

Abstract
We synthesized homologated truncated 4′-thioadenosine analogues 3 in which a methylene (CH2) group was inserted in place of the glycosidic bond of a potent and selective A3 adenosine receptor antagonist 2. The analogues were designed to induce maximum binding interaction in the binding site of the A3 adenosine receptor. However, all homologated nucleosides were devoid of binding affinity at all subtypes of adenosine receptors, indicating that free rotation through the single bond allowed the compound to adopt an indefinite number of conformations, disrupting the favorable binding interaction essential for receptor recognition.
Keywords: homologation, A3 adenosine receptor, binding affinity, truncated 4′-thioadenosine
1. Introduction
Adenosine, an endogenous material controls many physiological functions through binding to the adenosine receptors (ARs) which consist of four subtypes, A1, A2A, A2B, and A3.1 Binding of adenosine to the adenosine receptors changes the levels of the second messengers such as diacylglycerol (DAG) and cyclic 3′,5′-monophosphate (cAMP) for signal transduction.1 Therefore, adenosine receptors are regarded as promising therapeutic targets for the treatment of many types of diseases related to the signal transduction.2
Among four subtypes of ARs, A3 AR is most recently identified receptor and many efforts have been made to modulate A3 AR for the treatment of several diseases such as cancer, cardiac ischemia, asthma, glaucoma, and inflammation.3 4′-Thionucloesides were recently discovered to be good templates for the design of A3 AR ligands.4,5 Compound 14,5 was found to be very potent A3 AR agonist (Ki = 0.38 nM) and showed very potent in vivo antitumor activity in several types of tumor xenograft models. The mechanism study revealed that compound 1 induced cell-cycle arrest by lowering levels of c-myc and cyclin D1 in concentration- and time-dependent manners at lower concentrations and apoptosis through PRRP cleavage at higher concentrations.6 Compound 1 also inhibited Wnt signaling pathway at 10 nM of concentration.6
Molecular modeling study indicated that the NH of methylamide of 1 serves as a hydrogen bonding donor required for the receptor activation.7 On the basis of these findings, it was hypothesized that the truncated analogue 2 of compound 1 in which a hydrogen bonding donor NH essential for the agonist activity was removed, could be worked as an A3 AR antagonist. Compound 2 turned out to be a potent and selective antagonist at human (Ki = 4.16 nM) as well as rat (Ki = 3.90 nM) A3 AR.8
Introduction of a C-C single bond in the structure increases the free rotation. The increase of the free rotation allows the molecule to adopt many conformations, making it possible to induce maximum binding interaction in the binding site of the receptor. On the basis of this hypothesis, we designed and synthesized the homologated analogue 3 in which the methylene (CH2) was inserted between the glycosidic bond of a potent and selective A3 AR antagonist 2 for expecting maximum biding interaction in the binding site of the receptor, as potential A3 AR antagonists. Herein, we report the synthesis of the homologated truncated 4′-thioadenosine analogues 3 from D-mannose and their binding affinities at the human ARs.
2. Results and discussion
Our synthetic strategy was to synthesize the homologated glycosyl donor and then to condense with purine base. The homologated glycosyl donor 7 was first synthesized from D-mannose as shown in Scheme 1. D-Mannose was converted to the diol 4 according to our previously published procedure.8 D-Mannose was converted to the diacetonide 4 by treating with 2,2-dimethoxypropane and acetone under acidic conditions. Reduction of 4 with sodium borohydride followed by mesylation of the resulting diol afforded the dimesylate 5. Treatment of 5 with anhydrous sodium sulfide in DMF at 80 °C yielded the thiosugar 6, which was hydrolyzed with 60% acetic acid to give diol 6. Oxidative cleavage of the diol 6 with 1.2 equiv of Pb(OAc)4 produced the aldehyde 7, but use of excess Pb(OAc)4 gave the anomeric acetate, resulting from the oxidative decarboxylation.8 Reduction of the aldehyde 7 with NaBH4 afforded the primary alcohol 8. Treatment of 8 with phosphorous oxychloride yielded the glycosyl donor 9, which is ready for the condensation with purine bases.
Scheme 1.
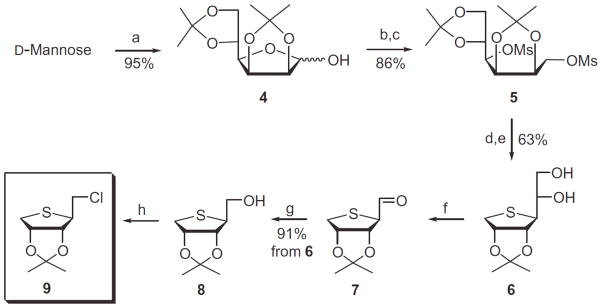
Reagents and conditions: (a) 2,2-dimethoxypropane, camphosulfonic acid, acetone, rt; (b) NaBH4, EtOH, rt; (c) MsCl, Et3N, CH2C12, rt; (d) Na2S, DMF, 80 °C; (e) 60% AcOH, rt; (f) Pb(OAc)4, EtOAc, 0 °C; (g) NaBH4, EtOH, 0 °C; (h) POCl3, CH3CN, rt.
The final nucleosides 3a-d and 3e-h were synthesized from direct SN2 displacement of the chloride 9 with 6-chloropurine and 2,6-dichloropurine anions, as shown in Scheme 2. Condensation of 7 with 6-chloropurine and 2,6-dichloropurine in the presence of NaH in DMF afforded the 6-chloropurine derivative 10 and the 2,6-dichloropurine derivative 11, respectively without concomitant formation of the corresponding N7-isomers. The structural assignment of N9-isomer and N7-isomer was accomplished by the comparison of UV and 13C NMR data reported in the literature.8,11 Removal of the isopropylidene group of 10 using 2 N HCl gave the 2-H analogue 12. Treatment of 6-chloropurine derivative 12 with 3-halobenzyl amines yielded the N6-(3-halobenzyl)amine derivatives 3a-d. Similarly, 2,6-dichloropurine derivative 11 was converted to the 2-Cl analogues 3e-h via 13.
Scheme 2.

Reagents and conditions: (a) NaH, 6-chloropurine or 2,6-dichloropurine, DMF, rt; (b) 2 N HCl, THF, rt; (c) 3-halobenzylamine, Et3N, EtOH.
All AR binding experiments were performed using adherent mammalian CHO (Chinese hamster ovary) cells stably transfected with cDNA encoding the appropriate human ARs (A1 AR and A3 AR in CHO cells and A2A AR in HEK-293 cells).9 Binding was carried out using 1 nM [3H]CCPA, 10 nM [3H]CGS21680, or 0.5 nM [125I]I-AB-MECA as radioligands for A1, A2A, and A3 ARs, respectively. Values are expressed as mean ± sem, n = 3–4 (outliers eliminated), and normalized against a non-specific binder, 5′-N-ethylcarboxamidoadenosine (NECA, 10 μM). If a percentage is given, it represents the percent inhibition at a fixed concentration of 10 μM. As shown in Table 1, all homologated compounds synthesized in this study were totally devoid of binding affinities at three subtypes of ARs. This result indicated that the homologation of the rotatable methylene (CH2) between 1′ and N9 positions disrupted favorable binding interactions at the ARs.
Table 1.
Binding affinities of known A3AR antagonist 2 and homologated 4′-thioadenosine derivatives 3a-d and 3e-h at three subtypes of ARs.
| |||
|---|---|---|---|
| Compounds | Ki (hA1AR) | Ki (hA2AAR) | Ki (hA3AR) |
| nM or % displ. at 10 μM | nM or % displ. at 10 μM | nM or % displ. at 10 μM | |
| 3a (R = H, X = F) | 0.3 ± 0.3% | 14.0 ± 5.6% | 8.7 ± 6.0% |
| 3b (R = H, X = Cl) | 3.3 ± 3.3% | 13.0 ± 8.2% | 8.9 ± 1.8% |
| 3c (R = H, X = Br) | 6.6 ± 3.8% | 25.8 ± 6.3% | 6.1 ± 3.2% |
| 3d (R = H, X = I) | 9.6 ± 4.7% | 18.2 ± 7.1% | 13.9 ± 5.5% |
| 3e (R = Cl, X = F) | 7.9 ± 4.0% | 12.0 ± 12.0% | 17.6 ± 4.6% |
| 3f (R = Cl, X = Cl) | 3.2 ± 1.9% | 11.0 ± 6.5% | 13.6 ± 2.9% |
| 3g (R = Cl, X = Br) | 23.6 ± 3.4% | 2.0 ± 1.2% | 21.0 ± 4.7% |
| 3h (R = Cl, X = I) | 16.4 ± 4.6% | 18.4 ± 4.3% | 10.0 ± 5.0% |
| 2 | 2490 ± 940 | 341 ± 75 | 4.16 ± 0.50 |
Molecular docking study
Since A2A is the only subtype of adenosine receptors (ARs) whose X-ray crystal structure is available (PDB code: 3EML),10 compounds 2 and 3h were docked into the A2A AR structure, considering the flexibility of the binding site residues (Figure 2).
Figure 2.

Predicted binding modes of compounds 2 and 3h in the A2A AR crystal structure. (A) Compound 2 (in magenta) interacts with the binding site residues. (B) Compound 3h (in orange) shows various binding modes. The key residues are displayed in capped-stick with carbon atoms in white. Hydrogen bonds are drawn in black dashed lines and non-polar hydrogens are undisplayed for clarity.
The docking results of compounds 2 and 3h were significantly different as shown in Figure 2. Compound 1 (Ki = 341 nM for hA2A) showed the reasonable binding mode. Its N6-amino group formed an H-bond with Glu169, and the 2′- and 3′-OH groups were involved in H-bonding with Ser277 and His278. In addition, the adenine ring made the π-π stacking with Phe168 (Figure 2A).
In contrast with compound 2, compound 3h showed various binding modes, losing important interactions (Figure 2B). It may retain or lose the H-bonding interaction with Glu169 and/or the π-π stacking with Phe168. Furthermore, it is more likely to lose H-bonding of the OH groups in the thio-ribose with Ser277/His278. It might be due to the rotatable extra methylene group (between the thio-ribose and the adenine ring), and that might explain why compound 3h lost its binding affinity on A2A AR. Structural and conformational factors in the hydrophilic ribose binding region are known to be critical for nucleoside recognition in the ARs.7
3. Conclusions
In this study, the synthesis of homologated truncated 40-thioadenosine derivatives from D-mannose and assay of their binding affinities at the ARs were accomplished. The CH2 homologation inserted in place of the glycosyl bond was introduced to allow maximum binding interaction through the single bond rotation, but the rotatable extra methylene group resulted in losing important interactions as illustrated in various predicted binding modes.
Experimental Section
1H NMR spectra (CDCl3, CD3OD, or DMSO-d6) were recorded on Varian Unity Inova 400 MHz. Chemical shifts were reported in ppm units with TMS as the internal standard. 13C NMR spectra (CDCl3, CD3OD, or DMSO-d6) were recorded on Varian Unity Inova 100 MHz. Optical rotations were determined on Jasco β in methanol. UV spectra were recorded on U-3000 made by Histachi in methanol. Elementary analyses were measured on EA1110. The crude products were purified using a silica gel 60 (230–400 mesh, Merck). Reagents were purchased from Aldrich Chemical Company. All the anhydrous solvents were distilled over CaH2 or P2O5 or Na/benzophenone prior to the reaction.
Chemical Synthesis
(2,2-Dimethyl-tetrahydro-thieno[3,4-d][1,3]dioxol-4-yl)-methanol (8)
To a stirred solution of 68 (20 g, 90.8 mmol) in ethyl acetate (500 mL) was added Pb(OAc)4 (48.3 g, 109 mmol) at 0°C and the reaction mixture was stirred for 10 min at which time TLC indicated the absence of starting material. The reaction mixture was filtered and the filtrate was diluted with EtOAc and the organic layer was washed with saturated aqueous NaHCO3 solution, dried over anhydrous MgSO4, and evaporated. The residue was used next step without further purification. To a stirred solution of aldehyde (5, 5.6 g, 30.0 mmol) in MeOH (70 mL) was carefully added sodium borohydride (1.3 g, 33.6 mmol) in several portions at 0 °C and the reaction mixture was stirred for 30 min at the same temperature and neutralized with glacial AcOH. After the removal of the solvent, the mixture was partitioned between EtOAc (150 mL) and brine (100 mL). The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate = 2:1) to give 8 (5.2 g, 91%) as a colorless syrup: 1H NMR (CDCl3) δ 1.27 (s, 3 H, CH3), 1.47 (s, 3 H, CH3), 2.68 (br s, 1 H, OH), 2.84 (dd, 1 H, J = 2.0, 12.6 Hz), 3.06 (dd, 1 H, J = 5.2, 13.0 Hz), 3.38 (m, 1 H), 3.56 (dd, 2 H, J = 2.8, 8.8 Hz, HOCH2), 4.66 (dd, 1 H, J = 1.6, 5.6 Hz, HOCHH), 4.86 (td, 1 H, J = 1.6, 4.8 Hz, HOCHH).; 13C NMR (CDCl3) δ 24.75, 26.66, 37.42, 56.62, 63.32, 83.70, 85.94, 111.21.; FAB-MS m/z 191 (M++1).
4-Chloromethyl-2,2-dimethyl-tetrahydro-thieno[3,4-d][1,3]dioxole (9)
To a stirred solution of 8 (8.5 g, 40.7 mmol) in anhydrous acetonitrile (100 mL) was added POCl3 (4.47ml, 48.9 mmol) at 0°C and the reaction mixture was stirred for 30 min at room temperature. The reaction mixture was dried in vacuo and co-evaporated with toluene two times to give 9. The residue was used next step without further purification.: 1H NMR (CDCl3) δ 1.27 (s, 3 H, CH3), 1.46 (s, 3 H, CH3), 2.88 (m, 2 H), 3.08 (dd, 1 H, J = 4.8, 13.2 Hz), 3.44 (m, 1 H), 3.65 (dd, 1 H, J = 4.0, 8.2Hz), 4.81 (dd, 1 H, J = 0.8, 5.6 Hz), 4.89 (m, 1 H).; 13C NMR (CDCl3) δ 24.73, 26.54, 37.87, 45.72, 55.32, 83.58, 86.18, 111.31.
General Procedure for the Condensation
To a solution of 6-chloropurine (21.69 mmol) or 2,6-dichloropurine (21.69 mmol) in a solution of anhydrous DMF (50 mL) were added NaH (21.69 mmol) and the mixture was stirred at room temperature until the solution became clear. A solution of 9 (18.07 mmol) in anhydrous DMF (10 mL) was added to the resulting solution at room temperature and stirred for overnight at room temperature. The reaction mixture was diluted with EtOAc (100ml) and washed with water several times, dried with MgSO4 and evaporated. The residue was purified by flash silica gel column chromatography (Hexane:EtOAc = 2:1) to give the condensed product 10 and 11, respectively.
6-Chloro-9-(2,2-dimethyl-tetrahydro-thieno[3,4-d][1,3]dioxol-4-ylmethyl)-9H-purine (10)
Yield = 67%; white foam; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.032° (c 0.25, MeOH); FAB-MS m/z 327 (M++1); 1H NMR (CDCl3) δ 1.19 (s, 3 H, CH3), 1.38 (s, 3 H, CH3), 2.88(m, 2H, CH2), 3.72(td, 1H, J = 1.2, 2.4 Hz), 4.26 (dd, 1 H, J = 8.0, 16.0 Hz, CHHN), 4.59 (dd, 1 H, J = 6.8, 14.0 Hz, CHHN), 4.66 (dd, 1 H, J = 2.0, 5.0 Hz), 4.79 (m, 1 H), 8.28 (s, 1 H), 8.67 (s, 1 H); 13C NMR (CDCl3) δ 24.70, 26.49, 36.99, 46.07, 54.13, 83.20, 86.02, 111.88, 131.18, 145.53, 151.07, 151.84, 152.16; Anal. Calcd for C13H15ClN4O2S: C, 47.78; H, 4.63; N, 17.14; S, 9.81. Found: C, 47.82; H, 4.65; N, 17.20; S, 9.85.
2,6-Chloro-9-(2,2-dimethyl-tetrahydro-thieno[3,4-d][1,3]dioxol-4-ylmethyl)-9H-purine (11)
Yield = 72%; white foam; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.052° (c 0.30, MeOH); FAB-MS m/z 361 (M+); 1H NMR (CDCl3) δ 1.27 (s, 3 H, CH3), 1.47 (s, 3 H, CH3), 2.98(m, 2H, CH2), 3.74(td, 1H, J = 2.0, 6.2 Hz), 4.21 (dd, 1 H, J = 8.8, 14.2 Hz, CHHN), 4.47 (dd, 1 H, J = 6.8, 14.2 Hz, CHHN), 4.69 (dd, 1 H, J = 2.0, 5.6 Hz), 4.89 (m, 1 H), 8.23 (s, 1 H); 13C NMR (CDCl3) δ 24.82, 26.62, 37.04, 46.18, 54.13, 83.32, 86.22, 112.20, 146.13, 145.53, 152.14, 153.32, 153.37; Anal. Calcd for C13H14Cl2N4O2S: C, 43.22; H, 3.91; N, 15.51; S, 8.88. Found: C, 43.29; H, 3.96; N, 15.63; S, 8.82.
General Procedure for the Removal of the Isopropylidene Group
To a stirred solution of 10 (12.4 mmol) or 11 (12.4 mmol) in THF (50 mL) was added 2N HCl at room temperature and the mixture was stirred for 24 h at same temperature. The mixture was neutralized with NH4OH and evaporated. The residue was purified by flash silica gel column chromatography (CH2Cl2:EtOAc:MeOH = 10:10:1) to give the products, 12 and 13, respectively.
2-(6-Chloro-purin-9-ylmethyl)-teteahydro-thiophene-3,4-diol (12)
Yield = 94%; white foam; FAB-MS m/z 287 (M++1); 1H NMR (DMSO-d6) δ 2.62 (m, 1 H), 2.90 (dd, 1H, J = 4.0, 10.0 Hz), 3.70(m, 1H), 3.80(brs, 1H), 4.12 (brs, 1 H), 4.40(dd, 1H, J = 7.2, 13.6 Hz), 4.60 (dd, 1 H, J = 7.2, 13.6 Hz), 5.03 (brs, 2 H), 8.70 (s, 1 H), 8.78 (s, 1 H); 13C NMR (DMSO-d6) δ 33.04, 47.31, 48.00, 73.54, 77.40, 130.68, 147.60, 148.95, 151.51, 151.96..
2-(2,6-Chloro-purin-9-ylmethyl)-teteahydro-thiophene-3,4-diol (13)
Yield = 92%; white foam; FAB-MS m/z 321 (M+); 1H NMR (DMSO-d6) δ 2.61 (dd, 1 H, J = 4.0, 10.8 Hz), 2.92 (dd, 1H, J = 4.4, 10.8 Hz), 3.67(m, 1H), 3.80(dd, 1H, J = 3.2, 6.6 Hz), 4.11 (m, 1 H), 4.37(dd, 1H, J = 7.2, 14.0 Hz), 4.55 (dd, 1 H, J = 6.8, 14.2 Hz), 5.04 (brs, 2 H), 8.72 (s, 1 H); 13C NMR (DMSO-d6) δ 33.19, 47.52, 73.58, 77.74, 130.40, 147.75, 148.65, 149.57, 153.53, 153.75.
General Procedure for the N6-Substitution Reaction
To a stirred solution of 6-chloropurine (12) or 2,6-dichloropurine derivative (13, 0.45 mmol) and an appropriate amines · HCl or free amines (0.90 mmol) in EtOH (10 mL) was added Et3N (1.35 mmol) and the solution was stirred overnight at room temperature. After removing the solvent under reduced pressure, the residue was purified by flash silica gel column chromatography (CH2Cl2:EtOAc:MeOH = 10:10:1) to give the N6-substituted amine derivatives 3e-h.
N6-(3-Fluoro-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3a)
Yield = 75%; white solid; mp 117–120°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.016° (c 0.30, MeOH); 1H NMR (DMSO-d6) δ 2.61(dd, 1H, J = 5.2, 10.8 Hz), 2.88(dd, 1H, J = 5.2, 10.4 Hz), 3.68(m, 1H), 3.78(m, 1H), 4.14(m, 1H), 4.19(dd, 1H, J =7.6, 14.0 Hz), 4.47(dd, 1H, J = 6.0, 14.2 Hz), 4.66(brs, 2H), 5.02(d, 1H, J = 4.8 Hz), 5.09(d, 1H, J = 5.6 Hz), 7.10(t, 1H, J = 8.0 Hz), 7.36(d, 1H, J = 7.6 Hz), 7.73(s, 1H), 8.16(s, 1H), 8.21(s, 1H), 8.36(brs, 1H). 13C NMR (DMSO-d6) δ 32.82, 42.26, 46.81, 48.77, 73.56, 76.88, 94.70, 118.94, 126.69, 130.45, 135.32, 135.76, 141.04, 142.94, 148.94, 152.34, 154.17; FAB-MS m/z 376 (M++1); Anal. Calcd for C17H18FN5O2S: C, 54.39; H, 4.83; N, 18.65; S, 8.54. Found: C, 54.32; H, 4.96; N, 18.77; S, 8.62.
N6-(3-Chloro-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3b)
Yield = 83%; white solid; ; mp 143–147°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.023° (c 0.32, MeOH); 1H NMR (DMSO-d6) δ 2.61(dd, 1H, J = 5.2, 10.4 Hz), 2.87(dd, 1H, J = 5.2, 10.4 Hz), 3.68(m, 1H), 3.78(m, 1H), 4.16(m, 1H), 4.19(dd, 1H, J =7.6, 14.0 Hz), 4.46(dd, 1H, J = 8.0, 14.0 Hz), 4.69(brs, 2H), 5.01(d, 1H, J = 4.8 Hz), 5.08(d, 1H, J = 5.6 Hz), 7.25(t, 1H, J = 8.0 Hz), 7.35(d, 1H, J = 7.6 Hz), 7.39(d, 1H, J = 7.6 Hz), 7.54(s, 1H), 8.16(s, 1H), 8.22(s, 1H), 8.38(brs, 1H). 13C NMR (DMSO-d6) δ 32.81, 42.40, 46.81, 48.76, 73.56, 76.83, 118.93, 121.52, 126.28, 129.46, 129.86, 130.42, 141.06, 143.09, 148.97, 152.34, 154.18; FAB-MS m/z 392 (M++1); Anal. Calcd for C17H18ClN5O2S: C, 52.10; H, 4.63; N, 17.87; S, 8.18. Found: C, 52.33; H, 4.64; N, 17.90; S, 8.16.
N6-(3-Bromo-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3c)
Yield = 78%; white solid; mp 134–139°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.018° (c 0.25, MeOH); 1H NMR (DMSO-d6) δ 2.62(dd, 1H, J = 5.2, 10.6 Hz), 2.87(dd, 1H, J = 4.8, 10.2 Hz), 3.67(m, 1H), 3.78(m, 1H), 4.14(m, 1H), 4.19(dd, 1H, J =7.6, 14.2 Hz), 4.47(dd, 1H, J = 6.4, 14.0 Hz), 4.70(brs, 2H), 5.02(d, 1H, J = 5.2 Hz), 5.08(d, 1H, J = 5.6 Hz), 7.31(m, 3H), 7.39(s, 1H), 8.16(s, 1H), 8.21(s, 1H), 8.37(brs, 1H). 13C NMR (DMSO-d6) δ 32.18, 42.45, 46.81, 48.77, 73.56, 76.84, 118.96, 125.87, 126.56, 126.95, 130.10, 132.86, 141.06, 142.83, 148.99, 152.34, 154.21; FAB-MS m/z 436 (M+); Anal. Calcd for C17H18BrN5O2S: C, 46.80; H, 4.16; N, 16.05; S, 7.35. Found: C, 46.89; H, 4.21; N, 16.07; S, 7.41.
N6-(3-Iodo-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3d)
Yield = 75%; white solid; mp 172–175°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.024° (c 0.35, MeOH); 1H NMR (DMSO-d6) δ 2.61(dd, 1H, J = 5.2, 10.4 Hz), 2.87(dd, 1H, J = 5.2, 10.6 Hz), 3.67(m, 1H), 3.78(m, 1H), 4.14(m, 1H), 4.19(dd, 1H, J =7.6, 18.0 Hz), 4.46(dd, 1H, J = 6.4, 14.0 Hz), 4.71(brs, 2H), 5.02(d, 1H, J = 4.8 Hz), 5.08(d, 1H, J = 5.6 Hz), 7.00–7.19(m, 3H), 7.32(m, 1H), 8.16(s, 1H), 8.21(s, 1H), 8.36(brs, 1H). 13C NMR (DMSO-d6) δ 32.81, 42.51, 46.80, 48.76, 73.56, 76.83, 118.94, 123.15, 130.08, 130.16, 141.03, 143.28, 148.98, 152.34, 154.25, 160.95, 163.37; FAB-MS m/z 484 (M++1); Anal. Calcd for C17H18IN5O2S: C, 42.25; H, 3.75; N, 14.49; S, 6.63. Found: C, 42.34; H, 3.78; N, 14.51; S, 6.65.
2-Chloro-N6-(3-Fluoro-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3e)
Yield = 82%; white solid; mp 164–167°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.024° (c 0.50, MeOH); 1H NMR (DMSO-d6) δ 2.61(dd, 1H, J = 4.8, 10.8 Hz), 2.89(dd, 1H, J = 4.8, 10.4 Hz), 3.65(m, 1H), 3.78(m, 1H), 4.16(m, 2H), 4.43(dd, 1H, J =6.4, 14.0 Hz), 4.60(brd, 2H, J = 5.2 Hz), 5.03(d, 1H, J = 4.8 Hz), 5.09(d, 1H, J = 6.0 Hz), 7.12(t, 1H, J = 7.6 Hz), 7.36(d, 1H, J = 7.6 Hz), 7.59(d, 1H, J = 8.0 Hz), 7.74(s, 1H), 8.17(s, 1H), 8.83(brs, 1H). 13C NMR (DMSO-d6) δ 32.93, 42.54, 47.06, 48.36, 73.58, 77.14, 94.73, 118.06, 126.87, 130.52, 135.56, 136.08, 141.67, 141.94, 149.95, 152.95, 154.70; FAB-MS m/z 410 (M++1); Anal. Calcd for C17H17ClFN5O2S: C, 49.82; H, 4.18; N, 17.09; S, 7.82. Found: C, 49.94; H, 4.25; N, 17.12; S, 7.85.
2-Chloro-N6-(3-Chloro-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3f)
Yield = 77%; white solid; mp 154–158°C; UV (MeOH) λmax 271 nm (pH 7); [α]25D 0.031° (c 0.40, MeOH); 1H NMR (DMSO-d6) δ 2.62(dd, 1H, J = 4.8, 10.6 Hz), 2.90(dd, 1H, J = 4.8, 10.8 Hz), 3.65(m, 1H), 3.78(m, 1H), 4.11–4.20(m, 2H), 4.44(dd, 1H, J =6.0, 14.0 Hz), 4.63(brd, 2H, J = 6.5 Hz), 5.04(d, 1H, J = 4.8 Hz), 5.09(d, 1H, J = 5.6 Hz), 7.27(t, 1H, J = 7.6 Hz), 7.35(d, 1H, J = 7.6 Hz), 7.42(d, 1H, J = 7.6 Hz), 7.55(s, 1H), 8.17(s, 1H), 8.84(brs, 1H). 13C NMR (DMSO-d6) δ 32.93, 42.65, 47.06, 48.36, 73.59, 77.15, 118.07, 121.56, 126.45, 129.71, 130.14, 130.51, 141.69, 142.09, 149.97, 152.95, 154.72; FAB-MS m/z 426 (M+); Anal. Calcd for C17H17Cl2N5O2S: C, 47.89; H, 4.02; N, 16.43; S, 7.52. Found: C, 47.92; H, 4.05; N, 16.50; S, 7.55.
N6-(3-Bromo-benzylamino)-2-Chloro-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3g)
Yield = 73%; white solid; mp 165–169°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.035° (c 0.50, MeOH); 1H NMR (DMSO-d6) δ 2.62(dd, 1H, J = 4.4, 10.6 Hz), 2.89(dd, 1H, J = 5.2, 10.6 Hz), 3.66(m, 1H), 3.79(m, 1H), 4.13–4.21(m, 2H), 4.44(dd, 1H, J =6.4, 13.8 Hz), 4.65(brd, 2H, J = 5.2 Hz), 5.04(d, 1H, J = 4.8 Hz), 5.10(d, 1H, J = 5.6 Hz), 7.02–7.30(m, 4H), 8.17(s, 1H), 8.84(brs, 1H). 13C NMR (DMSO-d6) δ 32.95, 42.76, 47.08, 48.38, 73.62, 77.18, 118.01, 123.32, 130.21, 130.29, 141.67, 142.32, 149.98, 152.99, 154.81, 160.96, 163.37; FAB-MS m/z 472 (M++1); Anal. Calcd for C17H17BrClN5O2S: C, 43.37; H, 3.64; N, 14.88; S, 6.81. Found: C, 43.40; H, 3.70; N, 14.90; S, 6.85.
2-Chloro-N6-(3-Iodo-benzylamino)-9-ylmethyl-(4-thio-β-D-ribofuranosyl)adenine (3h)
Yield = 79%; white solid; mp 174–180°C; UV (MeOH) λmax 271nm (pH 7); [α]25D 0.045° (c 0.33, MeOH); 1H NMR (DMSO-d6) δ 2.62(dd, 1H, J = 4.8, 10.6 Hz), 2.89(dd, 1H, J = 4.8, 10.4 Hz), 3.65(m, 1H), 3.78(m, 1H), 4.13–4.20(m, 2H), 4.44(dd, 1H, J =6.0, 14.0 Hz), 4.64(brd, 2H, J = 4.8 Hz), 5.04(d, 1H, J = 4.8 Hz), 5.09(d, 1H, J = 5.6 Hz), 7.27–7.40(m, 4H), 8.17(s, 1H), 8.84(brs, 1H). 13C NMR (DMSO-d6) δ 32.94, 42.71, 47.07, 48.36, 73.60, 77.17, 118.07, 126.04, 126.81, 127.22, 130.19, 132.93, 141.69, 141.84, 149.98, 152.96, 154.75; FAB-MS m/z 518 (M++1); Anal. Calcd for C17H17ClIN5O2S: C, 39.43; H, 3.31; N, 13.53; S, 6.19. Found: C, 39.52; H, 3.35; N, 13.58; S, 6.22.
Molecular modeling
Ligand structures were generated with Concord and energy minimized using MMFF94s force field and MMFF94 charge until the rms of Powell gradient was 0.05 kcal mol−1A−1 in SYBYL 8.1.1 (Tripos Inc., St. Louis, MO, USA). The X-ray crystal structure of human A2A adenosine receptor (PDB code: 3EML) was prepared using Biopolymer Structure Preparation Tool in SYBYL. The docking study was performed using GOLD v.4.1.2 (Cambridge Crystallographic Data Centre, Cambridge, UK), which employs a genetic algorithm (GA) and allows for full ligand flexibility and partial protein flexibility. The binding site was defined as the 9 Å around the co-crystallized ligand (ZM-241385). The side chains of the eight residues (i.e. Thr88, Phe168, Glu169, Trp246, Leu249, Asn253, Ser277, and His278) in the crystal ligand binding site were set to be flexible with ‘crystal mode’. GoldScore scoring function was used and other parameters were set as suggested by the GOLD authors except the number of GA runs as 30. All computation calculations were undertaken on Intel® Xeon™ Quad-core workstation with Linux Cent OS release 4.6.
Figure 1.
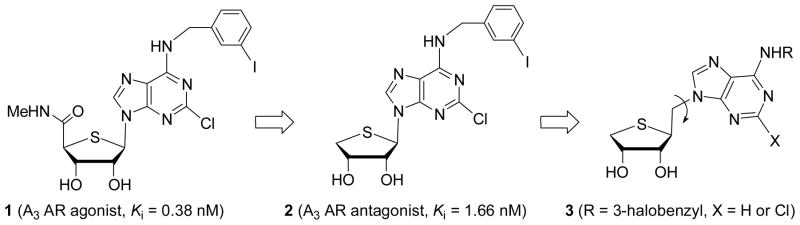
The rationale for the design of the target nucleoside 3
Acknowledgments
This work was supported by the grant from the Korea Research Foundation (NRF-2008-314-E00304) and by the NIDDK, NIH Intramural Research Program, and H. W. Lee acknowledges the research professor fellowship from the Ewha Womans University (2009).
References
- 1.Klotz KN. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:382–391. doi: 10.1007/s002100000315. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson KA, Gao ZG. Nature Rev Drug Disc. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baraldi PG, Cacciari B, Romagnoli R, Merighi S, Varani K, Borea PA, Spalluto G. Med Res Rev. 2000;20:103–128. doi: 10.1002/(sici)1098-1128(200003)20:2<103::aid-med1>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 4.Jeong LS, Jin DZ, Kim HO, Shin DH, Moon HR, Gunaga P, Chun MW, Kim Y-C, Melman N, Gao Z-G, Jacobson KA. J Med Chem. 2003;46:3775–3777. doi: 10.1021/jm034098e. [DOI] [PubMed] [Google Scholar]
- 5.Jeong LS, Lee HW, Jacobson KA, Kim HO, Shin DH, Lee JA, Gao ZG, Lu C, Duong HT, Gunaga P, Lee SK, Jin DZ, Chun MW, Moon HR. J Med Chem. 2006;49:273–281. doi: 10.1021/jm050595e. [DOI] [PubMed] [Google Scholar]
- 6.Kim SJ, Min HY, Chung HJ, Park EJ, Hong JY, Kang YJ, Shin DH, Jeong LS, Lee SK. Cancer Lett. 2008;264:309–315. doi: 10.1016/j.canlet.2008.01.037. [DOI] [PubMed] [Google Scholar]
- 7.Kim SK, Gao ZG, Jeong LS, Jacobson KA. J Mol Graph Model. 2006;25:562–577. doi: 10.1016/j.jmgm.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Jeong LS, Choe SA, Gunaga P, Kim HO, Lee HW, Lee SK, Tosh DK, Patel A, Palaniappan KK, Gao ZG, Jacobson KA, Moon HR. J Med Chem. 2007;50:3159–3162. doi: 10.1021/jm070259t. [DOI] [PubMed] [Google Scholar]; (b) Jeong LS, Pal S, Choe SA, Choi WJ, Jacobson KA, Gao ZG, Klutz AM, Hou X, Kim HO, Lee HW, Tosh DK, Moon HR. J Med Chem. 2008;51:6609–6613. doi: 10.1021/jm8008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. J Med Chem. 2002;45:4471. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. Biochem Pharmacol. 2003;65:1675. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao ZG, Jeong LS, Moon HR, Kim HO, Choi WJ, Shin DH, Elhalem E, Comin MJ, Melman N, Mamedova L, Gross AS, Rodriguez JB, Jacobson KA. Biochem Pharmacol. 2004;67:893. doi: 10.1016/j.bcp.2003.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paquette LA, Dong S. J Org Chem. 2005;70:5655. doi: 10.1021/jo0506985. [DOI] [PubMed] [Google Scholar]