

Abstract
Purpose
High-dose interleukin-2 (IL-2) induces responses in 15% to 20% of patients with advanced melanoma; 5% to 8% are durable complete responses (CRs). The HLA-A2–restricted, modified gp100 peptide (210M) induces T-cell immunity in vivo and has little antitumor activity but, combined with high-dose IL-2, reportedly has a 42% (13 of 31 patients) response rate (RR). We evaluated 210M with one of three different IL-2 schedules to determine whether a basis exists for a phase III trial.
Patients and Methods
In three separate phase II trials, patients with melanoma received 210M subcutaneously during weeks 1, 4, 7, and 10 and standard high-dose IL-2 during weeks 1 and 3 (trial 1), weeks 7 and 9 (trial 2), or weeks 1, 4, 7, and 10 (trial 3). Immune assays were performed on peripheral-blood mononuclear cells collected before and after treatment.
Results
From 1998 to 2003, 131 patients with HLA-A2–positive were enrolled. With 60-month median follow-up time, the overall RR for 121 assessable patients was 16.5% (95% CI, 10% to 26%); the RRs were 23.8% in trial 1 (42 patients), 12.5% in trial 2 (40 patients), and 12.8% in trial 3 (39 patients). There were 11 CRs (9%) and nine partial responses (7%), with 11 patients (9%) progression free at ≥ 30 months. Immune studies including assays of CD3-ζ expression and numbers of CD4+/CD25+/FoxP3+ regulatory T cells, CD15+/CD11b+/CD14− immature myeloid-derived cells, and CD8+gp100 tetramer-positive cells in the blood did not correlate with clinical benefit.
Conclusion
The results again demonstrate efficacy of high-dose IL-2 in advanced melanoma but did not demonstrate the promising clinical activity reported with vaccine and high-dose IL-2 in any of three phase II trials.
INTRODUCTION
The median survival time for patients with advanced melanoma remains dismal at 6 to 10 months.1 Chemotherapy with dacarbazine has a response rate (RR) of only 5% to 20%, with most responses being short lived. Chemotherapy combined with biologic agents provides no survival benefit.1-3 In a report on 270 patients with melanoma, high-dose bolus interleukin-2 (IL-2) therapy produced a 16% objective RR with long-lasting responses in select patients (5% to 8%); thus, the US Food and Drug Administration approved high-dose IL-2 in patients with melanoma. Previous attempts to enhance high-dose IL-2 efficacy in patients with melanoma have been largely unsuccessful.4-9
A number of immunogenic peptides derived from tissue (lineage) -restricted antigens, cancertestis antigens, and rarely mutated cancer-specific antigens have been identified in melanoma, and many have the ability to induce antimelanoma T-cell responses.10-16 A peptide derived from gp100 (a lineage-restricted antigen) has been synthesized in a mutated (heteroclitic) form for the purpose of enhancing HLA-A2.1 binding. This peptide, gp100:209-217(210M) binds with a higher affinity to HLA-A2 than the native peptide and induces better T-cell stimulation in vitro and in vivo.17 Initial clinical trials at the National Cancer Institute (NCI) with 210M demonstrated induction in peripheral blood of peptide- and tumor-specific cytotoxic T-lymphocyte responses in 10 of 11 patients with HLA-A2.1–positive melanoma, although no definitive antitumor activity was observed.18,19 However, clinical experience combining 210M followed by IL-2 (high-dose bolus) demonstrated tumor responses in 13 (42%) of 31 patients.18 Patients were treated with different schedules of peptide vaccines and IL-2. In responding patients receiving IL-2 early in their course of treatment, circulating tumor-specific T cells were not generated, questioning their role as a biomarker of clinical activity.
We evaluated different schedules of peptide vaccine plus high-dose IL-2 in three separate, single-arm, and single-stage phase II trials to define which schedule was most promising. The three schedules were chosen based on our discussion with the investigators at the NCI-Surgery Branch and the similarity to the regimens used by them, as previously reported.18 Our primary intent was to determine whether more extensive testing of any of the schedules was justified.
PATIENTS AND METHODS
Patient Eligibility
Patients with advanced melanoma were HLA-A2 positive by serology, flow cytometry, or molecular techniques (HLA-A2.1). Patients had an Eastern Cooperative Oncology Group performance status of 0 to 1 and adequate organ function for high-dose IL-2 therapy. Normal exercise stress test and forced expiratory volume in 1 second more than 2.0 L or 75% predicted were required. Patients with brain metastases or prior IL-2 therapy were ineligible. The protocol was approved by the institutional review boards at each of the participating centers, and written informed consent was required.
Therapeutic Agents
gp100:209-217(210M) peptide
gp100:209-217(210M) (NSC 683472) is an HLA-A2 restricted 9-mer (IMDQVPFSV) peptide from gp100 provided by Cancer Therapy Evaluation Program (CTEP) under Investigational New Drug Application BB6123 in vials containing 1 mg/mL of sterile water for injection with 0.1 N hydrogen chloride to adjust the pH.
Montanide ISA-51 adjuvant
Montanide ISA-51 (NSC 675756) is an oil-based adjuvant similar to incomplete Freund’s. The product is manufactured by Seppic, Inc (Fairfield, NJ) and was provided by CTEP.
IL-2
Commercial recombinant human IL-2 (Proleukin; Chiron, Emeryville, CA), lyophilized in vials containing 22 million U (approximately 1.3 mg), was reconstituted with 1.2 mL of sterile water for injection.
Treatment Plan
Patients were injected subcutaneously with 210M and Montanide ISA-51. Vaccine was administered on days 1, 22, 43, and 65 for all three clinical trials. High-dose IL-2 (600,000 U/kg every 8 hours) was administered up to 14 times intravenous bolus over a 5-day inpatient course, starting 1 day after vaccine. Details are provided in Figure 1.
Fig 1.
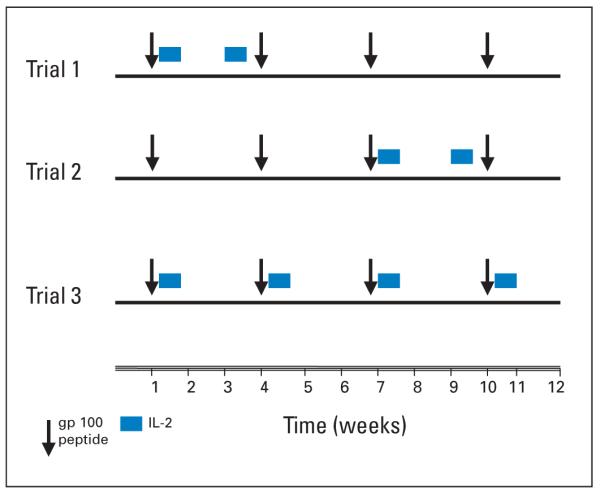
Treatment schema (initial 12-week course). gp100 (210M) was administered in incomplete Freund adjuvant every 3 weeks (weeks 1, 4, 7, and 10) for all three clinical trials. High-dose interleukin-2 (IL-2; 600,000 U/kg every 8 hours) was administered up to 14 times intravenous bolus over 5 days, starting the day after vaccine, on weeks 1 and 3 for trial 1, on weeks 7 and 9 for trial 2, and on weeks 1, 4, 7, and 10 for trial 3 (maximum).
Patients underwent tumor response assessments at week 12 by WHO criteria.20 Patients with stable disease, partial response (PR), or complete response (CR) could receive another course of treatment, whereas patients with disease progression were removed from protocol. All responses were confirmed at 3 months, and the RR was based on best response any time after treatment had been initiated. Patients could receive up to three 12-week courses of IL-2 and 36 weeks of vaccine. Vaccine alone could be administered without further IL-2. Progression-freesurvival (PFS) was defined as the time from trial entry to the time of documented objective disease progression or death. Overall survival (OS) was defined as the time from trial entry until death.
Treatment modifications were made by withholding doses of IL-2 for life-threatening toxicities including refractory hypotension, hypoxia, anuria, acidosis, and mental status changes. Toxicity was managed based on guidelines of the Cytokine Working Group (CWG).21 No dose modifications were required for the vaccinations because there were no systemic or severe local skin reactions.
Immunologic Assessments
Blood for immunologic assays was collected at two time points, within 2 weeks before therapy and during week 12 of therapy. Peripheral-blood mononuclear cells (PBMCs) were isolated and cryopreserved. A single center performed the analysis on cryopreserved PBMCs. Normal controls were run with each experiment. Studies were performed blinded to the clinical results.
Flow Cytometry Analysis
Reagents
CD3-peridinin-chlorophyll-protein complex (PerCP), CD4PerCP, CD8PerCP, CD11bPE, IgG2aPE, CD14-fluorescein isothiocyanate (FITC), gp100, and influenza (flu) HLA-A2 tetramers were conjugated to phycoerythrin (PE). CD15-PC5, CD3-ζ chain-FITC, and normal mouse IgG1-FITC were purchased from BD Pharmingen (Franklin Lakes, NJ), Beckman Coulter (Fullerton, CA), Immunotech (Boston, MA), and Santa Cruz Biotechnology (Santa Cruz, CA), respectively.
ζ-chain analysis
PBMCs were stained with CD3PerCP, washed, permeabilized, stained for CD3-ζ chain-FITC or an isotype control from Caltag Laboratories (San Francisco, CA), and then analyzed. The isotype control set the boundary of zeta positivity.
Surface staining of PBMC with CD14-FITC, CD11b-PE, CD15-PC5, and CD15-isotype control was at 4°C, and staining of CD8PerCP, gp100 PE, Flu PE, and CD15-isotype control was at room temperature. Cells were washed and then analyzed on FC500 cytometer (Beckman Coulter).
Lymphocytes were first stained using CD4-FITC and CD25-APC (eBioscience, San Diego, CA). After fixation and permeabilization, FoxP3-PE was added. Cells were washed and fixed with 1% formaldehyde in phosphatebuffered saline. For analysis, CD4+ and CD25+ cells were gated and analyzed for FoxP3. Isotype controls for all antibodies were included.
Statistical Analysis
The primary end point of the trial was RR including both PRs and CRs. The design defined a minimum 30% RR (12 of 40 patients; 95% CI, 16.3% to 43.7%) as demonstrating promise for proceeding to a phase III trial. The recommendation of a 30% RR in this specific population of trials (rather than powered to demonstrate superiority to a historical control response) was based on recommendations from a number of investigators from NCI-CTEP and the CWG. The RR would assist in estimating the number of patients required for a phase III trial with adequate power. The trial was not designed to definitively prove that the peptide vaccine combined with high-dose IL-2 was or was not an improvement compared with IL-2 alone. CIs computed in the article are based on the binomial distribution.
To examine immune cell populations and clinical efficacy (CR, PR, or PFS > 12 months), repeated measures analysis of variance methods were used for paired (pre- and post-treatment) data within each subject. These methods take the paired nature of the data into account, resulting in a test statistic that is equivalent to a paired t test. The data are assumed to be normally distributed because the immune cells are measured as percentage of cells expressing the phenotype. The data are interval data (eg, bounded by 0 to 100), and not discrete samples from Poisson, Bernoulli, or binomial distributions. These tests were used to compare data both within response groups (before and after treatment) and between response groups (pretreatment and post-treatment responders and nonresponders). We tested time (before and after treatment), response, and the time by response interaction in these models. We used the log-rank test statistic to examine the difference in survival between the cohorts. Rather than show all of the pair-wise CIs, we chose to show them within response group differences along with the actual data. All analyses were performed using SAS version 9.1 (SAS Institute, Cary, NC).
RESULTS
Patients With Melanoma
A total of 131 patients were enrolled from September 1998 to November 2003 at eight institutions. Clinical data were available for 121 patients. One patient did not have melanoma, and nine patients were lost to follow-up and excluded from the analysis. The characteristics of the 121 patients are listed in Table 1. More than 65% of patients had M1c disease. All three cohorts were similar in patient characteristics, except that trial 2 enrolled fewer patients who had received adjuvant interferon and more patients with lactate dehydrogenase (LDH) elevations. Neither of these differences was statistically significant.
Table 1.
Patient Characteristics
| Trial 1 (n = 42) |
Trial 2 (n = 40) |
Trial 3 (n = 39) |
All Patients (N = 121) |
|||||
|---|---|---|---|---|---|---|---|---|
| Characteristic | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % |
| Sex | ||||||||
| Male | 24 | 25 | 24 | 72 | ||||
| Female | 18 | 15 | 15 | 49 | ||||
| Age, years | ||||||||
| Median | 52 | 52 | 48 | 50 | ||||
| Range | 21-76 | 20-72 | 20-69 | 20-76 | ||||
| ECOG PS | ||||||||
| 0 | 34 | 35 | 29 | 99 | ||||
| 1 | 8 | 5 | 10 | 22 | ||||
| LDH | ||||||||
| Elevated | 10 | 31 | 13 | 72 | 9 | 47 | 34 | 46 |
| Normal | 21 | 66 | 5 | 28 | 10 | 53 | 40 | 54 |
| Unknown | 11 | 22 | 20 | 53 | ||||
| AJCC staging | ||||||||
| M1a | 5 | 11 | 2 | 5 | 6 | 15 | 13 | 11 |
| M1b | 6 | 14 | 10 | 25 | 11 | 28 | 27 | 22 |
| M1c | 31 | 74 | 28 | 70 | 22 | 56 | 81 | 67 |
| Prior therapy | ||||||||
| Interferon alfa | 15 | 36 | 9 | 23 | 16 | 41 | 40 | 34 |
| Chemotherapy | 5 | 12 | 5 | 12.5 | 5 | 13 | 15 | 13 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; PS, performance status; LDH, lactate dehydrogenase; AJCC, American Joint Committee on Cancer.
Treatment Administration and Toxicities
Toxicity of high-dose IL-2 was primarily assessed by reduction in the number of IL-2 doses administered. Toxicity was not affected by the 210M peptide administration.21,22 Vascular leak syndrome and mental status changes were the primary reasons for withholding doses of IL-2. Three patients died of progressive melanoma during the initial 12 weeks of the study. Fourteen patients did not receive IL-2 therapy (12 patients on trial 2), many as a result of a decline in performance status from disease progression during the initial weeks of vaccination, which prevented IL-2 administration. These patients were included in the analysis. Of patients who received ≥ one dose of IL-2, those on trial 3 received the most doses, with a median of 35 of 56 possible doses (range, eight to 51 doses) for the 12 weeks. Patients receiving ≥ one dose of IL-2 in both trials 1 and 2 received a median of 20 of 28 IL-2 doses. The number of doses ranged from nine to 27 in trial 2 and from 11 to 27 in trial 1. The only direct toxicities caused by the peptide vaccine were local swelling and discomfort. No episodes of uveitis, thyroiditis, or other autoimmune sequelae were recorded, and vitiligo was observed in some responders but not systematically collected.23 The number of patients who received additional 12-week courses of vaccine and/or IL-2 is presented in Table 2.
Table 2.
Subsequent Vaccine and IL-2 Therapy in Courses 2 and 3
| No. of Patients |
|||
|---|---|---|---|
| Course of Treatment | Trial 1 (n = 42) |
Trial 2 (n = 40) |
Trial 3 (n = 39) |
| Second course of vaccine | 17 | 9 | 9 |
| Second course of IL-2 | 17 | 7 | 4 |
| Third course of vaccine | 15 | 5 | 1 |
| Third course of IL-2 | 8 | 3 | 0 |
NOTE. Number of patients receiving therapy in subsequent 12-week courses. Vaccine could be administered without IL-2.
Abbreviation: IL-2, interleukin-2.
Tumor Responses, Durability of Responses, OS, and PFS
With a median follow-up time of 60 months (range, 30 to 74 months), there were 20 clinical responses (16%) among 121 assessable patients (Table 3). Trial 1 had the highest RR of 23.8% (10 of 42 patients), trial 2 had an RR of 12.5% (five of 40 patients), and trial 3 had an RR of 12.8% (five of 39 patients). None of the schedules (trials) reached the 30% RR targeted in the trial. Overall, nine of the responses (7%) were PRs, and 11 (9%) were CRs. Nine of the 11 patients who achieved CR remain progression free (PF) at 30+ to 73+ months (30+, 30+, 38+, 40+, 42+, 46+, 46+, 46+, and 73+ months), whereas two patients experienced progression at 17 and 51 months. Only one of the nine patients who achieved PR remains PF at 30+ months. Five of the patients with PR experienced disease progression in less than 12 months.
Table 3.
HD IL-2 Plus gp100 209-2 M Peptide Vaccine Trial
| Response by WHO Criteria |
|||||
|---|---|---|---|---|---|
| Cohort | No. of Patients Assessed | No. of CR | No. of PR | RR (%) | 95% CI* (%) |
| Overall | 121 | 11 | 9 | 16.5 | 10 to 26 |
| Trial 1 | 42 | 6 | 4 | 23.8 | 12 to 40 |
| Trial 2 | 40 | 4 | 1 | 12.5 | 4 to 27 |
| Trial 3 | 39 | 1 | 4 | 12.8 | 4 to 27 |
| HD IL-2 database | 270 | 17 | 26 | 15.9 | 12 to 21 |
| NCI-surgery branch trial | 31 | 13† | 41.9 | 26 to 59 | |
Abbreviations: HD IL-2, high-dose interleukin-2; CR, complete response; PR, partial response; RR, response rate; NCI, National Cancer Institute.
CIs based on the binomial distribution.
CR + PR.
The median PFS time was 3 months (time of initial tumor evaluation) for each of the three trials (log-rank test for difference in PFS between trials, P = .165; Appendix Fig A1, online only). Eleven patients (9%) remain PF with more than 30 months follow-up. Patients who are PF include seven patients in trial 1, three in trial 2, and one in trial 3.
At the time of this analysis, there were 18 patients (15%) who remained alive for 30+ to 70+ months. The median OS time of the entire group was similar in each trial (range, 13.7 to 15.4 months) or slightly under 15 months combined (log-rank test for difference in OS between trials, P = .723; Appendix Fig A2, online only).
Immunologic Studies and Clinical Correlations
Paired samples from peripheral blood with more than 80% viability were obtained before therapy and on week 12 in only 53 of the 121 patients because of early disease progression, poor compliance, and poor viability in some specimens. The characteristics of these patients are presented in Table 4. Expression of the TCR-ζ chain, an indicator of T-cell signaling dysfunction; CD4+/CD25+/FoxP3+, an indicator of regulatory T cells (Tregs); CD11b+/CD15+/CD14−, an indicator of myeloid-derived suppressor cells (MDSCs); and percentage of CD8+ tetramers positive for gp100 peptide and flu were assayed.24-29 Among the 53 patients, 10 had responses, including six CRs and four PRs. Furthermore, 13 patients had PFS ≥ 12 months (most with response). As shown in Figure 2, with means, CIs, and statistical findings (in legend), there was no difference in the percentage of PBMCs before and after treatment for any of the immune populations (CD3-ζ chain, CD4+/CD25+/FoxP3+,orCD11b+/CD15+/CD14−) in patients with or without clinical responses and patients with or without ≥ 12 months of PFS. Additionally, mean fluorescent intensity for each marker was not different (data not shown). Some patients had impressive increases in their CD8+ gp100 tetramers but without a correlation with either clinical response or 12-month PFS.
Table 4.
Patient Characteristics of Subset Tested for Immune Parameters
| Patients With Immune Tests (n = 53) |
Patients Not Immune Tested (n = 68) |
All Patients (N = 121) |
||||
|---|---|---|---|---|---|---|
| Characteristic | No. | % | No. | % | No. | % |
| Sex | ||||||
| Male | 31 | 41 | 72 | |||
| Female | 22 | 27 | 49 | |||
| Age, years | ||||||
| Median | 50 | 50 | 50 | |||
| Range | 21-69 | 20-76 | 20-76 | |||
| ECOG PS | ||||||
| 0 | 43 | 56 | 99 | |||
| 1 | 10 | 12 | 22 | |||
| Trial | ||||||
| Trial 1 | 21 | 39 | 21 | 31 | 42 | |
| Trial 2 | 13 | 25 | 27 | 40 | 40 | 33 |
| Trial 3 | 19 | 36 | 20 | 29 | 39 | 32 |
| AJCC staging | ||||||
| M1a | 4 | 8 | 9 | 13 | 13 | 11 |
| M1b | 15 | 28 | 12 | 18 | 27 | 22 |
| M1c | 34 | 64 | 47 | 69 | 81 | 67 |
| Prior therapy | ||||||
| Interferon alfa | 21 | 40 | 21 | 31 | 42 | 34 |
| Chemotherapy | 5 | 9 | 10 | 15 | 15 | 13 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; PS, performance status; AJCC, American Joint Committee on Cancer.
Fig 2.

Effects of gp100 (210M) plus high-dose interleukin-2 (IL-2) on immune populations. Peripheral-blood mononuclear cells were obtained before treatment and during week 12 of treatment and assayed by flow cytometry. Responding patients are defined in the Results. Dot plots for data are shown with the mean pre- and post-treatment difference (horizontal line) and 95% CI (vertical line) for both responding and nonresponding patients. Left-column responders are based on PFS, and right-column responders are based on objective response. The results are based on percentage of mononuclear cells. (A) Percentage of CD11b+/CD15+/CD14− preversus post-treatment expression in responders versus nonresponders by progression-free survival (PFS; P = .5052) or by objective responses (P = .4269). (B) Percentage of CD4+/CD25+/FoxP3+ preversus post-treatment levels in responders versus nonresponders by PFS (P = .6844) or by objective responses (P = .6340). (C) Percentage of CD3-ζ preversus post-treatment levels of expression in responders versus nonresponders by PFS (P = .7522) or by objective responses (P = .1445). (D) Percentage of CD8+ T cells expressing gp100-209 tetramers as the log of percentage of CD8+ T cells. There is no difference between preversus post-treatment levels of gp100 tetramer–positive cells in responders versus nonresponders by PFS (P = .7839) or by objective responses (P = .1502). (E) CD8+ T cells expressing influenza tetramers as the log of percentage of CD8+ T cells. There is no difference between pre- and post-treatment levels of flu-positive cells in any of the cohorts.
DISCUSSION
In advanced melanoma, high-dose IL-2 has an RR of 16%, with a 5-year PFS rate of more than 5% (14 of 270 patients).3 In 1998, Rosenberg et al18 reported on 31 patients with melanoma receiving both 210M and high-dose IL-2, with an RR of 42% (13 of 31 patients). We report the clinical results of three separate phase II trials with 210M peptide vaccine and high-dose IL-2 against melanoma. None of the three trials reached the target clinical activity (30% RR) to proceed to an adequately powered phase III trial. Why do our results differ from the NCI trial?18 Patient characteristics such as performance status, LDH level, prior chemotherapy, and disease organ sites may have significantly differed in the two studies and are not available for the NCI-SB trial.30 The NCI-SB has shown that certain characteristics are associated with responses to IL-2, such as subcutaneous or cutaneous disease.31 The CWG administered smaller doses of IL-2 (600,000 U/kg in CWG study v 720,000 U/kg in NCI-SB study), but the number of doses administered was greater in the CWG trial, with 20 doses over a 3-week period versus 13 to 14 doses for the NCI-SB. This more than compensated for the difference per dose. Finally, patients who travel to the NCI are a self-selected group of patients, which could impact on patient characteristics.
The overall RR of 16.5% was similar to HD IL-2 alone.3 Ten patients remain PF with ≥ 30 months of follow-up, which is similar to numbers previously reported (8.2% in this study v 5.1% historical). This remarkable benefit did not seem to be improved by the addition of an immunologically active peptide vaccine. Patients with prior adjuvant interferon alfa treatment had an RR similar to patients who had not received interferon alfa (20% v 14.2%, respectively; P = .7303). A number of the patients, mostly in trial 2, were removed from treatment before receiving IL-2 as a result of early disease progression. These patients were included in this analysis, but this may not be true in reports of therapies requiring a delay (cell harvest and expansion) before initiation of treatment. Attempts to combine the 210M peptide with a low-dose IL-2 regimen in patients with metastatic melanoma showed a lack of clinical activity.32
Unfortunately, our attempts to define immune correlates of response were unsuccessful in the 53 patients for whom pre- and post-treatment specimens were available. Ten of these patients had objective clinical responses. The populations of immune cells assessed were chosen because all four populations play a potential role in an antitumor immune response. The gp100-specific CD8 T cells represent the vaccine-induced effector T cells, whereas the CD4+/CD25+/FoxP3+ Tregs suppress effective T-cell responses by effector T cells, and the MDSCs (CD11b+/CD15+/CD14−) represent a population that also suppresses T-cell responses. Finally, low CD3-ζ expression is a potential marker of ineffective T-cell function with poor signaling through its T-cell receptor. One might hypothesize that modulation of these populations may be necessary for effective clinical responses.24-29 There was insufficient power to detect differences between responders and nonresponders in any single trial.
The main limitation in the immune testing is the amount of intrasubject variation inherent in the assays. Using pre- and post-treatment paired samples is generally an analytic approach for data with this type of variation. The results have limitations because the trials are not randomized and each cohort was relatively small. This set of phase II trials cannot definitively establish whether or not vaccine adds to the benefit of IL-2, but we did not reach the 30% RR that was targeted a priori.
Powering this type of study effectively is difficult. Serum LDH levels were elevated in only 18 of those patients tested and do not explain the variability of the groups. A great need exists to learn whether any biomarker might predict clinical outcome.32 Our results should not deter others from pursuing biomarkers that can predict response to immunotherapy. Functional studies were not feasible because of the number of cells collected and concern for loss of function with processing.33 The timing of the sample collections may be critical, and although samples were all obtained at week 12, the time after IL-2 dosing was variable. Two different approaches might have made the immune assays more informative. Most important is the need to examine the tumor site, not the peripheral blood, for immunologic changes. Changes in the blood may not reflect changes in tumor. Also, assays that include T-cell function may have been more useful, such as enzyme-linked immunosorbent spot assays for cytokine release or flow cytometry for intracellular cytokine expression.
Given our results, the combination of 210M peptide vaccine and high-dose IL-2 does not seem to represent a significant advance. This does not substitute for a phase III trial of high-dose IL-2 with or without peptide vaccination, which is ongoing and will accrue 160 patients. On the basis of our results (trial 1), this phase III trial could be underpowered to detect a difference. We must find better treatments for patients with melanoma. Peptide vaccines alone are likely not going to be the answer. Advances in immunization methods, understanding mechanisms of escape from immunotherapy, and improved assays for immune activation have all taken place since this trial.24-29 Presently, attempts to overcome host immune suppression induced by tumor is a major target of tumor immunology. Attempts include elimination of Tregs, induction of the differentiation of MDSCs, and utilization of ex vivo autologous tumor-specific T cells (adoptive cellular therapy) with nonmyeloablative chemotherapy and cell infusion with high-dose IL-2.33-37 These approaches are under investigation and will hopefully fulfill their promise to improve therapy of patients with advanced melanoma.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Supported by a research grant from Chiron Pharmaceuticals (Emeryville, CA); support from Cancer Therapy Evaluation Program–National Cancer Institute, which held the Investigation New Drug Applications for gp100:209-217(210M) (NSC 683472) and Montanide ISA-51 (NSC 675756); and K24 Grant No. 5K24-CA097588 (J.A.S.).
Appendix.
The Appendix is included in the full-text version of this article, available online at www.jco.org. It is not included in the PDF version (via Adobe® Reader®).
Footnotes
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO’s conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Michael B. Atkins, Novartis/Chiron (C); Janet Dutcher, Novartis/Chiron (C); Marc S. Ernstoff, Novartis/Chiron (C) Stock Ownership: None Honoraria: Lawrence Flaherty, Novartis/Chiron; Michael B. Atkins, Novartis/Chiron; Joseph I. Clark, Novartis/Chiron; Janet Dutcher, Novartis/Chiron; Kim A. Margolin, Novartis/Chiron; Jarod Gollob, Novartis/Chiron; Marc S. Ernstoff, Novartis/Chiron Research Funding: Jeffrey A. Sosman, Novartis; Lawrence Flaherty, Novartis/Chiron; Michael B. Atkins, Novartis/Chiron; Joseph I. Clark, Novartis/Chiron; Janet Dutcher, Novartis/Chiron; Kim A. Margolin, Novartis/Chiron; Jarod Gollob, Novartis/Chiron; Marc S. Ernstoff, Novartis/Chiron Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS Conception and design: Jeffrey A. Sosman
Provision of study materials or patients: Jeffrey A. Sosman, Walter J. Urba, Lawrence Flaherty, Michael B. Atkins, Joseph I. Clark, Janet Dutcher, Kim A. Margolin, James Mier, Jarod Gollob, John M. Kirkwood, David J. Panka, Nancy A. Crosby, Kevin O’Boyle, Marc S. Ernstoff
Collection and assembly of data: Jeffrey A. Sosman, Carole Carrillo, David J. Panka
Data analysis and interpretation: Jeffrey A. Sosman, Bonnie LaFleur
Manuscript writing: Jeffrey A. Sosman, Walter J. Urba, Michael B. Atkins, Kim A. Margolin, Bonnie LaFleur, David J. Panka, Marc S. Ernstoff
Final approval of manuscript: Jeffrey A. Sosman, Carole Carrillo, Walter J. Urba, Lawrence Flaherty, Joseph I. Clark, Janet Dutcher, Kim A. Margolin, James Mier, Jarod Gollob, David J. Panka, Nancy A. Crosby, Kevin O’Boyle, Bonnie LaFleur, Marc S. Ernstoff
Presented in part at the 41st Annual Meeting of the American Society of Clinical Oncology, May 13-17, 2005, Orlando, FL.
Authors’ disclosures of potential conflicts of interest and author contributions are found at the end of this article.
REFERENCES
- 1.Eggermont AM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: What have we learned in 30 years? Eur J Cancer. 2004;40:1825–1836. doi: 10.1016/j.ejca.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 2.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 3.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 4.Sosman JA, Weiss GR, Margolin KA, et al. Phase IB clinical trial of anti-CD3 followed by high-dose bolus interleukin-2 in patients with metastatic melanoma and advanced renal cell carcinoma: Clinical and immunologic effects. J Clin Oncol. 1993;11:1496–1505. doi: 10.1200/JCO.1993.11.8.1496. [DOI] [PubMed] [Google Scholar]
- 5.Whitehead RP, Friedman KD, Clark DA, et al. Phase I trial of simultaneous administration of interleukin 2 and interleukin 4 subcutaneously. Clin Cancer Res. 1995;1:1145–1152. [PubMed] [Google Scholar]
- 6.Sosman JA, Fisher SG, Kefer C, et al. A phase I trial of continuous infusion interleukin-4 (IL-4) alone and following interleukin-2 (IL-2) in cancer patients. Ann Oncol. 1994;5:447–452. doi: 10.1093/oxfordjournals.annonc.a058878. [DOI] [PubMed] [Google Scholar]
- 7.Sparano JA, Fisher RI, Sunderland M, et al. Randomized phase III trial of treatment with high-dose interleukin-2 either alone or in combination with interferon alfa-2a in patients with advanced melanoma. J Clin Oncol. 1993;11:1969–1977. doi: 10.1200/JCO.1993.11.10.1969. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Lotze MT, Yang JC, et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst. 1993;85:622–632. doi: 10.1093/jnci/85.8.622. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Aebersold P, Cornetta K, et al. Gene transfer into humans: Immunotherapy of patients with advanced melanoma, using tumorinfiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 10.Boon T. Tumor antigens recognized by cytolytic T lymphocytes: Present perspectives for specific immunotherapy. Int J Cancer. 1993;54:177–180. doi: 10.1002/ijc.2910540202. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami Y, Eliyahu S, Delgado CH, et al. Identification of a human melanoma antigen recognized by tumor infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2 restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawakami Y, Eliyahu S, Jennings C, et al. Recognition of multiple epitopes in the human melanoma antigen gp 100 by tumor infiltrating T-lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3461–3968. [PubMed] [Google Scholar]
- 15.Rivoltini L, Loftus DJ, Barracchini K, et al. Binding and presentation of peptides derived from melanoma antigens MART-1 and glycoprotein-100 by HLA-A2 subtypes: Implications for peptide-based immunotherapy. J Immunol. 1996;156:3882–3891. [PubMed] [Google Scholar]
- 16.Rosenberg SA. Progress in human tumor immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 17.Parkhurst MR, Salgaller ML, Southwood S, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 18.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller AB, Hoogstraten B, Staquet M, et al. Reporting results of cancer treatment. Cancer. 1981;47:207–214. doi: 10.1002/1097-0142(19810101)47:1<207::aid-cncr2820470134>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: Moving beyond current vaccines. Nat Med. 1998;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margolin KA, Rayner AA, Hawkins MJ, et al. Interleukin-2 and lymphokine-activated killer cell therapy of solid tumors: Analysis of toxicity and management guidelines. J Clin Oncol. 1989;7:486–498. doi: 10.1200/JCO.1989.7.4.486. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberg SA, Yang JC, Topalian SL. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin-2. JAMA. 1994;271:907–913. [PubMed] [Google Scholar]
- 23.Rosenberg SA, White DE. Vitiligo in patients with melanoma: Normal tissue antigens can be targets for cancer immunotherapy. J Immunother Emphasis Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- 24.Mizoguchi H, O’Shea JJ, Longo DL, et al. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science. 1992;258:1795–1798. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 25.Ochoa AC, Longo DL. Alteration of signal transduction in T cells from cancer patients. Important Adv Oncol. 1995;55:43–54. [PubMed] [Google Scholar]
- 26.Zou W. Regulatory T cells, tumor immunity, and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 27.Fehervari Z, Sakaguchi S. CD4+ Tregs and immune control. J Clin Invest. 2004;114:1209–1217. doi: 10.1172/JCI23395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 30.Manola J, Atkins M, Ibrahim J, et al. Prognostic factors in metastatic melanoma: A pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol. 2000;18:3782–3793. doi: 10.1200/JCO.2000.18.22.3782. [DOI] [PubMed] [Google Scholar]
- 31.Phan GQ, Attia P, Steinberg SM, et al. Factors associated with response to high-dose Interleukin-2 in patients with metastatic melanoma. J Clin Oncol. 2001;19:3477–3482. doi: 10.1200/JCO.2001.19.15.3477. [DOI] [PubMed] [Google Scholar]
- 32.Roberts JD, Niedzwiecki D, Carson WE, et al. Phase 2 study of the g209-2M melanoma peptide vaccine and low-dose interleukin-2 in advanced melanoma: Cancer and Leukemia Group B 509901. J Immunother. 2006;29:95–101. doi: 10.1097/01.cji.0000195295.74104.ad. [DOI] [PubMed] [Google Scholar]
- 33.Keilholz U, Weber J, Finke JH, et al. Immunologic monitoring of cancer vaccine therapy: Results of a workshop sponsored by the Society for Biological Therapy. J Immunother. 2002;25:97–138. doi: 10.1097/00002371-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Dannull J, Su Z, Rizzieri D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gabrilovich DI, Ishida T, Nadaf S, et al. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res. 1999;5:2963–2970. [PubMed] [Google Scholar]
- 36.Kusmartsev S, Cheng F, Yu B, et al. All-transretinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 37.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.