

Abstract
Trehalose synthase (TreS) catalyzes the reversible conversion of maltose into trehalose in mycobacteria as one of three biosynthetic pathways to this nonreducing disaccharide. Given the importance of trehalose to survival of mycobacteria, there has been considerable interest in understanding the enzymes involved in its production; indeed the structures of the key enzymes in the other two pathways have already been determined. Herein, we present the first structure of TreS from Mycobacterium smegmatis, thereby providing insights into the catalytic machinery involved in this intriguing intramolecular reaction. This structure, which is of interest both mechanistically and as a potential pharmaceutical target, reveals a narrow and enclosed active site pocket within which intramolecular substrate rearrangements can occur. We also present the structure of a complex of TreS with acarbose, revealing a hitherto unsuspected oligosaccharide-binding site within the C-terminal domain. This may well provide an anchor point for the association of TreS with glycogen, thereby enhancing its role in glycogen biosynthesis and degradation.
Keywords: drug design, enzyme inhibition, GH13 glycoside hydrolase, trehalose synthase, tuberculosis
Introduction
Trehalose is a nonreducing disaccharide of glucose, which is used by many lower organisms such as mycobacteria for key functions such as energy storage, signaling, protein-protection and bacterial cell wall components (Elbein 1974; Takayama and Armstrong 1976; Crowe et al. 1984). In mycobacteria, trehalose is also part of a toxic lipid in the cell wall known as trehalose-6,6′-dimycolate or cord factor, which has been identified as the main virulence factor of tuberculosis (Barry and Mdluli 1996; Barry et al. 1998; Daffe and Draper 1998). Accordingly, considerable attention has been directed toward the possibility that enzymes involved in the production of trehalose may serve as drug targets. However, the discovery of what appeared to be three independent biosynthetic pathways for trehalose in Mycobacterium smegmatis (Pan et al. 2004; Murphy et al. 2005), coupled with the absence of structural data for the TreS enzyme, has greatly hindered efforts to develop new antituberculosis drugs. These three proposed biosynthetic trehalose pathways involve the enzymes: (i) Ots-A/B, (ii) TreY-TreZ and (iii) TreS (De Smet et al. 2000). In the well-characterized Ots-A/B-pathway, trehalose is generated in a two-step enzymatic reaction. In the first step trehalose-6-phosphate-synthase (Ots-A in Escherichia coli) condenses uridine diphosphate glucose (UDP-glucose) with glucose-6-phosphate to produce trehalose-6-phosphate, which is dephosphorylated in the second step by trehalose-6-phosphatase (Ots-B in E. coli) to generate free trehalose (Kaasen et al. 1992). In the TreY-TreZ pathway, glycogen or malto-oligosaccharides serve as starting substrates. In an initial step, the maltosyl moiety at the end of a substrate is isomerized by malto-oligosyltrehalose synthase (TreY) generating a trehalosyl moiety, which is hydrolytically released by the second enzyme malto-oligosaccharyltrehalose trehalohydrolase (TreZ) to produce free trehalose (Maruta et al. 1996a, b, c). Significantly, TreS simply interconverts maltose and trehalose (Figure 1) with an equilibrium favoring trehalose by 4.1:1 (Tewari and Goldberg 1991; Tsusaki et al. 1996). While structural data are available for the enzymes in the Ots-A/B and TreY-TreZ pathways for trehalose synthesis, no such structure has been described for TreS.
Fig. 1.

(a) Interconversion of α-1-4 linked maltose and α-1-1 linked trehalose catalyzed by TreS. Numbers indicate carbon atom positions in the sugar ring. (b) Chemical structure of the inhibitor acarbose, a pseudo-tetrasaccharide. Asterisks indicate features that distinguish acarbose from the related substrate maltotetraose. These features include an unsaturated ring, a methyl group in place of a hydroxymethyl group and an N-linked “glycosidic” bond. Ring components have been numbered with respect to the N-linkage of this inhibitor.
Interconversion of trehalose and maltose by the enzymatic activity of TreS occurs as a two-step, double-displacement mechanism (Koshland 1953; Boyer 1960; Zhang et al. 2011). In the first step, the catalytic nucleophile, Asp230, attacks the anomeric center of the nonreducing sugar of maltose in an acid-catalyzed process, cleaving the glycosidic bond via an oxocarbenium ion-like transition state, to form a covalent β-glycosyl-enzyme intermediate. In the second step, the glucose released reorients within the active site and the 1-hydroxyl then attacks the anomeric center of the glycosyl-enzyme, forming the trehalose and regenerating free enzyme. Importantly, TreS interconverts maltose and trehalose by an intramolecular mechanism and therefore does not need external glucose or any other factor to carry out its activity (Zhang et al. 2011).
Of further interest are other studies indicating that TreS from M. smegmatis also possesses an amylase activity, albeit several orders of magnitude lower than its isomerase activity. This results in the release of maltose (thus also trehalose) from glycogen, and this TreS amylase activity can be competitively inhibited by the potent glucosidase-amylase inhibitor acarbose (Figure 1; (Pan et al. 2008)). More recently, TreS has been linked to a novel biosynthetic pathway in mycobacteria that generates glycogen from trehalose via four enzymatic steps mediated by TreS, maltokinase (Pep2), maltosyltransferase (GlgE) and branching enzyme (GlgB) (Elbein et al. 2010; Kalscheuer et al. 2010). Very recent studies have confirmed that flux through TreS is principally in this direction, consistent with the demonstration that TreS produces only alpha-maltose, the anomer that is the required substrate for the subsequent enzyme, maltokinase (Miah et al. 2013). Interestingly, inactivation of GlgE leads to rapid cell death in Mycobacterium tuberculosis due to a self-poisoning accumulation of maltose-1-phosphate, which is further amplified by the natural stress response in which trehalose is accumulated. GlgE, therefore, represents a promising target for new antituberculosis drugs (Kalscheuer and Jacobs 2010; Kalscheuer et al. 2010). However, since maltose uptake in mycobacteria is very poor compared with that of trehalose, it seems likely that inhibitors will need to be administered as “pro-drug” trehalose analogs that can be transformed into effective GlgE inhibitors by TreS and Pep2 (Zhang et al. 2011). Thus, while these recent studies cast doubt on a significant role for TreS in trehalose biosynthesis, it is likely to play an important role in attempts to develop useful GlgE inhibitors.
Clearly, a high-resolution structure of TreS is essential to understanding not only the isomerase and putative amylase activities of this enzyme, but also its potential in drug development. To that end, we have solved two structures: that of wild-type trehalose synthase (TreS) from M. smegmatis, an organism that is a close relative and benign model for the infective organism M. tuberculosis, and of the acarbose-complexed enzyme. The TreSs from these two organisms share 83% sequence identity and belong to the oligo-1,6-glucosidase subfamily 31 of GH13 family enzymes, Figure 2 (Kuriki and Imanaka 1999; Oslancova and Janecek 2002; Stam et al. 2006). These studies reveal an unexpected tetrameric structure for this enzyme and a novel active site configuration that suggests that the enzyme exists in both stable active and inactive conformations. In addition, a well-defined acarbose/carbohydrate-binding site is identified in TreS that is remote from the active site region, suggesting a role in the tethering of TreS to glycogen, as is often seen in enzymes involved in glycogen biosynthesis and degradation.
Fig. 2.

Sequence alignment between TreS of M. smegmatis (GenBank accession ID: YP_006571064) and M. tuberculosis (GenBank accession ID: EFI32604). Colored boxes highlight the domain organization found in M. smegmatis (Figure 3). Green indicates domain A, yellow indicates domain B, red indicates domain C and blue indicates the extended active site loop found in domain A. Catalytic residues are shown with red letters in black boxes. Leu344 is highlighted in green and is boxed as well.
Results and discussion
Structural features of the TreS fold
Our studies have focused on determining the high-resolution structures of TreS in its native state and in complex with the competitive α-glucosidase inhibitor acarbose (Table I). These structures should help us to shed light on the enzymatic mechanism of TreS and the putative role of acarbose in inhibiting amylase activity. Superposition of these two structures revealed a Cα root mean square deviation (RMSD) of 0.3 Å, indicating an excellent match in overall fold. Notably, the two protein molecules in the asymmetric units of both structures showed disordered peptide segments at their N-terminal ends. These included residues 1–28 (1–29 in the complexed structure) of molecule A and residues 1–16 of molecule B. At the C-terminal end, both structures miss the last seven residues (587–593) in molecule A and the last six residues (588–593) in molecule B. Another loop in domain C (residue 514–522) was only disordered in molecule A.
Table I.
Structure determination statisticsa
| Wild-type TreS | TreS/acarbose complex | |
|---|---|---|
| Wavelength (Å) | 0.9795 | 0.9795 |
| Resolution range (Å)a | 34.92–1.84 (1.906–1.84) | 34.82–1.85 (1.916–1.85) |
| Space group | I 41 | I 41 |
| Unit cell (Å) | 127.55, 127.55, 217.76 | 127.18, 127.18, 216.95 |
| Total reflections collected | 614,630 | 1,005,262 |
| Unique reflectionsa | 139,127 (14,429) | 143,064 (13,711) |
| Multiplicity | 4.42 | 7.03 |
| Rmerge (%)a | 6.2 (52.2) | 8.2 (68.9) |
| Completeness (%)a | 92.85 (96.41) | 97.99 (94.25) |
| Mean I/sigma (I)a | 14.52 (3.53) | 17.09 (3.33) |
| R-factora | 0.1630 (0.2096) | 0.1650 (0.2140) |
| R-freea | 0.1954 (0.2570) | 0.1981 (0.2626) |
| Total number of atoms | 10,273 | 10,192 |
| Protein atoms | 9209 | 9193 |
| Ions/ligands | 11/0 | 11/44 |
| Water | 1053 | 944 |
| Protein residues | 1120 | 1119 |
| RMS (bonds) | 0.007 | 0.007 |
| RMS (angles) | 1.11 | 1.11 |
| Average B-factor | 32.5 | 33.7 |
| Protein atoms | 31.8 | 33.2 |
| Ligands | – | 46.9 |
| Ions | 26.6 | 28.2 |
| Solvent | 38.7 | 38.2 |
aValues in parenthesis refer to the highest resolution shell.
TreS belongs to glycoside hydrolase family GH13, a grouping that also includes human pancreatic α-amylase. As illustrated in Figure 3D, our structure shows that TreS shares the common GH13 domain organization, consisting of three major domains: A, B and C. The N-terminal domain A forms the central core of the enzyme and is composed of a (β/α)8 triosephosphate isomerase (TIM) barrel structure which harbors the active site (Banner et al. 1975; Zhang et al. 2011). Domain B is inserted between β-strand 3 and α-helix 3 of the TIM barrel and contains one α-helix, three antiparallel β-strands and a calcium-ion-binding site that stabilizes the interface between domain A and domain B. Variations of this latter feature are frequently seen in other structures of amylolytic enzymes (Qian et al. 1993). The C-terminal domain C comprises a seven-stranded antiparallel β-sandwich. The function of domain C is unknown in TreS, but a number of GH13 enzymes have been shown to harbor a carbohydrate-binding domain in this part of the enzyme (Janecek et al. 2003; Christiansen et al. 2009).
Fig. 3.
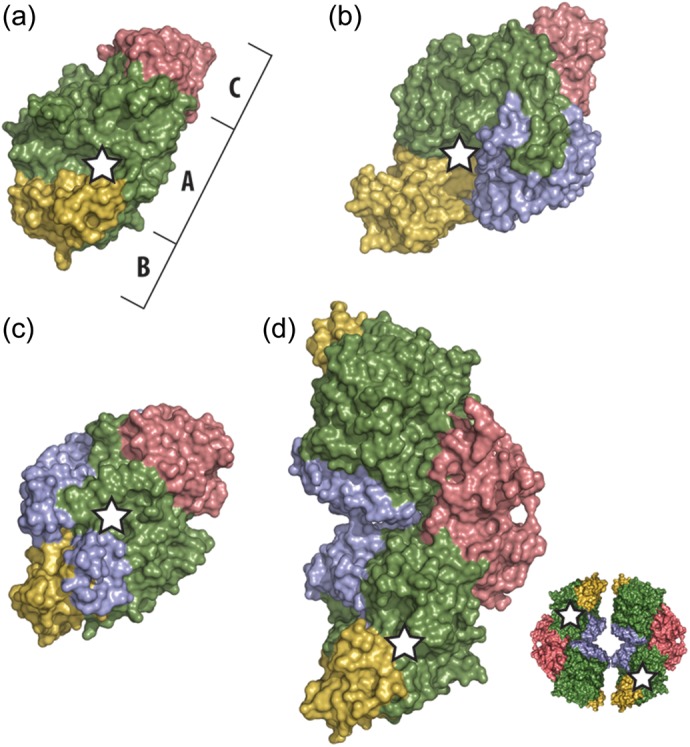
Illustration of the domain architecture of our TreS structure and representative GH13 family members. Protein surfaces of domains A, B and C are shown in green, yellow and red, respectively. Additional domains are shown in blue. A white star indicates the position of the catalytic center. (a) human pancreatic α-amylase (PDB ID: 1hny); (b) malto-oligosyltrehalose synthase TreY (PDB ID: 1iv8); (c) trehalulose synthase MutB (PDB ID: 2pwe); (d) the asymmetric unit dimer of TreS from our studies of the M. smegmatis enzyme (PDB ID: 3zo9). A schematic of the expected tetrameric assembly by two TreS homodimers is shown in the lower right of frame (D).
The folded conformation of TreS differs in three major aspects from that of a typical GH13 family enzyme. First, the central N-terminal domain A varies from the classical (β/α)8 TIM barrel by having an extended loop between β-strand 7 and α-helix 7 (designated L7, residues 338–384; Figure 4). This additional L7 polypeptide chain segment is composed of two α-helices along with an extended loop that contains the second aspartic residue, Asp342, of the active site (Zhang et al. 2011). Furthermore, unlike related α-amylase structures, this loop also contributes to the coordination of not only the Cl− ion located within the active site but also an additional nearby Cl− ion of unknown function (Figure 4).
Fig. 4.
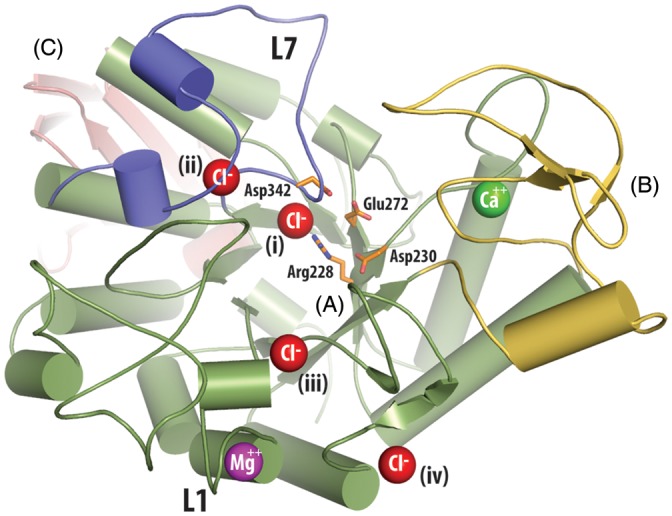
Ion binding in the structure of TreS. Domains are indicated by large capital letters and follow the same coloring scheme as Figure 2. For clarity, loops have been smoothed and only one enzyme molecule of the asymmetric unit is shown. Ion positions are indicated by colored spheres (red = Cl−, green = Ca2+, magenta = Mg2+). Chloride (i) is located in a homologous position compared with the allosterically activating chloride ion of the human α-amylases (Brayer et al. 1995; Maurus et al. 2005). Chloride (ii) and (iii) are located in loops L7 (residues 338–384) and L1 (residues 43–77), respectively, which surround the active site. Chloride (iv) binding would appear to be due to crystal packing interactions and is not present in the second molecule of this enzyme in the asymmetric unit. The sidechains of the catalytic triad composed of Asp230 (nucleophile), Glu272 (acid/base catalyst), and Asp 342 (substrate binding), along with the conserved active site residue Arg228, are shown using orange stick representations.
Secondly, it seems apparent from the contents of the asymmetric unit that TreS is, at a minimum, a dimeric enzyme in contrast to most enzymes of the GH13 family, which are found to be monomeric (Lee et al. 2005). This observed TreS asymmetric unit dimer is tightly held together with numerous hydrogen bonds and van der Waals contacts (Figure 3). Surface analyses with PISA (Krissinel and Henrick 2007; Krissinel 2010) show that this dimeric arrangement produces a buried surface of 962 Å2, corresponding to 4.5% of the solvent-accessible surface per TreS monomer. Furthermore, upon symmetry expansion of the unit cell, we have identified the compact formation of two TreS homodimers into a tetrameric assembly (Figure 3D). According to this analysis, a tetrameric arrangement of TreS produces a buried surface of 2283 Å2 that corresponds to 10.5% of the solvent-accessible surface per monomer, with a total of 17 hydrogen bonds and 10 salt-bridges across the interfaces. Additionally, PISA molecular assembly analysis (Krissinel and Henrick 2007; Krissinel 2010) suggests that TreS is thermodynamically stable in both the dimeric and tetrameric states, pointing out there may be a dynamic equilibrium between these two structural forms.
Interestingly, and in contrast, gel filtration results have indicated that TreS can form hexamers in solution (Pan et al. 2004). In that regard, it is noteworthy that the structure of TreS presented here has a disordered N-terminal end. If this same conformational instability is also present in solution, it could contribute to an increased hydrodynamic radius of the enzyme, thereby resulting in apparent larger molecular weights in gel filtration experiments. Based on our observations of a tightly integrated tetrameric structure, it would seem unlikely that TreS can in fact form a hexameric assembly.
Thirdly, the structure of TreS has five well-defined ion-binding sites. One is a hepta-coordinated calcium-ion-binding site located ∼13 Å from the active site at the interface between domain A and domain B (Figure 4). The coordinating residues involved include Asn132, Asp200, Tyr234, Leu235, Glu237 and two water molecules. This parallels other GH13 amylases such as human pancreatic α-amylase, which have a homologous calcium-ion-binding site that is involved in maintaining structural integrity (Vallee et al. 1959; Levitzki and Steer 1974; Lifshitz and Levitzki 1976). Interestingly, calcium ions are usually absent in the oligo-1,6-glucosidase subfamily to which TreS belongs, since an atom of another residue, usually a lysine, occupies the calcium position in this enzyme subfamily (Oslancova and Janecek 2002). In addition, for TreS we have identified a hexa-coordinated magnesium ion located in a solvent-accessible loop (L1, residues 50–60, Figure 4) of domain A, ∼25 Å from the catalytic center. This location is homologous to that of a calcium-ion site found in trehalulose synthase MutB (Ravaud et al. 2007). In TreS, interactions with this magnesium ion come from Asp51, Asn53, Asp55, Ile57, Asp59 and one water molecule, suggesting that this magnesium ion may be structurally required, similar to the calcium in MutB. Both TreS calcium and magnesium interactions satisfy the expected distances to their electron donors according to literature values (Dokmanic et al. 2008).
Four additional chloride ion-binding sites were detected in TreS. One of these sites is homologous to the allosterically activating chloride site in the human α-amylases (Maurus et al. 2005) and is located ∼8 Å from the TreS catalytic center. The coordination sphere of this chloride site (labeled (i) in Figure 4) is composed of the conserved active site residue Arg228, the backbone amides of Glu343 and Arg339, plus one water molecule. A further chloride ion site is located in the vicinity of each of the extended loops L1 and L7, both of which project out of the catalytic domain A. These loops surround the active site and their chloride ion-binding sites are within ∼13 and ∼16 Å, respectively, of the TreS enzymatic center. The chloride ion of loop L7 (labelled (ii) in Figure 4) is coordinated by residues His341 and Arg374, as well as the backbone amides of His341, Asn340, Arg375 and one water molecule. The chloride site of loop L1 (labeled (iii) in Figure 4) is composed of Arg47 and backbone amides of Leu46 and Arg47 plus three water molecules. While the chloride ion labeled (iv) is clearly present in our structure, its binding site is positioned in a highly solvent-exposed location and likely only forms due to crystal packing interactions.
Functional aspects of the TreS catalytic site
A comparison of the TreS active site structure with those of the related GH13 family enzymes human α-amylase, TreY and MutB is presented in Figure 5. A distinctive feature of the catalytic center of GH13 enzymes is a highly conserved triad of carboxylic acid-containing residues: a nucleophilic Asp; an acid/base catalyst Glu; a substrate-coordinating Asp residue (Svensson 1994). In TreS these three residues are indeed found in homologous positions within the TIM barrel scaffold with Cα RMSDs of 0.30, 1.13 and 1.27 Å, respectively, compared with the same residues in human α-amylase, TreY and MutB.
Fig. 5.

(a) Superposition of active site residues and loop L7 of TreS (orange), human pancreatic α-amylase (blue), trehalulose synthase MutB (green) and malto-oligosyltrehalose synthase TreY (cyan), with residue numbering according to the TreS structure. With the exception of the nucleophilic Asp230 in TreS, the catalytic residues for the other α-glucosidase enzymes are in comparable positions within the superposed structures (arrow indicates conformational change in TreS). The active site conformation of the bound inhibitor acarbose (gray sticks) in the active site of human α-amylase structure (blue) is shown (Maurus et al. 2005) in an overlay to evaluate the ligand-binding pocket in comparison with that of TreS. Importantly, the active site loop L7 of TreS (orange) protrudes into the catalytic center, suggesting that the structure we have determined is that of an inactive form of the enzyme. In addition, the side chain of Leu344 at the extremity of this loop projects into the active site to a position that would be <2 Å from the nucleophilic aspartate in the amylase structure. This feature presumably accounts for the displacement of the Asp230 catalytic nucleophile of TreS. (b) A cross sectional surface representation of TreS in the vicinity of the active site pocket. The narrow entrance to the active site from the surface of TreS is indicated by a dashed arrow and is delimited in this structure by the conformation of loop L7 and the positioning of the side chain of Leu344 in particular. A further consequence of the placement of Leu344 is a steric conflict with the side chain of the catalytic nucleophile Asp230, which as a consequence adopts a buried conformation, therefore becoming solvent and substrate inaccessible.
Notably, however, the side chain of the catalytic Asp230 nucleophile of TreS adopts a novel conformation wherein its side chain χ1 torsional angle is flipped ∼90°, compared with the equivalent residue in the other GH13 family structures (Figure 5). In these other structures, the carboxyl group of the Asp230 equivalent typically interacts with the catalytic partner acid/base glutamate residue in readiness to attack the glycosidic bond of an incoming substrate. Instead, the alternate conformation of Asp230 in TreS places its carboxylic acid group within hydrogen bonding distance of a nearby conserved Asp128 on the neighboring β-strand 3, an interaction that has not been seen previously in other members of the GH13 family.
This unexpected conformation of the side chain of Asp230 appears to be prompted by the unusual placement of the side chain of Leu344, which projects into the active site region of TreS. Leu344 placement is the result of another novel feature of the TreS structure wherein the active site loop L7, residues 338–384, adopts a different conformation within the active site of TreS compared with TreY, MutB or human α-amylase. Interestingly, the essential substrate-coordinating aspartate (Asp342 in TreS) sits at one end of this refolded loop but retains a position comparable with that seen in related GH13 enzymes. By contrast, in the other GH13 family enzyme structures examined, the L7 loop is folded differently to form a part of the S-1 binding site with the equivalent of Leu344 projected away from the active site toward α-helix 7 of the TIM barrel.
Our analyses suggest that TreS exists in both active and inactive forms, with our structure being representative of the inactive conformation. This idea is reinforced by a modeled overlay of the inhibitor acarbose as bound in the active site of human α-amylase (Maurus et al. 2005) and based on corresponding catalytic site amino acids in the active site of TreS. As illustrated in Figure 5, in this model the Leu344 side chain of TreS would block inhibitor binding at the −1 subsite, and thus would also prohibit substrate access. Even if substrate were to enter, the side chain of Leu344 in this inactive form of TreS would spatially conflict with the expected orientation of the catalytic nucleophile Asp230, prohibiting it from nucleophilic attack.
A comparison with the active site of MutB is instructive, since this is also a GH13 enzyme that isomerizes a disaccharide, namely sucrose to isomaltulose plus trehalulose. Both the catalytic and the conserved residues responsible for substrate binding in MutB adopt very similar positions in the active site of TreS, with the exception of the displaced Asp230 in TreS (Figure 6). Binding of the glucose moiety of the sucrose substrate in MutB is mediated by hydrogen bonds and hydrophobic interactions with a conserved tyrosine and phenylalanine, and two equivalent aromatic residues, Tyr93 and Phe194, are found in homologous positions in TreS. Likewise, an important hydrogen bonding arginine in MutB also coincides with Arg421 in a homologous position in TreS. Moreover, the active site pocket in MutB is narrow and enclosed, a feature also seen in TreS, consistent with the need for such enzymes to tightly control substrate movement such that the reaction remains intramolecular and hydrolysis is minimized. Clearly, it would be desirable to structurally characterize complexes with bound substrate or the covalent intermediate. Unfortunately, all attempts to date to obtain such structures of TreS have been unsuccessful.
Fig. 6.
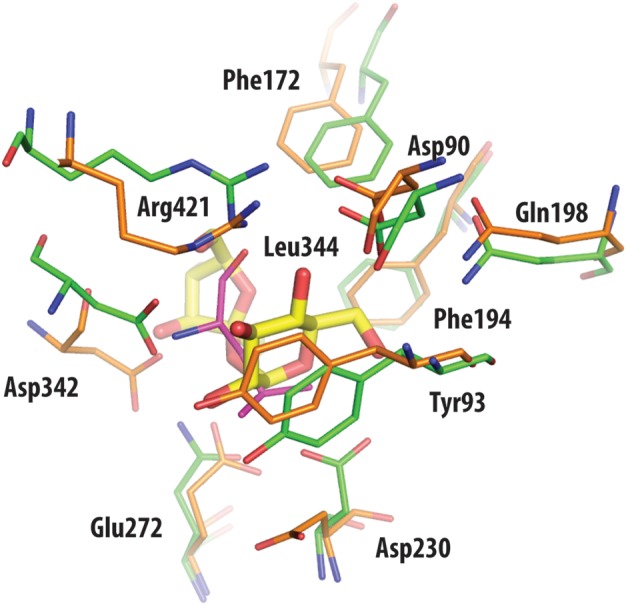
Superposition of active site residues of TreS (orange) and trehalulose synthase MutB (green), with residue numbering according to the TreS structure. With the exception of the side chain of the nucleophilic Asp230 in TreS, other catalytic residues are in comparable positions between the superimposed structures. Also drawn in yellow is the disaccharide substrate sucrose of MutB, as found bound in the active site of this enzyme. Leu344 of TreS (shown in magenta) overlaps with the sucrose molecule of MutB, hence prohibiting ligand access to the catalytic Asp230.
Defining the acarbose-binding site of TreS
Earlier results suggested that TreS exhibits an α-amylase activity, which can be competitively inhibited by the potent α-glucosidase inhibitor acarbose (Pan et al. 2008). To evaluate the binding of acarbose to TreS we soaked wild-type TreS crystals in 100 mM acarbose and solved the structure of the complex by X-ray diffraction methods to a resolution of 1.84 Å (Table I; PDB ID 3zoa). As shown in Figure 7, the resultant electron density observed in a Fobs − Fobs difference map very clearly resolved the bound conformation of acarbose. Furthermore, the directionality of the binding of this short polymeric inhibitor within its binding site on TreS could be unambiguously identified.
Fig. 7.

(a) Structure of the TreS/acarbose complex. The acarbose-binding site (within the white circle) is found in a crevice at the interface between two monomers of TreS (only a dimer is shown) and is located ∼40 Å from the active site of this enzyme (white asterisk). (b) The detailed structure of the observed acarbose-binding site and the overall fit of this inhibitor to a Fobs − Fobs electron density map (green mesh) contoured at the 3σ level. (c) LIGPLOT+ (Laskowski and Swindells 2011) representation of the interactions formed to acarbose in its binding site. Green dashed lines represent hydrogen bonds with residues (red letters) and water molecules (blue spheres). Residues involved in hydrophobic interactions are shown as red semicircles.
Surprisingly, bound acarbose was not found near the active site of TreS, but ∼40 Å away in a well-defined surface pocket between oligomerization interfaces of TreS (Figure 7). Forming a curved semicircle like conformation, bound acarbose interacts closely with a loop (553A–558A), particularly Thr555 and Tyr557, of domain C, while being further bracketed by another loop (534B–536B) of a related enzyme molecule, and helix (473A–486A) from opposite sides. A mainly hydrophobic area within the binding site is made up of Trp476, Met480, Phe577 and Trp579, which make contact with the −1 and +1 subsites of acarbose. Furthermore, LIGPLOT+ analysis (Laskowski and Swindells 2011) of the binding interfaces indicates that the binding pocket is composed of 13 residues located on two adjacent C-terminal domains and the preceding helix α8 of the TIM barrel of domain A (Figure 7C). Additionally, this assessment identified six hydrogen bonds between the ligand and its binding site. Four of these are direct interactions: Arg534 NH2-O2C, Arg534 NE-O2C, Thr555 OG1-N4B and Trp579N-04A, while another two are via water molecule 985 (W985-O2A and W985-O3A), which is coordinated by Gly574 and water 800. Furthermore, the protein-binding interface covers 505 Å2 or 63% of the total accessible surface area of the acarbose molecule.
Other studies have shown acarbose to bind in the active sites of α-glycosidases, where it acts as a transition state analog inhibitor (Mosi et al. 1998; Brayer et al. 2000). In this case, however, acarbose binds in a remote site that is unlikely to possess amylase activity given the lack of appropriate catalytic residues in its binding pocket. This casts some doubt on the proposed inherent amylase activity, which might perhaps have arisen from contamination. However, the results with acarbose are very interesting, since acarbose is seen to bind to the domain C of TreS, a region of many GH13 enzymes that has previously been suggested to function as a carbohydrate binding module (CBM). This situation is very similar to that observed in glycogen phosphorylase, another enzyme with a deep and contained active site pocket. In that case also acarbose was found to bind to an external-binding module (known then as the glycogen storage site) rather than within the active site (Goldsmith et al. 1987). In fact, such ancillary modules for carbohydrate binding are very common in glycoside hydrolases (Janecek et al. 2003), where they presumably target the enzyme to its appropriate substrate.
Structural analysis of the TreS acarbose-binding site shows similarities to other carbohydrate-binding sites (Tibbot et al. 2002; Boraston et al. 2004; Flint et al. 2004). A possible function for a CBM may lie in the report that TreS plays an important role in the formation of glycogen from trehalose (Elbein et al. 2010). This connection has been fleshed out recently in the form of the GlgE pathway, wherein it was demonstrated that TreS is directly involved in the formation of glycogen from trehalose via four enzymatic steps (Kalscheuer et al. 2010; Miah et al. 2013). It, therefore, makes sense that TreS would remain closely associated with this ultimate reservoir of glucose moieties.
Methods
Protein isolation and purification
TreS was purified from cell free extracts of M. smegmatis as previously described (Pan et al. 2004). Purified enzyme preparations showed one major band on sodium dodecyl sulfate gels at 68 kDa (Pan et al. 2008).
Crystallization and diffraction data collection
Mycobacterium smegmatis TreS was crystallized at a protein concentration of 20 mg/mL using the hanging drop vapor diffusion technique at 20°C. The protein solution, containing 40 mM sodium phosphate buffer at pH 6.0 was mixed with reservoir solution (0.1 M sodium cacodylate buffer at pH 6.5, 0.2 M MgCl2 and 10–14% polyethylene glycol (PEG) 1000) in a 1:1 ratio to a total volume of 4 μL on a siliconized cover slip. Crystals appeared after 1 day and grew to full size within 1 week. For cryoprotection, crystals were stabilized in 0.1 M sodium cacodylate buffer at pH 6.5, 0.2 M MgCl2, 22% PEG 1000 and 30% glycerol overnight at 4°C. Soaking experiments were carried out by adding 100 mM acarbose to the stabilization buffer and incubating crystals overnight at 4°C. All crystals were flash frozen in liquid nitrogen prior to data collection at the Stanford Synchrotron Radiation Lightsource, Stanford, CA. Diffraction data were collected at cryogenic temperature (100 K) with a PILATUS 6M detector at beamline BL-12. Diffraction data were integrated, scaled and reduced to structure factor amplitudes using the XDS software package (Kabsch 2010). Table I contains a summary of data collection and processing statistics. Data truncation was performed by using an I/sigma(I) criterion of 3 for the highest resolution shell. We also used a split half correlation CC(1/2) criterion of 50% to avoid discarding highly significant data (Karplus and Diederichs 2012).
Structure determinations
A preliminary molecular replacement solution for the M. smegmatis TreS structure proved particularly challenging and was found by using a derivative starting model of the GH13 α-glucosidase GSJ (PDB ID: 2ze0), the known enzyme structure of closest sequence homology (28% identity). Our derivative model was constructed by first aligning the TreS and GSJ amino-acid sequences and then deleting or pruning nonconserved residues from the TreS sequence using the program CHAINSAW (Stein 2008). Although the resulting starting model contained <50% of the initial TreS sequence, a unique molecular replacement solution was found using the automated molecular replacement mode in PHASER (McCoy 2007) with a log-likelihood gain of 220.5 and a TFZ score of 16.6. This preliminary model was then initially refined by simulated annealing employing PHENIX (Adams et al. 2010), with two molecules per asymmetric unit. The resulting electron density maps were in good agreement with existing parts of the molecule. However, for regions not included in the model to this point, the electron density was of poor quality and did not allow the construction of missing parts of the molecule. To improve the quality of the electron density map, maximum likelihood density modification (Terwilliger 2000) was carried out, which led to improved electron density maps. These eventually allowed the remaining amino acids to be built with confidence.
The structure of TreS complexed with the amylase inhibitor acarbose was solved by molecular replacement using the uncomplexed TreS structure for phasing. Unfortunately, initial Fobs − Fcalc difference maps did not reveal sufficiently good additional electron density to account for the inhibitor molecule. Notably, however, since the data sets from the uncomplexed and acarbose-complexed crystals had a 99% cross-correlation and showed <1% difference in unit cell dimension and resolution, we were able to calculate a bias free Fobs − Fobs map that clearly demonstrated the differences between these two data sets and produced excellent electron density about the bound acarbose inhibitor. Subsequent refinement of the complexed structure assessed an acarbose occupancy of 57% in the binding site. The average B-factor for the inhibitor at 47 Å2 was found to be ∼40% higher than for the protein (average B = 34 Å2).
Ion binding in both the TreS and TreS/acarbose complex structures was determined by evaluating Fobs − Fcalc difference density maps. Peaks larger than 5σ were screened for spherical electron density shape and specific environments that match ion-binding sites. Candidates were particularly closely evaluated for ligand distances and coordination to electron donors/acceptors with nearby protein or solvent atoms by comparison with literature values (Harding 1999, 2000, 2001, 2004, 2006). All ions added to this structure were subsequently refined using PHENIX (Adams et al. 2010). The backbone dihedral angles for both protein structures were distributed within the most favored (91.2–91.5%) and additionally allowed regions (8.7–8.5%) of the Ramachandran map. One Ramachandran outlier (0.1%) was observed in the complexed structure. Table I contains an extensive statistical analysis of the resultant TreS and TreS/acarbose complex structures obtained.
Funding
This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR—Reference Number (FRN): 111082). S.G.W. is supported by a Tier 1 Canada Research Chair. R.Z. is a recipient of British Columbia Innovation Scholarship.
Conflict of interest
None declared.
Abbreviations
CBM, carbohydrate binding module; PISA, Proteins, Interfaces, Structures and Assemblies software; RMSD, root mean square deviation; TreS, trehalose synthase.
Acknowledgements
We thank Robert Maurus and Leslie Williams for technical advice and helpful discussions. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. doi:10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI, Wilson IA, Corran PH, Furth AJ, Milman JD, Offord RE, et al. Structure of chicken muscle triose phosphate isomerase determined crystallographically at 2.5 angstrom resolution using amino acid sequence data. Nature. 1975;255:609–614. doi: 10.1038/255609a0. doi:10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- Barry CE, 3rd, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog Lipid Res. 1998;37:143–179. doi: 10.1016/s0163-7827(98)00008-3. doi:10.1016/S0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- Barry CE, III, Mdluli K. Drug sensitivity and environmental adaptation of mycobacterial cell wall components. Trends Microbiol. 1996;4:275–281. doi: 10.1016/0966-842x(96)10031-7. doi:10.1016/0966-842X(96)10031-7. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. doi:10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer PD. Mechanism of enzyme action. Annu Rev Biochem. 1960;29:15–44. doi: 10.1146/annurev.bi.29.070160.000311. doi:10.1146/annurev.bi.29.070160.000311. [DOI] [PubMed] [Google Scholar]
- Brayer GD, Luo Y, Withers SG. The structure of human pancreatic alpha-amylase at 1.8 A resolution and comparisons with related enzymes. Protein Sci. 1995;4:1730–1742. doi: 10.1002/pro.5560040908. doi:10.1002/pro.5560040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayer GD, Sidhu G, Maurus R, Rydberg EH, Braun C, Wang YL, Nguyen NT, Overall CH, Withers SG. Subsite mapping of the human pancreatic alpha-amylase active site through structural, kinetic, and mutagenesis techniques. Biochemistry (Mosc) 2000;39:4778–4791. doi: 10.1021/bi9921182. doi:10.1021/bi9921182. [DOI] [PubMed] [Google Scholar]
- Christiansen C, Abou Hachem M, Janecek S, Vikso-Nielsen A, Blennow A, Svensson B. The carbohydrate-binding module family 20—diversity, structure, and function. FEBS J. 2009;276:5006–5029. doi: 10.1111/j.1742-4658.2009.07221.x. doi:10.1111/j.1742-4658.2009.07221.x. [DOI] [PubMed] [Google Scholar]
- Crowe JH, Crowe LM, Chapman D. Preservation of membranes in anhydrobiotic organisms: The role of trehalose. Science. 1984;223:701–703. doi: 10.1126/science.223.4637.701. doi:10.1126/science.223.4637.701. [DOI] [PubMed] [Google Scholar]
- Daffe M, Draper P. The envelope layers of mycobacteria with reference to their pathogenicity. Adv Microb Physiol. 1998;39:131–203. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- De Smet KA, Weston A, Brown IN, Young DB, Robertson BD. Three pathways for trehalose biosynthesis in mycobacteria. Microbiology. 2000;146(Pt 1):199–208. doi: 10.1099/00221287-146-1-199. [DOI] [PubMed] [Google Scholar]
- Dokmanic I, Sikic M, Tomic S. Metals in proteins: Correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr D Biol Crystallogr. 2008;64:257–263. doi: 10.1107/S090744490706595X. doi:10.1107/S090744490706595X. [DOI] [PubMed] [Google Scholar]
- Elbein AD. The metabolism of alpha,alpha-trehalose. Adv Carbohydr Chem Biochem. 1974;30:227–256. doi: 10.1016/s0065-2318(08)60266-8. [DOI] [PubMed] [Google Scholar]
- Elbein AD, Pastuszak I, Tackett AJ, Wilson T, Pan YT. Last step in the conversion of trehalose to glycogen: A mycobacterial enzyme that transfers maltose from maltose 1-phosphate to glycogen. J Biol Chem. 2010;285:9803–9812. doi: 10.1074/jbc.M109.033944. doi:10.1074/jbc.M109.033944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Nurizzo D, Harding SE, Longman E, Davies GJ, Gilbert HJ, Bolam DN. Ligand-mediated dimerization of a carbohydrate-binding molecule reveals a novel mechanism for protein-carbohydrate recognition. J Mol Biol. 2004;337:417–426. doi: 10.1016/j.jmb.2003.12.081. doi:10.1016/j.jmb.2003.12.081. [DOI] [PubMed] [Google Scholar]
- Goldsmith EJ, Fletterick RJ, Withers SG. The three-dimensional structure of acarbose bound to glycogen phosphorylase. J Biol Chem. 1987;262:1449–1455. [PubMed] [Google Scholar]
- Harding MM. The geometry of metal-ligand interactions relevant to proteins. Acta Crystallogr D Biol Crystallogr. 1999;55:1432–1443. doi: 10.1107/s0907444999007374. doi:10.1107/S0907444999007374. [DOI] [PubMed] [Google Scholar]
- Harding MM. The geometry of metal-ligand interactions relevant to proteins. II. Angles at the metal atom, additional weak metal-donor interactions. Acta Crystallogr D Biol Crystallogr. 2000;56:857–867. doi: 10.1107/s0907444900005849. doi:10.1107/S0907444900005849. [DOI] [PubMed] [Google Scholar]
- Harding MM. Geometry of metal-ligand interactions in proteins. Acta Crystallogr D Biol Crystallogr. 2001;57:401–411. doi: 10.1107/s0907444900019168. doi:10.1107/S0907444900019168. [DOI] [PubMed] [Google Scholar]
- Harding MM. The architecture of metal coordination groups in proteins. Acta Crystallogr D Biol Crystallogr. 2004;60:849–859. doi: 10.1107/S0907444904004081. doi:10.1107/S0907444904004081. [DOI] [PubMed] [Google Scholar]
- Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr D Biol Crystallogr. 2006;62:678–682. doi: 10.1107/S0907444906014594. doi:10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- Janecek S, Svensson B, MacGregor EA. Relation between domain evolution, specificity, and taxonomy of the alpha-amylase family members containing a C-terminal starch-binding domain. Eur J Biochem. 2003;270:635–645. doi: 10.1046/j.1432-1033.2003.03404.x. doi:10.1046/j.1432-1033.2003.03404.x. [DOI] [PubMed] [Google Scholar]
- Kaasen I, Falkenberg P, Styrvold OB, Strom AR. Molecular cloning and physical mapping of the otsBA genes, which encode the osmoregulatory trehalose pathway of Escherichia coli: Evidence that transcription is activated by katF (AppR) J Bacteriol. 1992;174:889–898. doi: 10.1128/jb.174.3.889-898.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. doi:10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer R, Jacobs WR., Jr The significance of GlgE as a new target for tuberculosis. Drug News Perspect. 2010;23:619–624. doi: 10.1358/dnp.2010.23.10.1534855. doi:10.1358/dnp.2010.23.10.1534855. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R, Syson K, Veeraraghavan U, Weinrick B, Biermann KE, Liu Z, Sacchettini JC, Besra G, Bornemann S, Jacobs WR., Jr Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an alpha-glucan pathway. Nat Chem Biol. 2010;6:376–384. doi: 10.1038/nchembio.340. doi:10.1038/nchembio.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. doi:10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953;28:416–436. [Google Scholar]
- Krissinel E. Crystal contacts as nature's docking solutions. J Comput Chem. 2010;31:133–143. doi: 10.1002/jcc.21303. doi:10.1002/jcc.21303. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. doi:10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kuriki T, Imanaka T. The concept of the alpha-amylase family: Structural similarity and common catalytic mechanism. J Biosci Bioeng. 1999;87:557–565. doi: 10.1016/s1389-1723(99)80114-5. doi:10.1016/S1389-1723(99)80114-5. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. doi:10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lee H-s, Kim J-s, Shim K-h, Kim J-w, Park C-s, Park K-h. Quaternary structure and enzymatic properties of cyclomaltodextrinase from alkalophilic Bacillus sp. I-5. Biologia Bratislava. 2005;60:73–77. [Google Scholar]
- Levitzki A, Steer ML. The allosteric activation of mammalian alpha-amylase by chloride. Eur J Biochem. 1974;41:171–180. doi: 10.1111/j.1432-1033.1974.tb03257.x. doi:10.1111/j.1432-1033.1974.tb03257.x. [DOI] [PubMed] [Google Scholar]
- Lifshitz R, Levitzki A. Identity and properties of the chloride effector binding site in hog pancreatic alpha-amylase. Biochemistry (Mosc) 1976;15:1987–1993. doi: 10.1021/bi00654a028. doi:10.1021/bi00654a028. [DOI] [PubMed] [Google Scholar]
- Maruta K, Hattori K, Nakada T, Kubota M, Sugimoto T, Kurimoto M. Cloning and sequencing of trehalose biosynthesis genes from Arthrobacter sp. Q36. Biochim Biophys Acta. 1996a;1289:10–13. doi: 10.1016/0304-4165(95)00139-5. doi:10.1016/0304-4165(95)00139-5. [DOI] [PubMed] [Google Scholar]
- Maruta K, Hattori K, Nakada T, Kubota M, Sugimoto T, Kurimoto M. Cloning and sequencing of trehalose biosynthesis genes from Rhizobium sp. M-11. Biosci Biotechnol Biochem. 1996b;60:717–720. doi: 10.1271/bbb.60.717. doi:10.1271/bbb.60.717. [DOI] [PubMed] [Google Scholar]
- Maruta K, Mitsuzumi H, Nakada T, Kubota M, Chaen H, Fukuda S, Sugimoto T, Kurimoto M. Cloning and sequencing of a cluster of genes encoding novel enzymes of trehalose biosynthesis from thermophilic archaebacterium Sulfolobus acidocaldarius. Biochim Biophys Acta. 1996c;1291:177–181. doi: 10.1016/s0304-4165(96)00082-7. doi:10.1016/S0304-4165(96)00082-7. [DOI] [PubMed] [Google Scholar]
- Maurus R, Begum A, Kuo HH, Racaza A, Numao S, Andersen C, Tams JW, Vind J, Overall CM, Withers SG, et al. Structural and mechanistic studies of chloride induced activation of human pancreatic alpha-amylase. Protein Sci. 2005;14:743–755. doi: 10.1110/ps.041079305. doi:10.1110/ps.041079305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. doi:10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miah F, Koliwer-Brandl H, Rejzek M, Field RA, Kalscheuer R, Bornemann S. Flux through trehalose synthase flows from trehalose to the alpha anomer of maltose in mycobacteria. Chem Biol. 2013;20:487–493. doi: 10.1016/j.chembiol.2013.02.014. doi:10.1016/j.chembiol.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosi R, Sham H, Uitdehaag JCM, Ruiterkamp R, Dijkstra BW, Withers SG. Reassessment of acarbose as a transition state analogue inhibitor of cyclodextrin glycosyltransferase. Biochemistry (Mosc) 1998;37:17192–17198. doi: 10.1021/bi981109a. doi:10.1021/bi981109a. [DOI] [PubMed] [Google Scholar]
- Murphy HN, Stewart GR, Mischenko VV, Apt AS, Harris R, McAlister MS, Driscoll PC, Young DB, Robertson BD. The OtsAB pathway is essential for trehalose biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2005;280:14524–14529. doi: 10.1074/jbc.M414232200. doi:10.1074/jbc.M414232200. [DOI] [PubMed] [Google Scholar]
- Oslancova A, Janecek S. Oligo-1,6-glucosidase and neopullulanase enzyme subfamilies from the alpha-amylase family defined by the fifth conserved sequence region. Cell Mol Life Sci. 2002;59:1945–1959. doi: 10.1007/PL00012517. doi:10.1007/PL00012517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YT, Carroll JD, Asano N, Pastuszak I, Edavana VK, Elbein AD. Trehalose synthase converts glycogen to trehalose. FEBS J. 2008;275:3408–3420. doi: 10.1111/j.1742-4658.2008.06491.x. doi:10.1111/j.1742-4658.2008.06491.x. [DOI] [PubMed] [Google Scholar]
- Pan YT, Koroth Edavana V, Jourdian WJ, Edmondson R, Carroll JD, Pastuszak I, Elbein AD. Trehalose synthase of Mycobacterium smegmatis: Purification, cloning, expression, and properties of the enzyme. Eur J Biochem. 2004;271:4259–4269. doi: 10.1111/j.1432-1033.2004.04365.x. doi:10.1111/j.1432-1033.2004.04365.x. [DOI] [PubMed] [Google Scholar]
- Qian M, Haser R, Payan F. Structure and molecular model refinement of pig pancreatic alpha-amylase at 2.1 A resolution. J Mol Biol. 1993;231:785–799. doi: 10.1006/jmbi.1993.1326. doi:10.1006/jmbi.1993.1326. [DOI] [PubMed] [Google Scholar]
- Ravaud S, Robert X, Watzlawick H, Haser R, Mattes R, Aghajari N. Trehalulose synthase native and carbohydrate complexed structures provide insights into sucrose isomerization. J Biol Chem. 2007;282:28126–28136. doi: 10.1074/jbc.M704515200. doi:10.1074/jbc.M704515200. [DOI] [PubMed] [Google Scholar]
- Stam MR, Danchin EG, Rancurel C, Coutinho PM, Henrissat B. Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of alpha-amylase-related proteins. Protein Eng Des Sel. 2006;19:555–562. doi: 10.1093/protein/gzl044. doi:10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- Stein N. CHAINSAW: A program for mutating pdb files used as templates in molecular replacement. J Appl Cryst. 2008;41:641–643. doi:10.1107/S0021889808006985. [Google Scholar]
- Svensson B. Protein engineering in the alpha-amylase family: Catalytic mechanism, substrate specificity, and stability. Plant Mol Biol. 1994;25:141–157. doi: 10.1007/BF00023233. doi:10.1007/BF00023233. [DOI] [PubMed] [Google Scholar]
- Takayama K, Armstrong EL. Isolation, characterization, and function of 6-mycolyl-6′-acetyltrehalose in the H37Ra strain of Myocobacterium tuberculosis. Biochemistry (Mosc) 1976;15:441–447. doi: 10.1021/bi00647a032. doi:10.1021/bi00647a032. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. doi:10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari YB, Goldberg RN. Thermodynamics of hydrolysis of disaccharides. Lactulose, alpha-d-melibiose, palatinose, d-trehalose, d-turanose and 3-o-beta-d-galactopyranosyl-d-arabinose. Biophys Chem. 1991;40:59–67. doi: 10.1016/0301-4622(91)85029-p. doi:10.1016/0301-4622(91)85029-P. [DOI] [PubMed] [Google Scholar]
- Tibbot BK, Wong DWS, Robertson GH. Studies on the C-terminal region of barley α-amylase 1 with emphasis on raw starch-binding. Biologia Bratislava. 2002;57:229–238. [Google Scholar]
- Tsusaki K, Nishimoto T, Nakada T, Kubota M, Chaen H, Sugimoto T, Kurimoto M. Cloning and sequencing of trehalose synthase gene from Pimelobacter sp. R48. Biochim Biophys Acta. 1996;1290:1–3. doi: 10.1016/0304-4165(96)00023-2. doi:10.1016/0304-4165(96)00023-2. [DOI] [PubMed] [Google Scholar]
- Vallee BL, Stein EA, Sumerwell WN, Fischer EH. Metal content of alpha-amylases of various origins. J Biol Chem. 1959;234:2901–2905. [PubMed] [Google Scholar]
- Zhang R, Pan YT, He S, Lam M, Brayer GD, Elbein AD, Withers SG. Mechanistic analysis of trehalose synthase from Mycobacterium smegmatis. J Biol Chem. 2011;286:35601–35609. doi: 10.1074/jbc.M111.280362. doi:10.1074/jbc.M111.280362. [DOI] [PMC free article] [PubMed] [Google Scholar]