1364/BOE.3.001972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogilvie J. P., Débarre D., Solinas X., Martin J. L., Beaurepaire E., Joffre M., “Use of coherent control for selective two-photon fluorescence microscopy in live organisms,” Opt. Express 14(2), 759–766 (2006). 10.1364/OPEX.14.000759 [DOI] [PubMed] [Google Scholar]
- 26.Schelhas L. T., Shane J. C., Dantus M., “Advantages of ultrashort phase-shaped pulses for selective two-photon activation and biomedical imaging,” Nanomedicine 2(3), 177–181 (2006). 10.1016/j.nano.2006.07.007 [DOI] [PubMed] [Google Scholar]
- 27.Pillai R. S., Boudoux C., Labroille G., Olivier N., Veilleux I., Farge E., Joffre M., Beaurepaire E., “Multiplexed two-photon microscopy of dynamic biological samples with shaped broadband pulses,” Opt. Express 17(15), 12741–12752 (2009). 10.1364/OE.17.012741 [DOI] [PubMed] [Google Scholar]
- 28.Isobe K., Suda A., Tanaka M., Kannari F., Kawano H., Mizuno H., Miyawaki A., Midorikawa K., “Multifarious control of two-photon excitation of multiple fluorophores achieved by phase modulation of ultra-broadband laser pulses,” Opt. Express 17(16), 13737–13746 (2009). 10.1364/OE.17.013737 [DOI] [PubMed] [Google Scholar]
- 29.Moore D. S., “Optimal coherent control of sensitivity and selectivity in spectrochemical analysis,” Anal. Bioanal. Chem. 393(1), 51–56 (2009). 10.1007/s00216-008-2318-z [DOI] [PubMed] [Google Scholar]
- 30.Drobizhev M., Makarov N. S., Tillo S. E., Hughes T. E., Rebane A., “Two-photon absorption properties of fluorescent proteins,” Nat. Methods 8(5), 393–399 (2011). 10.1038/nmeth.1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastirk I., Dela Cruz J., Walowicz K., Lozovoy V., Dantus M., “Selective two-photon microscopy with shaped femtosecond pulses,” Opt. Express 11(14), 1695–1701 (2003). 10.1364/OE.11.001695 [DOI] [PubMed] [Google Scholar]
- 32.Tkaczyk E. R., Tkaczyk A. H., Mauring K., Ye J. Y., Baker J. R., Jr, Norris T. B., “Control of two-photon fluorescence of common dyes and conjugated dyes,” J. Fluoresc. 19(3), 517–532 (2009). 10.1007/s10895-008-0441-z [DOI] [PubMed] [Google Scholar]
- 33.Comstock M., Lozovoy V., Pastirk I., Dantus M., “Multiphoton intrapulse interference 6; binary phase shaping,” Opt. Express 12(6), 1061–1066 (2004). 10.1364/OPEX.12.001061 [DOI] [PubMed] [Google Scholar]
- 34.Meshulach D., Silberberg Y., “Coherent quantum control of two-photon transitions by a femtosecond laser pulse,” Nature 396(6708), 239–242 (1998). 10.1038/24329 [DOI] [Google Scholar]
- 35.Lozovoy V. V., Pastirk I., Walowicz K. A., Dantus M., “Multiphoton intrapulse interference. II. Control of two- and three-photon laser induced fluorescence with shaped pulses,” J. Chem. Phys. 118(7), 3187–3196 (2003). 10.1063/1.1531620 [DOI] [Google Scholar]
- 36.Walowicz K. A., Pastirk I., Lozovoy V. V., Dantus M., “Multiphoton Intrapulse Interference. 1. Control of Multiphoton Processes in Condensed Phases,” J. Phys. Chem. A 106(41), 9369–9373 (2002). 10.1021/jp0258964 [DOI] [Google Scholar]
- 37.Weiner A. M., “Femtosecond pulse shaping using spatial light modulators,” Rev. Sci. Instrum. 71(5), 1929–1960 (2000). 10.1063/1.1150614 [DOI] [Google Scholar]
- 38.Lozovoy V. V., Dantus M., “Systematic control of nonlinear optical processes using optimally shaped femtosecond pulses,” ChemPhysChem 6(10), 1970–2000 (2005). 10.1002/cphc.200400342 [DOI] [PubMed] [Google Scholar]
- 39.Dela Cruz J. M., Pastirk I., Lozovoy V. V., Walowicz K. A., Dantus M., “Multiphoton Intrapulse Interference 3: Probing Microscopic Chemical Environments,” J. Phys. Chem. A 108(1), 53–58 (2004). 10.1021/jp036150o [DOI] [Google Scholar]
- 40.Coello Y., Lozovoy V. V., Gunaratne T. C., Xu B., Borukhovich I., Tseng C.-, Weinacht T., Dantus M., “Interference without an interferometer: a different approach to measuring, compressing, and shaping ultrashort laser pulses,” J. Opt. Soc. Am. B 25(6), A140–A150 (2008). 10.1364/JOSAB.25.00A140 [DOI] [Google Scholar]
- 41.Xu B., Gunn J. M., Cruz J. M. D., Lozovoy V. V., Dantus M., “Quantitative investigation of the multiphoton intrapulse interference phase scan method for simultaneous phase measurement and compensation of femtosecond laser pulses,” J. Opt. Soc. Am. B 23(4), 750–759 (2006). 10.1364/JOSAB.23.000750 [DOI] [Google Scholar]
- 42.Merzlyak E. M., Goedhart J., Shcherbo D., Bulina M. E., Shcheglov A. S., Fradkov A. F., Gaintzeva A., Lukyanov K. A., Lukyanov S., Gadella T. W., Chudakov D. M., “Bright monomeric red fluorescent protein with an extended fluorescence lifetime,” Nat. Methods 4(7), 555–557 (2007). 10.1038/nmeth1062 [DOI] [PubMed] [Google Scholar]
- 43.Shaner N. C., Lin M. Z., McKeown M. R., Steinbach P. A., Hazelwood K. L., Davidson M. W., Tsien R. Y., “Improving the photostability of bright monomeric orange and red fluorescent proteins,” Nat. Methods 5(6), 545–551 (2008). 10.1038/nmeth.1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shcherbo D., Merzlyak E. M., Chepurnykh T. V., Fradkov A. F., Ermakova G. V., Solovieva E. A., Lukyanov K. A., Bogdanova E. A., Zaraisky A. G., Lukyanov S., Chudakov D. M., “Bright far-red fluorescent protein for whole-body imaging,” Nat. Methods 4(9), 741–746 (2007). 10.1038/nmeth1083 [DOI] [PubMed] [Google Scholar]
- 45.Salomonnson E., Mihalko L. A., Verkhusha V. V., Luker K. E., Luker G. D., “Cell-based and in vivo spectral analysis of fluorescent proteins for multiphoton microscopy,” J. Biomed. Opt. 17(9), 096001 (2012). 10.1117/1.JBO.17.9.096001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsurui H., Nishimura H., Hattori S., Hirose S., Okumura K., Shirai T., “Seven-color fluorescence imaging of tissue samples based on Fourier spectroscopy and singular value decomposition,” J. Histochem. Cytochem. 48(5), 653–662 (2000). 10.1177/002215540004800509 [DOI] [PubMed] [Google Scholar]
- 47.Zimmermann T., Rietdorf J., Pepperkok R., “Spectral imaging and its applications in live cell microscopy,” FEBS Lett. 546(1), 87–92 (2003). 10.1016/S0014-5793(03)00521-0 [DOI] [PubMed] [Google Scholar]
- 48.Clark I. B., Muha V., Klingseisen A., Leptin M., Müller H. A., “Fibroblast growth factor signalling controls successive cell behaviours during mesoderm layer formation in Drosophila,” Development 138(13), 2705–2715 (2011). 10.1242/dev.060277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boardman J. W., “Inversion Of Imaging Spectrometry Data Using Singular Value Decomposition,” in Geoscience and Remote Sensing Symposium, 1989. IGARSS'89. 12th Canadian Symposium on Remote Sensing., 1989 International (1989), 2069–2072 10.1109/IGARSS.1989.577779 [DOI] [Google Scholar]
- 50.Hiraoka Y., Shimi T., Haraguchi T., “Multispectral imaging fluorescence microscopy for living cells,” Cell Struct. Funct. 27(5), 367–374 (2002). 10.1247/csf.27.367 [DOI] [PubMed] [Google Scholar]
- 51.Kraus B., Ziegler M., Wolff H., Linear Fluorescence Unmixing in Cell Biological Research (2007). [Google Scholar]
- 52.Kramer-Hämmerle S., Ceccherini-Silberstein F., Bickel C., Wolff H., Vincendeau M., Werner T., Erfle V., Brack-Werner R., “Identification of a novel Rev-interacting cellular protein,” BMC Cell Biol. 6(1), 20 (2005). 10.1186/1471-2121-6-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garini Y., Young I. T., McNamara G., “Spectral imaging: principles and applications,” Cytometry A 69(8), 735–747 (2006). 10.1002/cyto.a.20311 [DOI] [PubMed] [Google Scholar]
- 54.Davis L. M., Shen G., “Extension of multidimensional microscopy to ultrasensitive applications with maximum-likelihood analysis,” Proc. SPIE 6443, 64430N–, 64430N-12. (2007). 10.1117/12.702217 [DOI] [Google Scholar]
- 55.Ducros M., Moreaux L., Bradley J., Tiret P., Griesbeck O., Charpak S., “Spectral unmixing: analysis of performance in the olfactory bulb in vivo,” PLoS ONE 4(2), e4418 (2009). 10.1371/journal.pone.0004418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu B., Coello Y., Lozovoy V. V., Dantus M., “Two-photon fluorescence excitation spectroscopy by pulse shaping ultrabroad-bandwidth femtosecond laser pulses,” Appl. Opt. 49(32), 6348–6353 (2010). 10.1364/AO.49.006348 [DOI] [PubMed] [Google Scholar]
- 57.Koester H. J., Baur D., Uhl R., Hell S. W., “Ca2+ fluorescence imaging with pico- and femtosecond two-photon excitation: signal and photodamage,” Biophys. J. 77(4), 2226–2236 (1999). 10.1016/S0006-3495(99)77063-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pestov D., Andegeko Y., Lozovoy V. V., Dantus M., “Photobleaching and photoenhancement of endogenous fluorescence observed in two-photon microscopy with broadband laser sources,” J. Opt. 12(8), 084006 (2010). 10.1088/2040-8978/12/8/084006 [DOI] [Google Scholar]