

Abstract
We hypothesized that quantitative tandem mass spectrometry-based proteomics at multiple time points, incorporating immunoenrichment prior to rapid microwave and magnetic (IM2) sample preparation, might enable correlation of the relative expression of CD47 and other low abundance proteins to disease progression in the experimental autoimmune encephalomyelitis (EAE) animal model of multiple sclerosis. To test our hypothesis, anti-CD47 antibodies were used to enrich for low abundance CD47 prior to microwave and magnetic (M2) proteomics in EAE. Decoding protein expression at each time point, with CD47-immunoenriched samples and targeted proteomic analysis, enabled peptides from the low abundance proteins to be precisely quantified throughout disease progression, including: CD47: 86-99, corresponding to the “marker of self” overexpressed by myelin that prevents phagocytosis, or “cellular devouring”, by microglia and macrophages; MBP: 223-228, corresponding to myelin basic protein; and MIF: 79-87, corresponding to a proinflammatory cytokine that inhibits macrophage migration. While validation in a larger cohort is underway, we conclude that IM2 proteomics is a rapid method to precisely quantify peptides from CD47 and other low abundance proteins throughout disease progression in EAE. This is likely due to improvements in selectivity and sensitivity, necessary to partially overcome masking of low abundance proteins by high abundance proteins and improve dynamic range.
Keywords: Isobaric chemical labeling, Microwave proteomics, Multiple sclerosis, Sample preparation, CD47
1 INTRODUCTION
Inflammatory demyelination during experimental autoimmune encephalomyelitis (EAE) and multiple sclerosis generates myelin debris in the brain. Many phagocytically active microglia and central nervous system (CNS)-infiltrating macrophages (MΦs) containing myelin debris have been identified at the site of active inflammatory lesions from multiple sclerosis patients [1] and in the murine EAE model [2]. However, it remains unresolved whether there are prognostic/predictive protein biomarkers and therapeutic targets that reflect these phagocytic phenomena that could help to improve early detection, diagnosis, prognosis and treatment of patients with multiple sclerosis.
CNS-infiltrating MΦs are most likely important in promoting disease progression, while phagocytosis of myelin debris by phagocytes may be beneficial for surveillance, scavenging and synaptic pruning [3–9]. Phagocytosis of intact myelin or myelin debris by MΦs is promoted by complement protein 3bi (C3bi) -receptor 3 (CR3) interactions and inhibited by CD47-signal regulatory protein alpha (SIRPα) interactions. In 1990, Bruck et al discovered that C3bi, bound to the Fc domains of anti-myelin debris-specific antibodies, opsonized myelin debris to accelerate phagocytosis by CR3+ MΦs [10–12]. In 2011, Gitik et al discovered that recombinant anti-CD47-antibodies opsonized CD47+myelin debris to accelerate FCγR-mediated phagocytosis by SIRPα+ MΦs [9]. Thus, the CD47 protein expressed by intact myelin or myelin debris may be an important clue to the molecular dynamics of CNS repair during demyelinating CNS diseases and serve as a potentially important biomarker or therapeutic target.
We hypothesized that quantitative tandem mass spectrometry (MS/MS)-based proteomics at multiple time points, incorporating immunoenrichment prior to rapid microwave and magnetic (IM2) sample preparation might enable correlation of the relative expression of CD47 and other low abundance proteins to disease progression in EAE. IM2 proteomics was inspired by reports of affinity proteomics [13, 14] where immunodepletion of high abundance proteins and/or immunoenrichment of low abundance proteins was used to overcome masking problems and improve dynamic range. To test our hypothesis, anti-CD47 antibodies were used to enrich for low abundance CD47 prior to microwave-assisted reduction/alkylation/digestion of proteins from brain tissue lysates bound to C8 magnetic beads. Then, microwave-assisted isobaric chemical labeling of released peptides was performed for all samples spanning disease progression and pooled reference material from the peak of disease. This was achieved in a total of 90 seconds prior to unbiased and targeted proteomic analysis. Decoding protein expression at each time point, with CD47-immunoenriched samples and targeted proteomic analysis, enabled peptides from the low abundance proteins to be precisely quantified throughout disease progression, including: CD47: 86-99; MBP: 223-228; and MIF: 79-87.
2 MATERIAL & METHODS
2.1 Murine Experimental Autoimmune Encephalomyelitis (EAE)
C57BL/6 female 5 week-old mice were purchased from the Jackson Laboratory (Stock number 000664; Bar Harbor, ME). Mice were maintained under specific pathogen-free conditions and all animal procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee of the University of Texas at San Antonio. Active induction of EAE was performed with a subcutaneous injection of each mouse with 300 μg of myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide (United Biochemical Research, Seattle, WA) in 50 μL of complete Freund’s adjuvant (CFA) containing Mycobacterium tuberculosis H37 RA (Difco Laboratories, Detroit, MI) at a final concentration of 5 mg/mL. Two intra-peritoneal (i.p.) injections of pertussis toxin (List Biological, Campbell, CA) at 200 ng per mouse were given at the time of immunization and 48 hours later. Animals were monitored and graded daily for clinical signs of EAE using the following scoring system [15]: 0, no abnormality; 1, limp tail; 2, moderate and hind limb weakness; 3, complete hind limb paralysis; 4, quadriplegia or premoribund state; 5, death. EAE scores are presented as the mean ± standard deviation and were confirmed by histopathology (data not shown). Mice were sacrificed at 5 disease time points, described by the number of days (d) post-immunization (−1 d (non-immunized), 0 d (3 hr post-immunization), 10 d, 20 d and 25 d) in biological quadruplicate (n = 4 per time point). Half of all brain tissue was snap-frozen in liquid nitrogen and stored at −80°C for IM2 proteomics and Western blotting while the remainder was used for cytokine measurement and immunofluorescence analysis for inflammatory infiltrates (data not shown).
2.2 Brain Tissue Lysate
Whole cell protein was extracted from brain tissue using the RIPA lysis Buffer Kit (Santa Cruz Biotechnology, Inc. Santa Cruz, CA.) according to the manufacturer’s protocol. Briefly, an appropriate amount of RIPA complete lysis buffer was added to cell pellet. The mixture was incubated on ice for 5 min, followed by centrifugation at 14 000 × g for 15 min at 4°C. The supernatant was collected as brain tissue lysate and stored at −80°C until further use. Protein concentration was determined using Invitrogen EZQ Protein Quantitation Kit (Invitrogen, Grand Island, NY). Protein from mice at the peak of disease (day 20) was pooled (n = 4) as reference material.
2.3 Immunoenrichment
Immunoenrichment of CD47 was performed by precipitation with anti-CD47 antibodies for this low abundance protein. To 500 μg of brain tissue lysate from control and EAE mice, 25 μL of anti-CD47 antibody (sc-12731, Santa Cruz Biotechnology, CA) was added and incubated overnight at 4°C on shaker. After incubation, 50 μL each of protein G plus agarose beads (sc-2002, Santa Cruz Biotechnology, CA) was added prior to overnight shaking at 4°C. Centrifugation was carried out at 3000 rpm for 30 seconds at 4°C and washed (4x) with 150 μL of RIPA lysis buffer (sc-24948, Santa Cruz Biotechnology, CA). Supernatant was discarded and the pellets were re-suspended in 50 μL of 1% SDS buffer, and boiled for 10 min at 95°C. Next, 50 μL of HPLC grade water is added to the tubes, vortexed and centrifuged to collect the eluent or supernatant. The total eluent collected was 100 μL. Protein concentration of eluents were estimated as described above.
2.4 Microwave & Magnetic (M2) Sample Preparation
C8 magnetic beads (BcMg, Bioclone Inc., San Diego, CA) were used in this study. Briefly, 50 mg of beads were suspended in 1 mL of 50% methanol. Immediately before use, 100 μl of the beads were washed 3 times with equilibration buffer (200 mM NaCl, 0.1% trifluoroacetic acid (TFA)). Brain tissue lysate (25–100 μg at 1 μg/μL) was mixed with pre-equilibrated beads and 1/3rd sample binding buffer (800 mM NaCl, 0.4% TFA) by volume. The mixture was incubated at room temperature for 5 min followed by removing the supernatant. The beads were washed twice with 150 μL of 40 mM triethylammonium bicarbonate (TEAB), and then 150 μL of 10 mM dithiolthreitol (DTT) was added followed by microwave heating for 10 s. DTT solution was then removed and 150 μL of 50 mM iodoacetamide (IAA) was added followed by microwave heating for 10 s. Next, beads were washed twice with 150 μL of 40 mM TEAB and resuspended in 150 μL of 40 mM TEAB. In vitro proteolysis was performed with 4 μL of trypsin in a 1:25 trypsin-to-protein ratio (stock = 1 μg/μL in 50 mM acetic acid) and microwave heated for 20 s in triplicate. The supernatant was transferred to a new tube for immediate use or stored at −80°C. Released tryptic peptides from digested brain tissue lysates, including the reference material described above, were modified at the N-terminus and at lysine residues with the tandem mass tagging (TMT)-6plex isobaric labeling reagents (Thermo scientific, San Jose, CA). Each biological replicate (n = 4–5) for each time point was encoded with one of the TMT-127-130 reagents, while pooled reference material from −1 d (non-immunized) or 20 d (disease peak) was encoded with the TMT-126 or TMT-131 reagents, respectively. Then, 41 μL of anhydrous acetonitrile was added to 0.8 mg of TMT labeling reagent and 25μg of brain tissue lysate was added and microwave-heated for 10 s. To quench the reaction, 8 μL of 5% hydroxylamine was added to the sample at room temperature. To normalize across the time-course of disease progression, TMT-encoded brain tissue lysates from individual mice, randomly labeled with the TMT-127-130 reagents to improve confidence in our results (Supplementary Table 1), were mixed with the reference materials encoded with the TMT-126 and TMT-131 reagents in 1126:1127:1128:1129:1130:1131 ratios. These sample mixtures, I–V, corresponding to 5 disease time points, were stored at −80°C until further use.
2.5 Capillary Liquid Chromatography-Fourier-Transform-Tandem Mass Spectrometry (LC/FT-MS/MS) with Protein Database Searching
Capillary LC/-FT-MS/MS was performed with a splitless nanoLC-2D pump (Eksigent, Livermore, CA), a 50 μm-i.d. column packed with 7 cm of 3 μm-o.d. C18 particles, and a hybrid linear ion trap-Fourier-transform tandem mass spectrometer (LTQ-ELITE; ThermoFisher, San Jose, CA) operated with a lock mass for calibration. The reverse-phase gradient was 2 to 62% of 0.1% formic acid (FA) in acetonitrile over 60 min at 350 nL/min. For unbiased analyses, the top 6 most abundant eluting ions were fragmented by data-dependent HCD in centroid mode (mass resolution 60 000 for precursor ions and 15 000 for product ions) with dynamic exclusion enabled and a 4 m/z isolation width. Probability-based (and error-tolerant) protein database searching of MS/MS spectra against the IPI mouse protein database (release 2010_jan10; 56 729 sequences) were performed with a 10-node MASCOT cluster (ver. 2.3.02, Matrix Science, London, UK) to identify and quantify proteins. Search criteria included: peak picking with Mascot Distiller; 10 ppm precursor ion mass tolerance, 0.8 Da product ion mass tolerance, 3 missed cleavages, trypsin, carbamidomethyl cysteines and oxidized methionines as variable modifications, an ion score threshold of 20 and TMT-6-plex for quantification. For targeted analyses, only ions corresponding to peptides observed for selected proteins were fragmented by HCD in profile mode (mass resolution 15 000 for product ions) with a 4 m/z isolation width, identification by protein database searching was not performed, and quantification was performed by calculating the signal-to-noise ratios of TMT reporter product ions.
2.6 Statistical Analysis
The IM2 proteomics results for each technical replicate estimate protein expression for individual mice, encoded in sample mixtures I–V, relative to pooled reference material at disease peak (day 20). The hypothesis that relative protein expression changes over time was tested using ANOVA, treating time as a multilevel factor. Relative peptide expression, corresponding to relative protein expression, at each time point (10 d, 20 d and 25 d) was contrasted with both control time points, −1 d (non-immunized) or 0 d (3 hr post-immunization), using a two-sample t-test without assuming equality of variance. All tests were two-sided with no adjustments for multiple testing. Pearson correlation coefficients were computed to identify the relationships between relative protein expression and EAE Score. Statistical analysis was performed with R v2.13 (R-Project, Vienna, Austria).
2.7 Western Blotting
Immunoenriched samples (25 μg at 1 μg/mL) were treated with βME (5% by volume) prior to boiling for 10 minutes and separating proteins in each sample with SDS-PAGE precast gels (456-9034, Bio-Rad, CA) for one-dimensional electrophoresis. Then proteins were transferred to a 0.2 μm nitro-cellulose membrane (162-0212, Bio-Rad, CA) followed by blocking with 5% milk in TBS for 1 hr. The membrane was then washed with TBS buffer for 5 min in triplicate and probed with (1:250) rat monoclonal IgG2a primary anti-CD47 antibody (sc-12731, Santa Cruz Biotechnology, CA) in TBS with overnight incubation. Detection was performed by 1 hr incubation with (1:2500) goat anti-rat IgG HRP-conjugated secondary antibody (sc-2006, Santacruz Biotechnology, CA) in TBS prior to chemiluminescence measurement using X-ray film and Luminol reagents (sc-2048, Santacruz Biotechnology, CA).
3 RESULTS
EAE was investigated by quantitative MS/MS-based proteomics at multiple time points, incorporating immunoenrichment prior to rapid microwave and magnetic (IM2) sample preparation, as a model for correlating the relative protein expression of low abundance proteins to disease progression in multiple sclerosis patients. Shown in Figure 1 is a schematic of the IM2 proteomics experimental design used in our study (top and middle) superimposed on a plot of EAE disease score vs. post-disease induction time (bottom). Anti-CD47 antibodies were used to enrich for low abundance CD47 prior to microwave-assisted reduction/alkylation/digestion of proteins from brain tissue lysates bound to C8 magnetic beads. Then, microwave-assisted isobaric chemical labeling of released peptides was performed for all 32 samples spanning disease progression and pooled reference material from the peak of disease. This was achieved in a total of 90 seconds prior to unbiased and targeted proteomic analysis. Isobaric-encoding enabled 6-plex quantitative proteomic analysis of 5 sample mixtures, corresponding to 5 disease time points, where each mixture included 4 mice per time point and 2 pooled reference materials. Disease progression in EAE was assessed by scoring clinical EAE disease severity and confirmed by histopathologic evaluation for CNS inflammation (data not shown). Clinical onset of disease was observed on days 8–10, followed by disease peak at days 19–20 and remission by day 23.
Figure 1.

Schematic of IM2 proteomics. The IM2 proteomics experimental design used in our study (top and middle) is superimposed on a plot of EAE disease score vs. post-disease induction time (bottom). Anti-CD47 antibodies were used to enrich for low abundance CD47 prior to microwave-assisted reduction/alkylation/digestion of proteins from brain tissue lysates bound to C8 magnetic beads. Then, microwave-assisted isobaric chemical labeling of released peptides was performed for all 32 samples spanning disease progression and pooled reference material from the peak of disease. This was achieved in a total of 90 seconds prior to unbiased and targeted proteomic analysis. Isobaric-encoding enabled 6-plex quantitative proteomic analysis of 5 sample mixtures (such as the those shown in boxes), corresponding to 5 disease time points, where each mixture included 4 mice per time point (red, blue yellow, green) and pooled reference materials (white and pink). Clinical onset of disease was observed on days 8–10, followed by disease peak at days 19–20 and remission at day 23.
Three different methods were compared to determine whether or not immunoenrichment and targeted proteomics enhanced the precision of peptide quantification from low abundance proteins throughout disease progression, including: CD47: 86-99; MBP: 223-228; and MIF: 79-87. These peptides were selected because they were not precisely quantified in our previous work by microwave and magnetic (M2) proteomics [19]). Figure 2 shows the amino acid sequence coverage and peptides that were typically observed for CD47 by unbiased M2 proteomics. Also illustrated is the expected trend in the precision of peptide quantification for the methods investigated in this work (A < B < C). In method A, no immunoenrichment of CD47 was performed prior to unbiased M2 proteomics. In methods B and C, immunoenrichment of CD47 was performed prior to unbiased (B) or targeted (C) M2 proteomics, respectively. Figure 3 shows a representative annotated MS/MS spectrum, with sequence-specific b- (N-terminal) and y- (C-terminal) type product ions for the triply-charged precursor ion corresponding to peptide CD47: 86-99 with the amino acid sequence ISVSDLINGIASLK. The relative abundance of TMT reporter ions, corresponding to the relative abundance of this peptide and its corresponding protein in representative individual mice, is shown in the inset. Figure 4 shows qualitative (Mascot score) and/or quantitative (relative abundance) measures for peptide CD47: 86-99 by the three proteomics methods described in Figure 2. Error bars for quantitative measures show the standard error of the mean, a measure of precision. Method A, no immunoenrichment and unbiased proteomic analysis, was incapable of identifying or quantifying CD47: 86-99 throughout disease progression. While CD47: 86-99 was identified in a few samples, it was never quantified by method A. Method B, immunoenrichment and unbiased proteomic analysis, was capable of identifying CD47: 86-99 throughout disease progression, but incapable of quantifying CD47: 86-99 without significant error. Only method C, incorporating both immunoenrichment and targeted proteomic analysis, was capable of precisely quantifying CD47: 86-99 throughout disease progression. (Western blotting of immunoenriched samples confirmed that CD47 was expressed throughout disease progression (Supplementary Figure 1). Western blotting without prior immunoprecipitation was not successful, supporting the notion that CD47 is a low abundance protein in the brain.)
Figure 2.
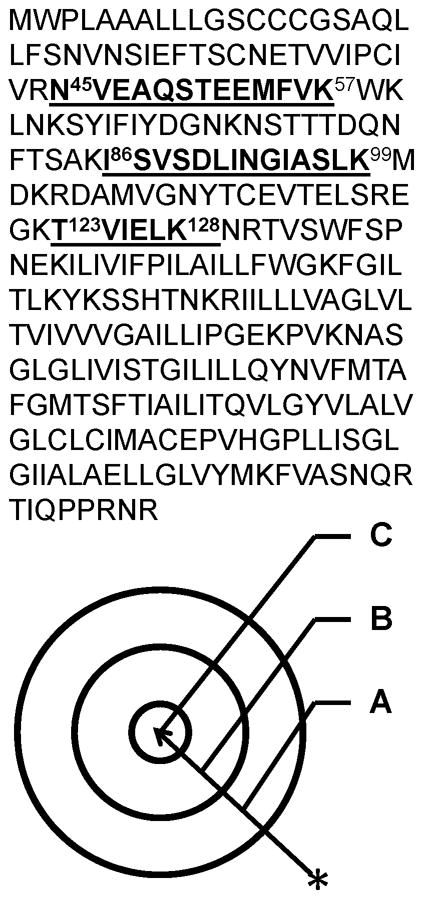
Method development. The amino acid sequence coverage and peptides that we typically observe for CD47 by unbiased M2 proteomics (top). Also illustrated is the expected trend in the precision of peptide quantification for the methods investigated in this work (A < B < C) (bottom). In method A, no immunoenrichment of CD47 was performed prior to unbiased M2 proteomics. In methods B and C, immunoenrichment of CD47 was performed prior to unbiased (B) or targeted (C) M2 proteomics, respectively.
Figure 3.

A representative annotated MS/MS spectrum from method C, with sequence-specific b- (N-terminal) and y- (C-terminal) type product ions, for the triply-charged precursor ion corresponding to peptide CD47: 86-99 with the amino acid sequence ISVSDLINGIASLK. The relative abundance of TMT reporter ions, corresponding to the relative abundance of this peptide and its corresponding protein in representative individual mice, is shown in the inset.
Figure 4.

Method comparison. Shown are qualitative (Mascot score) and/or quantitative (relative abundance) measures for peptide CD47: 86-99 by the three proteomics methods described in Figure 2. Error bars for quantitative measures show the standard error of the mean, a measure of precision. Method A, no immunoenrichment and unbiased proteomic analysis, was incapable of identifying or quantifying CD47: 86-99 throughout disease progression. While CD47: 86-99 was identified in a few samples, it was never quantified by method A. Method B, immunoenrichment and unbiased proteomic analysis, was capable of identifying CD47: 86-99 throughout disease progression, but incapable of quantifying CD47: 86-99 without significant error. Only method C, incorporating both immunoenrichment and targeted proteomic analysis, was capable of precisely quantifying CD47: 86-99 throughout disease progression.
Decoding protein expression at each time point, with CD47-immunoenriched samples and targeted proteomic analysis (method C), enabled peptides from low abundance proteins to be precisely measured throughout disease progression, including: CD47: 86-99, MBP: 223-228, and MIF: 79-87. Figure 5 shows the correlation of the relative expression of these peptides to EAE score throughout disease progression: CD 47:85-99 (5A); MBP 223-228 (5B); and MIF 79-87 (5C). Relative peptide expression, corresponding to relative protein expression, at each time point (10 d, 20 d and 25 d) was contrasted with both control time points, −1 d (non-immunized, red) or 0 d (3 hr post-immunization, blue), using a two-sample t-test without assuming equality of variance. The following time points were the most significant vs. the −1 d (red) or 0 d (blue) controls, respectively: CD47: 86-99 (20 d, P = 0.0925; 10 d, P = 0.0487;), MBP: 223-228 (0 d, P = 0.1671; 10 d, P = 0.1019), and MIF: 79-87 (20 d, P = 0.0119; 20 d, P = 0.0383).
Figure 5.

Correlation of the relative expression of three low abundance peptides to EAE score throughout disease progression: CD 47:85-99 (5A); MBP 223-228 (5B); and MIF 79-87 (5C). Relative peptide expression, corresponding to relative protein expression, at each time point (10 d, 20 d and 25 d) was contrasted with both control time points, −1 d (non-immunized, blue) or 0 d (3 hr post-immunization, red), using a two-sample t-test without assuming equality of variance.
4 DISCUSSION
Below, we provide an overview of our results by IM2 proteomics, a comparison to previous proteomics work, and an introduction to the biological functions of CD47, MBP and MIF. Immunoenrichment was performed solely for CD47 in these experiments. Therefore, co-purification of MBP and MIF may be due to non-specific or specific protein-protein interactions. While beyond the scope of this work, a search of the Ingenuity Knowledgebase showed no known interactions among these low abundance proteins. Lastly, we discuss how our results may provide some insights on the unanswered questions posed in the introduction.
4.1 IM2 Proteomics
This proof-of-principle study shows that quantitative MS/MS-based proteomics at multiple time points, incorporating immunoenrichment prior to rapid microwave and magnetic (IM2) sample preparation and targeted proteomic analysis, enables low abundance protein expression to be correlated to disease progression in EAE. Peptides from low abundance proteins were precisely quantified throughout disease progression, including: CD47: 86-99, corresponding to the “the marker of self” overexpressed by myelin that prevents phagocytosis, or “cellular devouring”, by MΦs; MBP: 223-228, corresponding to myelin basic protein; and MIF: 79-87, corresponding to a proinflammatory cytokine. As described above, clinical onset of disease was observed on days 8–10, followed by disease peak at days 19–20 and remission at day 23. Therefore, the most statistically significant times for changes in CD47: 86-99 (10 d) and MBP: 223-228 (10 d) expression coincided with clinical onset of disease (8–10 d), while the most statistically significant time for MIF: 79-87 (20 d) expression was closer in time to disease peak (20 d).
Only method C, incorporating both immunoenrichment and targeted proteomic analysis, was capable of precisely quantifying CD47: 86-99 throughout disease progression (Other combinations of methods, with intermediate levels of expected improvements in selectivity and sensitivity, were not investigated in this work because we were focused on developing the most precise method to quantify CD47. However, such methods, including no immunoenrichment and targeted proteomic analysis, may prove ideal for certain applications). This is likely due to the improved selectivity and sensitivity for method C compared to methods A and B, necessary to partially overcome masking of low abundance proteins by high abundance proteins and improve dynamic range. The improved selectivity of method C is most simply explained by the removal of ~ 1191 high abundance proteins [19] by immunoenrichment of low abundance CD47 prior to in vitro proteolysis. Masking of co-eluting low abundance peptides by high abundance peptides is primarily due to sequence-specific competition for charge by peptide precursor ions during the nanoelectrospray ionization process (a.k.a., ion suppression) [20]. The improved sensitivity of method C is most simply explained by the replacement of data-dependent fragmentation of peptides in centroid mode with dynamic exclusion enabled (unbiased proteomic analysis) by targeted proteomic analysis of peptides in profile mode. Unbiased proteomic analysis is ideal for maximizing the number of unique high quality MS/MS spectra, corresponding to unique peptide sequences, that can be collected in a given time (i.e., the duty cycle). However, undersampling can be a problem when complex mixtures are investigated with isobaric labeling strategies such as TMT, where quantification occurs in the MS/MS dimension. The reason for this is that MS/MS spectra are not always collected from the apex of the chromatographic peak for a given peptide [21]. Sometimes the quality of MS/MS spectra, not collected at the apex, can be sufficient to assign a peptide sequence with high confidence (i.e., identify) but insufficient to precisely measure (TMT) reporter product ions (i.e., quantify). In contrast, the high duty cycle of targeted proteomic analysis insures that the highest quality MS/MS spectra are collected from the apex. Collecting MS/MS spectra in profile mode, rather than centroid mode, provides additional insurance that the highest quality MS/MS spectra are collected for quantification purposes. Profile mode data can also provide oversampling benefits that are important for quantitative peak picking algorithms (unbiased proteomic analysis in profile mode is uncommon for isobaric labeling strategies because of large, unwieldy file sizes and the relatively low resolving power (e.g., 15 000) needed to distinguish low mass, singly-charged reporter ions to quantify in the MS/MS dimension). Lastly, the benefits of using signal-to-noise ratios to quantify low abundance peptides, as performed herein for TMT reporter product ions by method C, has been previously shown [22].
4.2 Comparison of IM2 Proteomics to Previous Proteomics Work
Few studies have focused on precisely quantifying peptides from low abundance proteins in EAE or multiple sclerosis specimens. Previously, we identified, but did not quantify, CD47 in the murine EAE model by laser capture microdissection (LCM) followed by fractionation by SDS-PAGE and capillary LC/MS/MS with protein database searching (data not shown). Likewise, CD47, MBP and MIF were recently identified, but not quantified, by LCM of post-mortem brain tissue specimens from multiple sclerosis patients followed by MS/MS-based proteomics [23]. LCM proteomics of large-scale murine EAE studies requires impractically lengthy sample preparation times to pool the relatively large number of small-sized mouse brain lesions needed for proteomic analysis. While our recently reported quantitative MS/MS method (M2 proteomics [19]) was superior to LCM proteomics for murine EAE studies, it was still difficult to precisely quantify low abundance proteins such as CD47 throughout disease progression. Unlike the other proteomics methods described above, IM2 proteomics precisely quantified peptides from low abundance proteins such as CD47 throughout disease progression in murine EAE. This is likely due to the improved selectivity and sensitivity of IM2 proteomics compared to previous work, necessary to partially overcome masking of low abundance proteins by high abundance proteins and improve dynamic range. Moreover, IM2 proteomics can be readily adapted to an automated, 384-well plate format for very high-throughput sample preparation strategies needed to study low abundance biomarkers in a large number of specimens. Compared to Western blotting, MS/MS-based methods such as IM2 proteomics are more selective, enabling precise and accurate quantification of proteins by specific amino acid residues and post-translational modifications. Moreover, methods that incorporate isobaric labeling strategies for encoding samples, such as IM2 proteomics, enable significant multiplexing advantages over Western blotting. For these reasons, we believe that IM2 proteomics of brain tissue lysates is superior to other methods for quantifying low abundance proteins in EAE studies.
4.3 CD47
Peptide CD47: 86-99, with the amino acid sequence ISVSDLINGIASLK, corresponds to CD47, the “marker of self” overexpressed by myelin that prevents phagocytosis, or “cellular devouring”, by harmful phagocytically active microglia and CNS-infiltrating MΦs [24–27]. CD47 acts as a ligand for signal regulatory protein alpha (SIRPα). It is a member of the immunoglobulin (Ig) superfamily and was originally identified in association with the integrin αvβ3, possessing a V-type Ig-like extracellular domain, five putative membrane-spanning segments and a cytoplasmic tail [28]. Integrin β3 subunit is associated with the extracellular region of CD47. Thus CD47 is also called integrin associated protein [29]. Most CD47-mediated cellular responses involve activation of integrins, particularly αvβ3 or αIIBβ3 but the mechanism involved in the activation is not clearly understood. Various groups hypothesize that a part of CD47 function is mediated by binding of the pertussis toxin-sensitive heterotrimeric G protein to the extracellular Ig-domain of CD47 [28, 30]. The unknown function and role of CD47 in integrin β3 signal transduction suggest that there could be various tissue-specific isoforms.[31]. The Ig-V domain has five segments and a cytoplasmic tail which are alternatively spliced to give rise to four isoforms. Isoform 2 is the most abundant and isoform 4, with the longest cytoplasmic extension, is the second most abundant in neurons, intestine and testis. There is no known motif for enzymatic activity or protein interaction for any of these cytoplasmic interactions [32].
4.4 MBP
Peptide MBP: 223-228, with the amino acid sequence NIVTPR, corresponds to myelin basic protein (MBP). MBP is the second most abundant protein in the central nervous system, comprising ~ 30% of the total protein in myelin [33]. MBP is a positively charged protein that binds to negatively charged lipids, present at the cytosolic surface of myelin. Alternative splicing and post-translational modifications generate numerous MBP isoforms [33–39]. One of the charged isoforms, known as C8, is overexpressed in multiple sclerosis patients and may be involved in its pathogenesis [40]. Another important post-translational modification is proteolytic processing of MBP by calcium-dependent enzymes, such as calpain [41–44].
4.5 MIF
Peptide MIF: 79-87, with the amino acid sequence LLCGLLSDR, corresponds to macrophage migration inhibitory factor (MIF). MIF is a proinflammatory cytokine involved in autoimmune disease that may counter the effects of interleukin-2 [45]. MIF has also been linked to various proliferative, anti-apoptotic, inflammatory and angiogenic survival pathways of p53, CD74, NFκB, Bcl2, IL-8 and VEGF [46, 47]. Akin to the anti-inflammatory actions of glucocorticoids, anti-MIF antibodies have also been shown to ameliorate EAE [48].
4.6 Conclusions and outlook
While more time points and mice are needed to clearly define the expression trajectories of these low abundance proteins, our results showed that the most statistically significant correlations for CD47 and MBP expression to EAE score coincided with clinical onset of disease, while the most statistically significant correlation for MIF expression to EAE score was closer in time to disease peak. This is in general agreement with previous work showing that CD47 was decreased in active inflammatory lesions [8, 49] (at disease peak), while phagocytically active MΦs containing myelin debris were increased in active inflammatory lesions [1, 2] (at disease peak). In one of these studies, CD47 messenger RNA (mRNA) was identified and quantified as the target of microRNA (miR) by LCM of post-mortem brain tissue specimens followed by quantitative PCR and immunohistochemical analysis for CD47 protein [8]. Expression of miR-34a, miR-155 and miR-326 was increased while CD47 protein was decreased in active inflammatory lesions. This was confirmed by another study quantifying CD47 mRNA [49]. Based on our own results and previous work, we posit that disease progression in EAE may reflect the transition of beneficial phagocytosis of CD47+ myelin debris to harmful phagocytosis of CD47+ intact myelin in active inflammatory lesions and back again. This transition may be concordantly regulated with the expression of CD47 and proteins involved in demyelination and inflammation such as MBP and MIF. The mechanism, if any, by which CD47+ intact myelin and CD47+ myelin debris are distinguished by phagocytically active MΦs remains an intriguing biological question. Nonetheless, discovering protein biomarkers of phagocytic phenomena that reflect distinct disease stages, such as disease onset, peak, and remission, is expected to provide critical information that may result in improved early detection, diagnosis, prognosis and treatment monitoring of patients with multiple sclerosis.
While validation in a larger cohort is underway, we conclude that IM2 proteomics is a rapid method to precisely quantify peptides from CD47 and other low abundance proteins throughout disease progression in EAE. This is likely due to improvements in selectivity and sensitivity, necessary to partially overcome masking of low abundance proteins by high abundance proteins and improve dynamic range. The future application of IM2 proteomics to longitudinal measurements of multiple low abundance peptides from brain protein biomarkers and therapeutic targets in blood is expected to be important for correlating to other quantitative measures of disease progression such as magnetic resonance imaging and/or the expanded disability status scale disease score in patients with multiple sclerosis. IM2 proteomics may also illuminate future investigations on the biochemistry of CD47 and myelin.
Supplementary Material
Western blotting of immunoenriched samples confirmed IM2 proteomics results showing that CD47 was expressed throughout disease progression.
TMT labeling scheme. To normalize across the time-course of disease progression, TMT-encoded brain tissue lysates from individual mice, randomly labeled with the TMT-127-130 reagents to improve confidence in our results, were mixed with the reference materials encoded with the TMT-126 and TMT-131 reagents in 1126:1127:1128:1129:1130:1131 ratios.
Acknowledgments
This work was supported by grants NIH5G12RR013646-12 (WEH, TGF), NS52177 (TGF), and NIH5U54RR022762-05 (WEH) from the National Institute of Health, and grant RG3701 from the National Multiple Sclerosis Society (TGF). We thank the RCMI program and facilities at UTSA for assistance. The authors also acknowledge the support of the Cancer Therapy and Research Center at the University of Texas Health Science Center San Antonio, a National Cancer Institute -designated Cancer Center (NIHP30CA054174).
References Cited
- 1.Bruck W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, Lassmann H. Ann Neurol. 1995;38:788–796. doi: 10.1002/ana.410380514. [DOI] [PubMed] [Google Scholar]
- 2.Bauer J, Sminia T, Wouterlood FG, Dijkstra CD. J Neurosci Res. 1994;38:365–375. doi: 10.1002/jnr.490380402. [DOI] [PubMed] [Google Scholar]
- 3.Neumann H, Kotter MR, Franklin RJ. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz M. J Cereb Blood Flow Metab. 2003;23:385–394. doi: 10.1097/01.WCB.0000061881.75234.5E. [DOI] [PubMed] [Google Scholar]
- 5.Aldskogius H. Microsc Res Tech. 2001;54:40–46. doi: 10.1002/jemt.1119. [DOI] [PubMed] [Google Scholar]
- 6.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Science. 2011 doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Cardona AE. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 8.Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E. Brain: a journal of neurology. 2009;132:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 9.Gitik M, Liraz-Zaltsman S, Oldenborg PA, Reichert F, Rotshenker S. J Neuroinflammation. 2011;8:24. doi: 10.1186/1742-2094-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruck W, Friede RL. Acta Neuropathol. 1990;80:415–418. doi: 10.1007/BF00307696. [DOI] [PubMed] [Google Scholar]
- 11.Kreutzberg GW. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 12.Bryn T, Mahic M, Enserink JM, Schwede F, Aandahl EM, Tasken K. J Immunol. 2006;176:7361–7370. doi: 10.4049/jimmunol.176.12.7361. [DOI] [PubMed] [Google Scholar]
- 13.Xie S, Moya C, Bilgin B, Jayaraman A, Walton SP. Expert Rev Proteomics. 2009;6:573–583. doi: 10.1586/epr.09.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joos T. Proteomics. 2009;9:1418–1419. doi: 10.1002/pmic.200990019. [DOI] [PubMed] [Google Scholar]
- 15.Nekrasova T, Shive C, Gao Y, Kawamura K, Guardia R, Landreth G, Forsthuber TG. J Immunol. 2005;175:2374–2380. doi: 10.4049/jimmunol.175.4.2374. [DOI] [PubMed] [Google Scholar]
- 16.Shive CL, Hofstetter H, Arredondo L, Shaw C, Forsthuber TG. Eur J Immunol. 2000;30:2422–2431. doi: 10.1002/1521-4141(2000)30:8<2422::AID-IMMU2422>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 17.Heeger PS, Forsthuber T, Shive C, Biekert E, Genain C, Hofstetter HH, Karulin A, Lehmann PV. J Immunol. 2000;164:5771–5781. doi: 10.4049/jimmunol.164.11.5771. [DOI] [PubMed] [Google Scholar]
- 18.Hofstetter HH, Forsthuber TG. Neurosci Lett. 2010;476:150–155. doi: 10.1016/j.neulet.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 19.Haskins WE. Electrophoresis. In Press. [Google Scholar]
- 20.Hirabayashi A, Ishimaru M, Manri N, Yokosuka T, Hanzawa H. Rapid Commun Mass Spectrom. 2007;21:2860–2866. doi: 10.1002/rcm.3157. [DOI] [PubMed] [Google Scholar]
- 21.Blackburn K, Mbeunkui F, Mitra SK, Mentzel T, Goshe MB. J Proteome Res. 2010;9:3621–3637. doi: 10.1021/pr100144z. [DOI] [PubMed] [Google Scholar]
- 22.Bakalarski CE, Elias JE, Villen J, Haas W, Gerber SA, Everley PA, Gygi SP. J Proteome Res. 2008;7:4756–4765. doi: 10.1021/pr800333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 24.Tsai RK, Discher DE. J Cell Biol. 2008;180:989–1003. doi: 10.1083/jcb.200708043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai RK, Rodriguez PL, Discher DE. Blood Cells Mol Dis. 2010;45:67–74. doi: 10.1016/j.bcmd.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van VQ, Lesage S, Bouguermouh S, Gautier P, Rubio M, Levesque M, Nguyen S, Galibert L, Sarfati M. EMBO J. 2006;25:5560–5568. doi: 10.1038/sj.emboj.7601415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 28.Brown E. J Clin Invest. 2001;107:1499–1500. doi: 10.1172/JCI13315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindberg FP, Gresham HD, Reinhold MI, Brown EJ. J Cell Biol. 1996;134:1313–1322. doi: 10.1083/jcb.134.5.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frazier WA, Gao AG, Dimitry J, Chung J, Brown EJ, Lindberg FP, Linder ME. J Biol Chem. 1999;274:8554–8560. doi: 10.1074/jbc.274.13.8554. [DOI] [PubMed] [Google Scholar]
- 31.Reinhold MI, Lindberg FP, Plas D, Reynolds S, Peters MG, Brown EJ. J Cell Sci. 1995;108(Pt 11):3419–3425. doi: 10.1242/jcs.108.11.3419. [DOI] [PubMed] [Google Scholar]
- 32.Brown EJ, Frazier WA. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 33.Boggs JM. Cell Mol Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura M, Sato M, Akatsuka A, Saito S, Ando K, Yokoyama M, Katsuki M. Brain Res. 1998;785:245–252. doi: 10.1016/s0006-8993(97)01383-8. [DOI] [PubMed] [Google Scholar]
- 35.Nakajima K, Ikenaka K, Kagawa T, Aruga J, Nakao J, Nakahira K, Shiota C, Kim SU, Mikoshiba K. J Neurochem. 1993;60:1554–1563. doi: 10.1111/j.1471-4159.1993.tb03321.x. [DOI] [PubMed] [Google Scholar]
- 36.Ottens AK, Golden EC, Bustamante L, Hayes RL, Denslow ND, Wang KK. J Neurochem. 2008;104:1404–1414. doi: 10.1111/j.1471-4159.2007.05086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Givogri MI, Bongarzone ER, Campagnoni AT. J Neurosci Res. 2000;59:153–159. [PubMed] [Google Scholar]
- 38.Akiyama K, Ichinose S, Omori A, Sakurai Y, Asou H. J Neurosci Res. 2002;68:19–28. doi: 10.1002/jnr.10188. [DOI] [PubMed] [Google Scholar]
- 39.Boggs JM, Yip PM, Rangaraj G, Jo E. Biochemistry. 1997;36:5065–5071. doi: 10.1021/bi962649f. [DOI] [PubMed] [Google Scholar]
- 40.Kim JK, Mastronardi FG, Wood DD, Lubman DM, Zand R, Moscarello MA. Mol Cell Proteomics. 2003;2:453–462. doi: 10.1074/mcp.M200050-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Schaecher K, Rocchini A, Dinkins J, Matzelle DD, Banik NL. J Neuroimmunol. 2002;129:1–9. doi: 10.1016/s0165-5728(02)00142-x. [DOI] [PubMed] [Google Scholar]
- 42.Shields DC, Banik NL. J Neurosci Res. 1999;55:533–541. doi: 10.1002/(SICI)1097-4547(19990301)55:5<533::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 43.Shields DC, Banik NL. Brain Res. 1998;794:68–74. doi: 10.1016/s0006-8993(98)00193-0. [DOI] [PubMed] [Google Scholar]
- 44.Shields DC, Tyor WR, Deibler GE, Hogan EL, Banik NL. Proc Natl Acad Sci U S A. 1998;95:5768–5772. doi: 10.1073/pnas.95.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denkinger CM, Metz C, Fingerle-Rowson G, Denkinger MD, Forsthuber T. Arch Immunol Ther Exp (Warsz) 2004;52:389–400. [PubMed] [Google Scholar]
- 46.Fingerle-Rowson G, Petrenko O, Metz C, Forsthuber T, Mitchell R, Huss R, Moll U, Müller W, Bucala R. Proc Natl Acad Sci U S A. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Binsky I, Haran M, Starlets D, Gore Y, Lantner F, Harpaz N, Leng L, Goldenberg DM, Shvidel L, Berrebi A, Bucala R, Shachar I. Proc Natl Acad Sci U S A. 2007;104:13408–13413. doi: 10.1073/pnas.0701553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG. J Immunol. 2003;170:1274–1282. doi: 10.4049/jimmunol.170.3.1274. [DOI] [PubMed] [Google Scholar]
- 49.Koning N, Bo L, Hoek RM, Huitinga I. Ann Neurol. 2007;62:504–514. doi: 10.1002/ana.21220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blotting of immunoenriched samples confirmed IM2 proteomics results showing that CD47 was expressed throughout disease progression.
TMT labeling scheme. To normalize across the time-course of disease progression, TMT-encoded brain tissue lysates from individual mice, randomly labeled with the TMT-127-130 reagents to improve confidence in our results, were mixed with the reference materials encoded with the TMT-126 and TMT-131 reagents in 1126:1127:1128:1129:1130:1131 ratios.