

Abstract
Mutations in the PSEN1 gene encoding Presenilin-1 (PS1) are the predominant cause of familial Alzheimer's disease (FAD), but the underlying mechanisms remain unresolved. To reconcile the dominant action of pathogenic PSEN1 mutations with evidence that they confer a loss of mutant protein function, we tested the hypothesis that PSEN1 mutations interfere with γ-secretase activity in a dominant-negative manner. Here, we show that pathogenic PSEN1 mutations act in cis to impair mutant PS1 function and act in trans to inhibit wild-type PS1 function. Coexpression of mutant and wild-type PS1 at equal gene dosage in presenilin-deficient mouse embryo fibroblasts resulted in trans-dominant-negative inhibition of wild-type PS1 activity, suppressing γ-secretase-dependent cleavage of APP and Notch. Surprisingly, mutant PS1 could stimulate production of Aβ42 by wild-type PS1 while decreasing its production of Aβ40. Mutant and wild-type PS1 efficiently coimmunoprecipitated, suggesting that mutant PS1 interferes with wild-type PS1 activity via physical interaction. These results support the conclusion that mutant PS1 causes wild-type PS1 to adopt an altered conformation with impaired catalytic activity and substrate specificity. Our findings reveal a novel mechanism of action for pathogenic PSEN1 mutations and suggest that dominant-negative inhibition of presenilin activity plays an important role in FAD pathogenesis.
Introduction
Presenilins are the catalytic subunits of γ-secretase, which proteolyzes type I transmembrane protein substrates, such as Notch and APP. The pathogenic mechanism by which PSEN1 and PSEN2 (PSEN) mutations cause familial Alzheimer's disease (FAD) is a major unresolved problem with important therapeutic implications. Any tenable hypothesis to explain FAD pathogenesis must account for several fundamental properties of PSEN mutations: PSEN mutations are dominant, cause FAD in the heterozygous state, are missense in nature, display extreme allelic heterogeneity (>180 PSEN1 mutations affecting ∼20% of PS1 residues have been identified in FAD), impair the activity of the mutant protein, and cause cerebral deposition of β-amyloid (Aβ) peptides (for review, see Shen and Kelleher, 2007).
PSEN mutations have been presumed to cause FAD by enhancing the mutant protein's production of Aβ42 (Hardy and Selkoe, 2002). Such a “gain-of-normal-function” (i.e., hypermorphic) mechanism is compatible with the dominance of PSEN mutations and their ability to promote Aβ deposition. However, increased Aβ42 production is not a consistent property of presenilins bearing FAD mutations (Shioi et al., 2007; Heilig et al., 2010). Moreover, this hypothesis does not account for the allelic heterogeneity of PSEN mutations, the failure of Aβ42 overproduction to cause neurodegeneration in mouse models (Irizarry et al., 1997; Takeuchi et al., 2000), and growing evidence that pathogenic PSEN mutations impair protein function.
As an alternative view of FAD pathogenesis, we have proposed that a dominant-negative (i.e., antimorphic) mechanism offers the most straightforward explanation for the properties of PSEN mutations (Shen and Kelleher, 2007; Kelleher and Shen, 2010). The allelic heterogeneity of PSEN mutations implies that they cause FAD by impairing protein function, consistent with their deleterious impact on presenilin activity. However, the missense nature of PSEN mutations argues against a simple loss-of-function disease mechanism (i.e., haploinsufficiency). Rather, the absence of inactivating mutations implies that the mutant protein must be expressed to exert its pathogenic effect. Collectively, these features are most compatible with a “gain-of-negative-function” (i.e., dominant-negative) mechanism in which mutant presenilin with impaired function interferes with the activity of wild-type (WT) presenilin. Interestingly, we recently described a PSEN1 mutation in FAD that causes nearly complete loss of the mutant protein's activity (Heilig et al., 2010), suggesting that inactive mutant PS1 may also interfere with WT PS1 activity in a manner that promotes Aβ deposition in affected individuals.
The functional impact of PSEN mutations has generally been interpreted in terms of their cis (i.e., intramolecular) effects on the activity of the mutant protein. However, PSEN mutations cause FAD in the heterozygous state, raising the possibility that they could also interfere in trans with the function of WT presenilin. Here, we test the hypothesis that pathogenic PSEN mutations can influence WT PS1 activity in a dominant-negative manner by comparing the cis and trans effects of PSEN1 mutations on γ-secretase activity. Our results demonstrate that mutant PS1 can inhibit the γ-secretase activity of coexpressed WT PS1 while also enhancing its ability to produce Aβ42. These findings identify a novel dominant-negative mechanism through which PSEN1 mutations can perturb γ-secretase function in FAD.
Materials and Methods
DNA constructs.
WT PS1 cDNA expression vector (pCZ-PS1) and expression vectors for PS1 bearing C410Y or Δex9 mutations were kindly provided by W. Xia (Brigham and Women's Hospital) and have been previously described (Citron et al., 1997). Additional PS1 mutations (L166P, R278I, L435F, G384A, and L392V) were introduced into the PS1 coding sequence by site-directed mutagenesis of pCZ-PS1 using the GeneTailor system (Invitrogen). 3xHA and 3xFLAG epitope tags were appended to the N terminus of PS1 by insertion of a consensus Kozak sequence (GCCACCATG) followed by epitope tag sequences into pCI-PS1. This strategy resulted in insertion of four additional amino acids (Val-Asp-Ala-Thr) at the junction between epitope tag sequences and the PS1 coding sequence. Correct introduction of mutations and epitope tags was verified by bidirectional sequencing. Recombinant myc-tagged APP C99 and NotchΔE constructs were kindly provided by A. Goate (Washington University) (Kopan et al., 1996; Wang et al., 2004).
Cell culture.
Psen-null mouse embryo fibroblasts (MEFs) lacking endogenous PS1 and PS2 (provided by B. DeStrooper, K.U. Leuven, Belgium) (Herreman et al., 2003) were maintained in DMEM (Invitrogen) supplemented with 10% FBS (Hyclone) and transiently transfected with expression vectors encoding PS1 variants and substrates using Lipofectamine 2000 (Invitrogen). The concentrations of expression vectors were quantitatively determined by spectrophotometry (Nanodrop) and gel electrophoresis. Cell lysates were collected 20–22 h after transfection in modified RIPA buffer (50 mm Tris pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mm DTT, CompleteMini protease inhibitor mixture, Roche). Insoluble material was removed by centrifugation (16,000 × g, 10 min., 4°C). Protein concentration was determined by Bradford assay (Bio-Rad). For experiments examining PS1 protein turnover, cells transiently transfected with WT or mutant PS1 were treated 24 h after transfection with 100 μg/ml cycloheximide (Sigma), and cell lysates were subsequently collected at 6, 12, and 24 h time points.
Gel electrophoresis and Western blotting.
Cell lysates were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose membranes. After blocking in TBST/5% Blotto (Santa Cruz Biotechnology), membranes were incubated overnight with primary antibodies and developed the following day with HRP-linked anti-mouse or anti-rabbit secondary antibodies and LumiGLO chemiluminescent reagent (Cell Signaling Technology). To control for loading, membranes were stripped and reprobed with anti-α-tubulin antibody. Band intensity was quantified using ImageJ software (National Institutes of Health), and results were normalized to α-tubulin levels. Antibodies used included mouse anti-PS1-CTF (Millipore Bioscience Research Reagents, clone 5232), rat anti-PS1-NTF (Millipore Bioscience Research Reagents, clone 1563), rabbit anti-cleaved Notch (Val1744) (Cell Signaling Technology), mouse anti-myc (Sigma, clone 9E10), and mouse anti-α-tubulin (Sigma, clone B512).
ELISA.
Psen-null MEFs were cotransfected with expression vectors encoding WT and/or mutant PS1 and APP C99. After transfection, MEFs were cultured in DMEM/10% FBS supplemented with 10 μm phosphoramidon (Sigma). Conditioned medium was collected 20–22 h after transfection and centrifuged to remove insoluble material (10,000 × g, 10 min, 4°C). Sandwich ELISA was performed as previously described (Sun et al., 2005) with the following modifications. ELISA plates (Corning #3577) were coated with monoclonal antibodies recognizing Aβ40 (11A50-B10) or Aβ42 (12F4) (Covance Research Products) and blocked with 1% BlockACE (AbD Serotec). For detection of total Aβ, plates were coated with monoclonal antibody recognizing residues 3–14 (6E10, Covance). A biotinylated monoclonal anti-Aβ secondary antibody recognizing an internal sequence of both Aβ40 and Aβ42 (4G8, Covance) was used for simultaneous capture of Aβ in conditioned medium. Samples were diluted 1:3 in PBST containing 4G8 (1:2000) for detection of Aβ40, or 1:1 in PBST containing 4G8 (1:200) for detection of Aβ42, and immediately applied to precoated, blocked plates. Bound biotinylated antibody was detected with a streptavidin-conjugated alkaline phosphatase reporter system (Promega) and AttoPhos reagent (GE Healthcare). Synthetic Aβ40 and Aβ42 peptide standards diluted in cell culture medium were routinely included as quantification standards.
Membrane preparation and coimmunoprecipitation.
HEK 293T cells were transiently transfected with expression vectors encoding WT PS1 and PS1-L435F modified with N-terminal 3xHA and 3xFLAG epitope tags using Lipofectamine 2000 (Invitrogen). All subsequent steps were performed on ice or at 4°C. At 24 h after transfection, cells were washed twice in PBS, scraped in breaking buffer (20 mm HEPES, pH 7.3, 50 mm NaCl, 250 mm sucrose, 1 mm EDTA, 1 mm PMSF, 10 μm leupeptin, 1 μm pepstatin A, 1 μm aprotinin), and collected by centrifugation at 800 × g for 5 min. The resulting pellet was resuspended in breaking buffer and lysed using a Dounce homogenizer. Nuclei were removed by centrifugation at 800 × g for 5 min. The resulting supernatant was then subjected to centrifugation at 180,000 × g for 30 min to collect cell membranes. Pellets were resuspended in breaking buffer to a final protein concentration between 25 and 50 μg/μl, aliquoted, and snap-frozen in liquid nitrogen for subsequent analysis. Membranes were solubilized in solubilization buffer (50 mm Bis-Tris, pH 7.2, 50 mm NaCl, 10% glycerol (w/v), 1 mm PMSF, 10 μm leupeptin, 1 μm pepstatin A, 1 μm aprotinin) containing CHAPSO detergent at 4 g/g protein for 1 h at 4°C. Samples were then centrifuged at 16,100 × g for 30 min to remove insoluble material. For coimmunoprecipitation of PS1-containing complexes, supernatants were immediately incubated with antibody-coated magnetic beads at 4°C for 1.5 h. Beads were prepared in advance by conjugation of mouse anti-FLAG (clone M2, Sigma) or chicken anti-HA (Aves Laboratories) antibodies according to the manufacturer's instructions (Dynabeads, Invitrogen). After incubation at 4°C, beads were washed 3 times in solubilization buffer, followed by a 5 min wash at room temperature in solubilization buffer containing 0.02% Tween 20. Immunoprecipitates were eluted in 1× SDS gel-loading buffer (Bio-Rad) by heating at 55°C for 10 min. Proteins were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and analyzed by Western blotting with mouse anti-FLAG (M2, Sigma), mouse anti-HA (HA-11, Santa Cruz Biotechnology), mouse anti-PS1-CTF (5232, Millipore Bioscience Research Reagents), rabbit anti-Pen2 (Zymed), or rabbit anti-Nct (1660, Sigma).
Statistical analysis.
Data are presented as mean ± SE. Statistical significance at the 95% confidence interval was determined by Student's t test (two-tailed, unequal variance).
Results
Development of a quantitative cell-based assay for the functional effects of pathogenic PSEN1 mutations
We sought to develop a cell-based assay that would enable accurate assessment of the impact of pathogenic mutations on γ-secretase activity. First, we expressed WT and/or mutant PS1 in Psen-null MEFs (Herreman et al., 2003) to eliminate any confounding contribution of endogenous PS1 or PS2 to measured γ-secretase activity. Second, N-terminally truncated forms of APP and Notch (APP C99 and NotchΔE) that constitute the substrates for γ-secretase-mediated cleavage were coexpressed with WT and/or mutant PS1 to measure γ-secretase activity directly (Kopan et al., 1996; Wang et al., 2004). Third, our analysis used WT APP C99 because pathogenic APP mutations can exert independent effects on APP processing, potentially confounding assessment of the effects of PS1 mutations (Cai et al., 1993). Fourth, a key element of our approach is the expression of limiting quantities of PS1 in the presence of excess substrate to approximate steady-state conditions and to avoid confounding effects of substrate depletion on estimation of γ-secretase activity. Cell-based assays of γ-secretase activity typically depend on measurement of product accumulated over many hours. Under these conditions, a mutant enzyme with reduced catalytic efficiency could nevertheless appear to have WT levels of activity by converting a limiting quantity of substrate entirely to product. As a result, overexpression of PS1 could lead to systematic underestimation of the impact of pathogenic mutations on PS1 and γ-secretase activity.
Maturation of the γ-secretase complex is associated with PS1 endoproteolysis by an intrinsic presenilinase activity, which cleaves the PS1 holoprotein between transmembrane domains 6 and 7 to produce N-terminal and C-terminal fragments (NTF and CTF). Presenilinase cleavage requires the essential cofactor Pen2 and is thought to be required for full catalytic activity of the complex (Levitan et al., 2001; Luo et al., 2003; Ahn et al., 2010). We detected a progressive increase in the expression levels of PS1 NTF when Psen-null MEFs were transfected with increasing amounts of vector encoding WT PS1 (Fig. 1A,B). At higher levels of PS1 expression, accumulation of PS1 holoprotein was also observed. This dose–response relationship was linear across a 64-fold range of the amount of PS1 vector transfected, although PS1 NTF expression began to plateau at the highest amount transfected. These results indicate that PS1 expression is not saturable and that cofactors required for PS1 endoproteolysis are not limiting within the range of PS1 expression tested.
Figure 1.

Development of a linear dose–response assay for γ-secretase activity. A, Psen-null MEFs were transfected with increasing amounts of vector encoding WT PS1, and Western analysis was performed with antibody recognizing the PS1 N terminus. untrans, Untransfected MEFs. B, Quantification of PS1-NTF levels demonstrates linear nonsaturating expression as a function of increasing vector transfected. C–F, Psen-null MEFs were cotransfected with increasing amounts of vector expressing WT PS1 and a constant amount of vector expressing APP C99-myc (C,D) or NotchΔE-myc (E,F). Western analysis was performed with antibody recognizing the C-terminal myc epitope (C) or the cleaved N terminus of NICD (E). *γ-Secretase-independent band. Quantification of AICD (D) and NICD (F) production demonstrates that the activity assays saturated with respect to PS1 expression at >25 ng of PS1 vector transfected. Data are expressed as percentage of maximum NTF level ± SEM (n = 3 independent experiments).
Cleavage of substrates by γ-secretase requires prior cleavage by an accessory protease to remove the N-terminal extracellular/luminal ectodomain (Vassar et al., 1999; Yan et al., 1999; Brown et al., 2000; Kopan and Ilagan, 2009). APP undergoes initial cleavage by β-secretase, which yields a C-terminal fragment termed β-CTF or C99, and subsequent cleavage by γ-secretase at the γ- and ε-cleavage sites within the transmembrane region releases Aβ and the APP intracellular domain (AICD), respectively. Notch is sequentially cleaved by the metalloprotease ADAM-10 and γ-secretase to release the Notch intracellular domain (NICD). We evaluated the dose–response relationships for γ-secretase-mediated cleavage of APP C99 to produce AICD, and NotchΔE (equivalent to ADAM-10-cleaved substrate) to produce NICD, as a function of increasing PS1 expression (Fig. 1C–F). Production of both AICD and NICD increased linearly across the lower amounts of PS1 vector transfected, but saturated at higher levels. These results suggest that substrate is present in excess at lower levels of PS1 expression but becomes limiting at the higher levels of PS1 expression tested. All subsequent experiments analyzing the effects of FAD mutations were conducted under conditions of limiting PS1 and excess substrate within the linear range of these dose–response curves for γ-secretase activity.
Assessment of the cis and trans effects of FAD PSEN1 mutations on PS1 activity
PSEN1 mutations cause FAD in the heterozygous state, and the mutant PSEN1 allele exerts its pathogenic effect in the presence of a WT PSEN1 allele. It is therefore important to consider the cis (intramolecular) and trans (intermolecular) effects of FAD mutations on PS1 function. Whereas the pathogenicity of PSEN1 mutations has typically been viewed in terms of their cis effects on the function of the mutant protein, we examined the possibility that PSEN1 mutations can act in trans to interfere with the activity of WT PS1. Our strategy is based on comparison of the effects of mutant PS1 on γ-secretase activity when expressed in the absence or presence of WT PS1. If PSEN1 mutations caused a simple gain or loss of protein function, and thus affected PS1 function only in cis, coexpressed mutant and WT PS1 would contribute to the total observed γ-secretase activity in an additive manner. In the case of a dominant-negative interaction, however, mutant PS1 would exert a negative effect in trans on the activity of WT PS1, resulting in an infra-additive summation of their individual γ-secretase activities. This phenomenon would be most clearly illustrated in situations where mutant PS1 decreases total γ-secretase activity below the level displayed by WT PS1 alone. This difference between the expected effects of simple and dominant-negative loss-of-function mutations on WT PS1 activity provides a biochemical counterpart to the classical genetic observation that the phenotypic effects of a dominant-negative (i.e., antimorphic) allele are more severe than those of a null (i.e., amorphic) allele (Muller, 1932).
We first tested a series of FAD mutations for their cis effects on PS1 endoproteolysis by expressing either WT or mutant PS1 individually in Psen-null MEFs. The mutations selected for analysis are distributed throughout the PS1 coding sequence and have been previously reported to impair γ-secretase-dependent processing of APP and/or Notch (Shen and Kelleher, 2007; Heilig et al., 2010). An artificial PS1 mutation in which one of the catalytic aspartate residues is replaced by alanine (D257A) was used as a negative control. Western analysis with antibodies recognizing PS1 NTF and CTF revealed that all mutations analyzed decrease presenilinase processing of the mutant protein relative to WT PS1 (Fig. 2A,B). NTF and CTF were not detectable for R278I and L435F, and were reduced to <10% of WT levels for C410Y. The mutations L166P, G384A, and L392V reduced presenilinase cleavage to 50–60% of WT levels. The Δex9 mutation is not subject to presenilinase cleavage because it lacks the cleavage site encoded by exon 9. To exclude the possibility that impaired production of NTF and CTF by mutant PS1 proteins might be the result of decreased holoprotein stability, we assessed the stability of WT and mutant PS1 bearing either the L435F or C410Y mutations in Psen-null MEFs following translational inhibition with cycloheximide. Consistent with prior results (Wang et al., 2004), no differences were observed in the stability of mutant PS1 holoprotein relative to WT PS1 (data not shown).
Figure 2.

FAD PSEN1 mutations inhibit presenilinase activity of mutant PS1 in cis and WT PS1 in trans. A, B, To assess the cis effects of PSEN1 mutations on presenilinase activity, Psen-null MEFs were transfected with vector encoding WT or mutant PS1. PS1 endoproteolysis was assessed by Western analysis with antibodies recognizing PS1 holoprotein and NTF, or CTF. Quantification of PS1-NTF levels demonstrates significantly reduced presenilinase activity with all mutations. untrans, Untransfected MEFs. C, D, To assess the trans effects of PSEN1 mutations on presenilinase activity, Psen-null MEFs were transiently transfected with vector encoding WT and mutant PS1 in a 1:1 ratio. PS1 endoproteolysis was assessed by Western analysis. Coexpression of several PS1 mutants (R278I, L435F, Δex9) with WT PS1 significantly reduced PS1-NTF levels compared with the level observed for WT PS1 alone, indicating dominant-negative inhibition of WT endoproteolysis, whereas coexpression of the remaining PS1 mutants did not interfere with WT endoproteolysis. Data are expressed as percentage of WT level ± SEM (n = 3 independent experiments). *p < 0.05 relative to WT.
We next examined whether PS1 mutations can interfere in trans with presenilinase cleavage of WT PS1. To model the 1:1 gene dosage ratio in FAD patients heterozygous for PSEN1 mutations, Psen-null MEFs were transfected with equal quantities of vectors expressing WT and mutant PS1. In control experiments, transfection of Psen-null MEFs with equal quantities of vector-expressing WT PS1 and empty vector was used to determine the level of γ-secretase activity contributed by WT PS1 alone, and to model the effect of a heterozygous PSEN1 null mutation causing complete loss of function. Coexpression of WT PS1 with each of the three mutants (L166P, G384A, and L392V) that displayed a partial reduction in intrinsic presenilinase activity resulted in PS1-NTF levels that were ∼150–170% of the level displayed by WT PS1 alone, consistent with an additive contribution of WT and mutant PS1 to total presenilinase activity (Fig. 2C,D). However, coexpression of WT PS1 with mutants displaying little or no intrinsic presenilinase activity (R278I, L435F, and Δex9) decreased PS1-NTF levels to ∼70% of the level displayed by WT PS1 alone, suggesting that these mutations interfere with the presenilinase activity of WT PS1 in a dominant-negative manner.
FAD PSEN1 mutations inhibit γ-secretase-mediated proteolysis of APP in cis and in trans
We first assessed the cis effects of PSEN1 mutations on γ-secretase-mediated cleavage of APP by expressing the APP C99 substrate together with either WT or mutant PS1 in Psen-null MEFs. The use of APP C99 as substrate eliminates the requirement for prior cleavage by β-secretase, and fusion of a myc epitope tag to the C terminus facilitated detection of AICD by Western analysis. All seven of the FAD mutations analyzed caused a significant loss of function in the APP cleavage assay, with the degree of impairment varying among mutations (Fig. 3A,B). Several mutations (L166P, R278I, C410Y, L435F, and G384A) imparted a severe impairment in AICD production to <10% of WT levels. For the L435F and R278I mutations, AICD production was at or below the limit of detection, comparable with the effect of the catalytically inactive D257A mutation, indicating complete inactivation of APP proteolysis. The L392V and Δex9 mutations caused moderate reductions in APP cleavage activity (∼55% and 80% of WT AICD levels, respectively).
Figure 3.

FAD PSEN1 mutations inhibit APP processing by mutant PS1 in cis and by WT PS1 in trans in a dominant-negative manner. A, B, To assess the cis effects of PSEN1 mutations on APP processing, Psen-null MEFs were transfected with vector encoding WT or mutant PS1 together with vector encoding APP C99-myc substrate. C99 and AICD were detected by Western analysis with anti-myc antibody. untrans, Untransfected MEFs. *γ-Secretase-independent band. Quantification of AICD production shows that all mutations tested significantly reduced intrinsic PS1 activity. C, D, To assess the trans effects of PSEN1 mutations on APP processing, Psen-null MEFs were transfected with vectors encoding WT and mutant PS1 in a 1:1 ratio together with vector encoding APP C99-myc substrate. Quantification of AICD levels shows that mutant PS1 antagonized WT PS1 activity, indicated most clearly by suppression of AICD production below the level displayed by WT alone in the case of several mutations (L166P, R278I, C410Y, L435F). E, F, Psen-null MEFs were cotransfected with a constant amount of vector encoding WT PS1 and increasing amounts of vector encoding either L435F or C410Y mutant PS1, together with constant amount of vector encoding APP C99-myc. PS1 was detected by Western analysis with antibody recognizing the C terminus; C99 and AICD were detected with anti-myc antibody. Quantification of AICD production shows that PS1 bearing the L435F or C410Y mutations produced similar gene dosage-dependent suppression of AICD production by WT PS1, consistent with dominant-negative inhibition of γ-secretase activity. Data are expressed as percentage of WT level ± SEM (n = 3–5 independent experiments). *p < 0.05 relative to WT.
We next sought to determine whether mutant PS1 could interfere with the ability of WT PS1 to process APP. To model heterozygosity for a pathogenic PSEN1 mutation in FAD patients, Psen-null MEFs were transiently transfected with APP-C99 substrate and equal amounts of vectors expressing WT and mutant PS1. In control experiments, empty vector was substituted for vector expressing mutant PS1 to determine the level of γ-secretase activity conferred by WT PS1 alone and to model heterozygosity for a PSEN1 null mutation. Strikingly, coexpression of mutant PS1 with WT PS1 suppressed AICD production below the level displayed by WT PS1 alone in the case of several pathogenic mutations (L166P, R278I, L435F, and C410Y) (Fig. 3C,D). Whereas an additive contribution of mutant and WT PS1 to total γ-secretase activity would be expected to yield ≥100% of the AICD level produced by WT PS1 alone, each of these mutants reduced total AICD production to <80% of the WT level. Interestingly, these four mutations also displayed the most severe loss of intrinsic catalytic activity toward APP when assayed individually (Fig. 3A,B). These results show that pathogenic PS1 mutations can act in trans to exert a dominant-negative effect on the γ-secretase activity of WT PS1.
PS1 bearing the G384A and L392V mutations contributed approximately additively to total AICD production when coexpressed with WT PS1. However, the Δex9 mutation also appeared to exert a trans-dominant-negative effect on APP processing by WT PS1. Specifically, PS1 with the Δex9 mutation displayed ∼80% of WT activity when expressed alone (Fig. 3B), but total activity when coexpressed with WT PS1 was only ∼140% of the level displayed by WT PS1 alone. This infra-additive contribution of mutant PS1 to AICD production also suggests dominant-negative suppression of WT PS1 activity, paralleling the reduction in WT PS1 NTF level observed when PS1 with the Δex9 mutation was coexpressed (Fig. 2C,D).
An important property of dominant-negative mutations is that their inhibitory effect on the WT gene product is a function of the dosage of the mutant allele and the resulting expression level of the mutant protein (Muller, 1932; Herskowitz, 1987). We cotransfected Psen-null MEFs with a constant amount of vector encoding WT PS1 and increasing amounts of vector encoding PS1 carrying the L435F or C410Y mutations (Fig. 3E). The total amount of vector transfected was maintained within the linear range for PS1 expression and APP C99 cleavage (Fig. 1A–D). For both the C410Y and L435F mutations, transfection of increasing amounts of vector led to a corresponding increase in the level of PS1 holoprotein. Consistent with the results in Figure 2C, D, overexpression of L435F mutant PS1 progressively impaired the endoproteolysis of coexpressed WT PS1, whereas overexpression of C410Y mutant PS1 had no effect on endoproteolysis of WT PS1. Nevertheless, both mutations caused a gene dosage-dependent suppression of APP processing by WT PS1, consistent with dominant-negative inhibition of γ-secretase activity. Moreover, the similar inhibitory profiles of the L435F and C410Y mutations with respect to APP processing despite their differential effects on WT PS1 endoproteolysis argue that the observed dominant-negative inhibition is the result of interference with the catalytic activity of WT PS1 rather than its expression or endoproteolysis.
FAD PSEN1 mutations inhibit γ-secretase-mediated proteolysis of Notch in cis and in trans
We next analyzed the cis effects of the same series of FAD mutations on γ-secretase-dependent processing of Notch by expressing the Notch substrate (NotchΔE) together with either WT or mutant PS1 in Psen-null MEFs. The use of NotchΔE as substrate eliminates the requirement for initial cleavage by ADAM-10, and release of NICD therefore depends on cleavage by γ-secretase alone. All seven pathogenic mutations tested significantly impaired the ability of PS1 to support γ-secretase-dependent Notch processing and NICD production (Fig. 4A,B). The rank order of inhibitory effects caused by mutations was similar for the APP and Notch substrates, with the R278I, C410Y, and L435F mutations again causing nearly complete loss of γ-secretase product generation. The L166P and G384A mutations produced less severe impairment of NICD production than observed with AICD production. The moderate inhibition of γ-secretase activity produced by the L392V and Δex9 mutations was similar for both substrates.
Figure 4.

FAD PSEN1 mutations inhibit Notch processing by mutant PS1 in cis and by WT PS1 in trans in a dominant-negative manner. A, B, To assess the cis effects of PSEN1 mutations on Notch processing, Psen-null MEFs were transfected with vector encoding WT or mutant PS1 together with vector encoding NotchΔE-myc substrate. NICD was detected by Western analysis with antibody recognizing the cleaved N terminus (V1744), and both full-length NotchΔE and NICD were detected with anti-myc antibody. Quantification of NICD production shows that all mutations tested significantly reduced intrinsic PS1 activity. untrans, Untransfected MEFs C, D, To assess the trans effects of PSEN1 mutations on Notch processing, Psen-null MEFs were transfected with vectors encoding WT and mutant PS1 in a 1:1 ratio together with vector encoding NotchΔE-myc substrate. Quantification of NICD levels shows none of the mutations was able to elevate NICD production above the level displayed by WT PS1 alone, with some mutations significantly suppressing NICD production below this level (e.g., C410Y, L435F), consistent with dominant-negative inhibition of Notch processing. Data are expressed as percentage of WT level ± SEM (n = 5–6 independent experiments). *p < 0.05 relative to WT.
To determine whether PSEN1 mutations act in trans to inhibit Notch processing by WT PS1, we transfected Psen-null MEFs with Notch substrate (NotchΔE) and equal amounts of vector-expressing WT and mutant PS1. When coexpressed with WT PS1 at equal gene dosage, none of the PS1 mutants elevated Notch cleavage activity above the level displayed by WT PS1 alone, even though several PS1 mutants retained substantial intrinsic Notch processing activity (Fig. 4C,D). For example, PS1 carrying the G384A, L392V, and Δex9 mutations produced NICD at 40–70% of the WT level when expressed alone (Fig. 4B), and yet total NICD production when these mutants were coexpressed with WT PS1 did not differ from the level produced by WT PS1 alone, indicating an infra-additive summation of activities. Moreover, several PS1 mutants suppressed total NICD production below the level generated by WT PS alone, clearly illustrating a negative effect of mutant PS1 on the ability of WT PS1 to support Notch processing. For example, coexpression of PS1 carrying the C410Y or L435F mutations with WT PS1 diminished total NICD production to ∼65% and 50%, respectively, of the level produced by WT PS1 alone. Thus, the FAD mutations analyzed appeared to exert a trans-dominant-negative effect on Notch processing by WT γ-secretase.
FAD PSEN1 mutations act in trans to inhibit Aβ40 production and stimulate Aβ42 production by WT PS1
To determine the cis effects of FAD mutations on the ability of PS1 to support Aβ production, mutant or WT PS1 was expressed in Psen-null MEFs together with APP-C99, and levels of secreted Aβ peptides were quantified using Aβ40- and Aβ42-specific ELISA. Production of Aβ40 was reduced by all mutations tested, with five of the pathogenic mutations (L166P, R278I, C410Y, L435F, G384A) decreasing Aβ40 production to <5% of WT levels (Fig. 5A). Production of Aβ42 was dramatically decreased by several mutations (L166P, R278I, C410Y, L435F) that also caused severe deficits in generation of AICD and Aβ40 (Fig. 5B). Indeed, the levels of Aβ40 and Aβ42 produced by PS1 bearing the L435F and R278I mutations were at or below the limit of detection, indicating virtually complete loss of proteolytic activity. In contrast, the L392V mutation, which caused a milder reduction in total APP cleavage activity and Aβ40 production, significantly increased Aβ42 production.
Figure 5.

FAD PSEN1 mutations inhibit Aβ production by mutant PS1 in cis and modulate Aβ production by WT PS1 in trans. A, B, To assess the cis effects of PSEN1 mutations on Aβ production, Psen-null MEFs were transfected with vector encoding WT or mutant PS1 together with vector encoding APP C99-myc. Production of Aβ40 and Aβ42 was assayed by sandwich ELISA with isoform-specific antibodies. All PS1 mutants exhibited significantly impaired production of Aβ40. PS1 bearing the L166P, R278I, C410Y, and L435F mutations displayed significantly reduced Aβ42 production, whereas PS1 carrying the L392V and Δex9 mutations showed elevated Aβ42 production. C, D, To assess the trans effects of PSEN1 mutations on Aβ production, Psen-null MEFs were transfected with vectors encoding WT and mutant PS1 in a 1:1 ratio together with vector encoding APP C99-myc. Coexpression of mutant PS1 antagonized Aβ40 production by WT PS1, illustrated most clearly by significant reduction of Aβ40 production below the level produced by WT PS1 alone in the case of several mutations (R278I, C410Y, L435F). In contrast, several PS1 mutants stimulated Aβ42 production by WT PS1, as indicated by their supra-additive effect on Aβ42 production (e.g., L166P, C410Y, L435F). E, To assess the cis effects of PSEN1 mutations on total Aβ production, Psen-null MEFs were transfected with vector encoding WT or mutant PS1 together with vector encoding APP C99-myc. Total secreted Aβ levels were measured in culture supernatants by sandwich ELISA detecting all Aβ species ranging from Aβ38 through Aβ46. All PSEN1 mutations tested significantly impaired total Aβ production by the mutant protein, with several pathogenic mutants producing <10% of WT levels. F, To assess the trans effects of PSEN1 mutations on total Aβ production, Psen-null MEFs were transfected with vectors encoding WT and mutant PS1 in a 1:1 ratio together with vector encoding APP C99-myc. The effects of mutant and WT PS1 coexpression on total Aβ production were qualitatively similar to those observed for Aβ40 production (C), indicating that Aβ40 is the major Aβ species produced by WT and mutant PS1 under these conditions. Data are expressed as percentage of WT level ± SEM (n = 4–6 independent experiments). *p < 0.05 relative to WT.
To determine whether mutant PS1 can act in trans to influence Aβ production by WT PS1, we analyzed Aβ40 and Aβ42 production in Psen-null MEFs cotransfected with APP-C99 and equal amounts of vectors expressing WT PS1 and mutant PS1. In control experiments, empty vector was substituted for vector expressing mutant PS1 to establish the level of activity displayed by WT PS1 alone. Mutant PS1 bearing several pathogenic mutations (R278I, C410Y, L435F) compromised the ability of WT PS1 to produce Aβ40, as indicated by total Aβ40 production that fell significantly below the level produced by WT PS1 alone (Fig. 5C). With two additional mutations (L392V, Δex9), an infra-additive contribution of mutant PS1 to total Aβ40 production similarly suggested trans-dominant inhibition of WT PS1 activity. The cis and trans effects of each mutation on total Aβ corresponded closely to the observed effects on Aβ40, confirming that Aβ40 is the major Aβ species produced under these conditions (Fig. 5E,F).
Remarkably, mutant PS1 appeared to stimulate Aβ42 production by WT PS1 in the case of several pathogenic mutations (L166P, C410Y, and L435F) (Fig. 5D). Coexpression of each of these PS1 mutants with WT PS1 resulted in Aβ42 levels that were greater than would be expected if mutant PS1 contributed to Aβ42 production in an additive manner. This trans-dominant stimulatory effect of mutant PS1 was most clearly illustrated by the L435F mutation, which suppressed Aβ42 production by the mutant protein to <10% of WT levels, and yet coexpression of L435F mutant PS1 with WT PS1 significantly enhanced Aβ42 production to 140% of the level generated by WT PS1 alone. In contrast, several mutations that potentiated Aβ42 production by mutant PS1 when tested alone (G384A, L392V, Δex9) nevertheless resulted in infra-additive Aβ42 production when coexpressed with WT PS1. For example, the L392V mutation elevated Aβ42 production by mutant PS1 when tested alone to 212% of WT level, but coexpression of L392V mutant PS1 and WT PS1 did not further augment total Aβ42 production. Collectively, these results suggest complex modulation of Aβ42 production when mutant PS1 is coexpressed with WT PS1, with some mutations exerting a trans-dominant stimulatory effect and others a trans-dominant inhibitory effect on WT PS1 activity. Due to combined effects on Aβ42 and Aβ40, co-expression of mutant and WT PS1 elevated the Aβ42/Aβ40 ratio relative to expression of WT PS1 alone, with this elevation ranging from 1.5- to 2.2-fold among the mutations tested. Notably, the trans-stimulatory effects on Aβ42 and trans-inhibitory effects on Aβ40 exerted by mutations (e.g., L166P, C410Y, and L435F) that severely impair catalytic activity of the mutant protein may help to explain how such mutations can nonetheless promote Aβ42 deposition in the FAD brain.
Physical interaction between mutant and WT PS1 provides a mechanistic basis for the dominant-negative effects of PSEN1 mutations
A physical interaction between WT and mutant PS1 proteins would provide the most straightforward explanation for the observed trans-dominant effects of PS1 mutants on WT PS1 and γ-secretase activity. We examined this possibility by performing coimmunoprecipitation experiments with differentially epitope tagged mutant and WT PS1. First, we confirmed that FLAG- and HA-tagged WT PS1 displayed normal γ-secretase-dependent processing of APP and Notch, demonstrating that the presence of N-terminal epitope tags did not compromise PS1 activity (Fig. 6A). Next, we determined whether HA-tagged WT or L435F mutant PS1 stably interacted with FLAG-tagged WT PS1. To maintain γ-secretase complex integrity, we prepared membrane fractions from cells coexpressing differentially tagged PS1 proteins and solubilized proteins in CHAPSO detergent immediately before carrying out coimmunoprecipitation experiments. Immunoprecipitation of FLAG-tagged WT PS1 resulted in coimmunoprecipitation of either HA-tagged WT or HA-tagged L435F mutant PS1, accompanied by the γ-secretase subunits Nct and Pen2 (Fig. 6B). The interaction between WT PS1 and differentially tagged WT or L435F mutant PS1 was also detected in reciprocal experiments in which immunoprecipitation was performed with anti-HA antibody (Fig. 6C). Interestingly, WT PS1 holoprotein and NTF coimmunoprecipitated with differentially tagged WT PS1, suggesting that the interaction occurs before PS1 endoproteolysis. By contrast, the L435F mutation abrogates endoproteolysis of mutant PS1, and mutant holoprotein alone was therefore observed in coimmunoprecipitates with WT PS1. These results demonstrate that WT and mutant PS1 can stably interact in the context of the γ-secretase complex.
Figure 6.
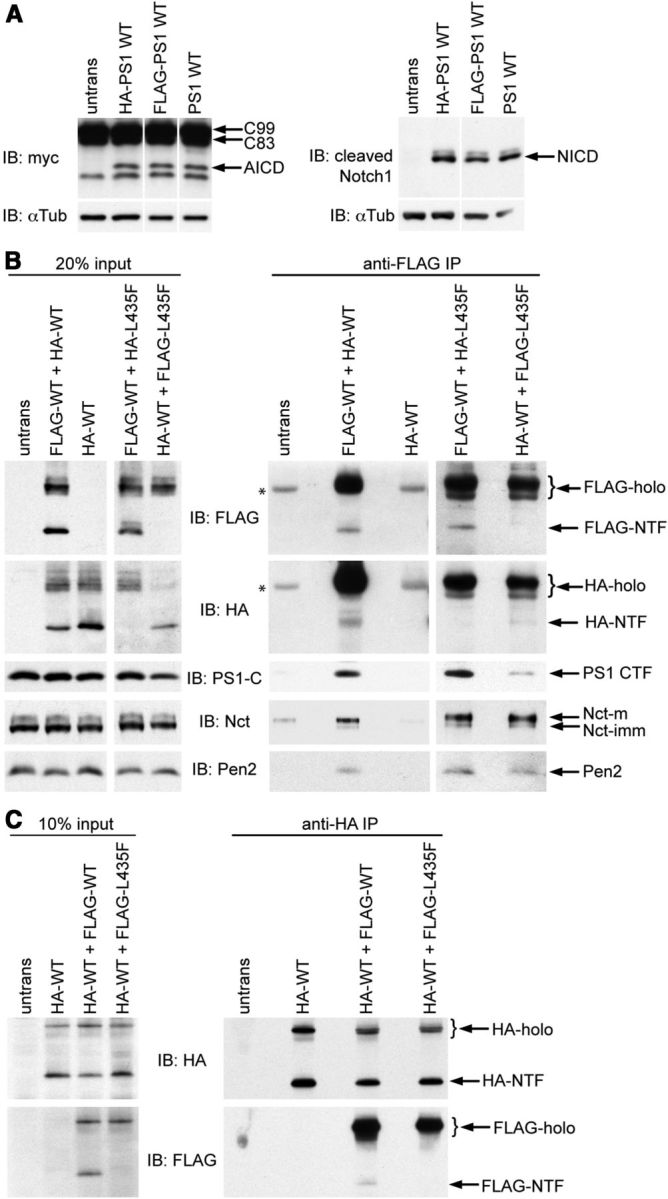
Physical interaction between mutant PS1 and WT PS1. A, To confirm that the epitope tags did not compromise γ-secretase activity, the ability of HA- and FLAG-tagged WT PS1 to reconstitute APP and Notch processing was assessed relative to untagged WT PS1. Psen-null MEFs were cotransfected with vector encoding untagged or tagged PS1 and vector encoding APP C99-myc or NotchΔE-myc substrate. AICD (left) and NICD (right) were detected by Western analysis with antibodies directed against the myc epitope or the cleaved N terminus of NICD. No differences in APP or Notch processing activity were observed between tagged and untagged PS1. B, WT and mutant PS1 bearing N-terminal FLAG or HA epitope tags were coexpressed in 293T cells. Expression levels of epitope-tagged PS1, PS1-CTF, and the endogenous γ-secretase components Nicastrin (Nct) and Pen2 were evaluated by Western analysis of cell lysates (left). Immunoprecipitation was performed on cell lysates with mouse anti-FLAG antibody. Western analysis of immunoprecipitates from cells coexpressing HA-PS1-WT and FLAG-PS1-WT (right) revealed coimmunoprecipitation of HA-PS1 holoprotein and NTF, PS1-CTF, Nct, and Pen2 with FLAG-PS1-WT (lane 2). Coimmunoprecipitation of HA-PS1 or γ-secretase components with anti-FLAG antibody was not observed in the absence of FLAG-PS1 (lanes 1 and 3). *Background bands resulting from interaction of primary antibodies with mouse IgG in immunoprecipitates. Analysis of immunoprecipitates from cells coexpressing FLAG-PS1-WT and HA-PS1-L435F (lane 4), or conversely HA-PS1-WT and FLAG-PS1-L435F (lane 5), revealed coimmunoprecipitation of mutant and WT PS1 in association with endogenous Nct and Pen-2. Epitope-tagged NTF is not detected for the PS1-L435F mutant because of its impaired presenilinase cleavage. C, To confirm the specificity of the interaction between WT and mutant PS1, the reciprocal experiment was performed with the epitopes used for immunoprecipitation and Western analysis reversed. Left, Expression of HA- and FLAG-tagged PS1 in cell lysates used for immunoprecipitation was evaluated by Western analysis. Right top, Immunoprecipitation was performed with chicken anti-HA antibody and confirmed by Western analysis with mouse anti-HA antibody. Right bottom, Western analysis with mouse anti-FLAG antibody revealed coimmunoprecipitation of HA-PS1-WT with either FLAG-PS1-WT holoprotein and NTF or FLAG-PS1-L435F holoprotein. FLAG-holo and HA-holo, PS1 holoprotein appended with the designated epitope tags; FLAG-NTF and HA-NTF, PS1 NTF appended with the designated epitope tags; PS1-C, antibody recognizing the PS1 C terminus; Nct-m and Nct-imm, the mature and immature forms of Nct, respectively; untrans, lysate from untransfected cells.
Discussion
Pathogenic PSEN1 mutations are invariably dominant and cause FAD in the heterozygous state. A complete picture of how PSEN1 mutations cause FAD therefore requires understanding how PS1 function is altered when mutant and WT PS1 are coexpressed as in FAD patients. However, the pathogenic effects of PSEN1 mutations have typically been viewed in terms of their impact on the activity of the mutant protein. Here, we examined the possibility that pathogenic PSEN1 mutations compromise the activity of WT PS1 by evaluating a series of FAD-causing PSEN1 mutations for their cis effects on mutant protein activity and their trans effects on coexpressed WT protein activity.
We assessed the cis effects of mutations by expressing either WT or mutant PS1 alone in Psen-null MEFs and comparing their abilities to reconstitute γ-secretase activity. To obtain accurate estimates of the intrinsic activity of mutant PS1, our analysis relied on limiting expression of PS1 in the presence of excess substrate, thereby avoiding factors that could lead to overestimation of mutant PS1 activity, such as saturation with excess PS1 or depletion of substrate. All seven pathogenic mutations analyzed significantly impaired presenilinase cleavage and γ-secretase-dependent proteolysis of Notch and APP substrates. Notably, several mutations caused severe deficiencies in the γ-secretase activity of the mutant protein, including marked diminution of Aβ40 and Aβ42 production. These findings illustrate that pathogenic PSEN1 mutations generally impair PS1 function and, in some cases, virtually abolish the ability of mutant PS1 to support γ-secretase activity (Heilig et al., 2010).
We evaluated the possibility that PSEN1 mutations perturb PS1 function in trans by measuring γ-secretase activity in Psen-null MEFs expressing WT PS1 in either the absence or the presence of mutant PS1. If WT and mutant PS1 contributed independently to total γ-secretase activity when coexpressed, additive summation of their individual activities would be expected. Remarkably, however, all seven clinical PSEN1 mutations tested interfered in trans with the γ-secretase activity of WT PS1, as indicated by less-than-additive contribution of mutant PS1 to total APP and/or Notch processing activity. The dominant-negative effect of PSEN1 mutations was demonstrated most clearly when coexpression of mutant PS1 with WT PS1 depressed γ-secretase activity below the level displayed by WT PS1 alone. For example, PS1 bearing the L166P, R278I, C410Y, or L435F mutations made a negative contribution to γ-secretase activity toward APP and/or Notch when coexpressed with WT PS1. In such cases, the contribution of mutant PS1 was effectively worse than nothing (i.e., empty vector). This behavior of PSEN1 mutations corresponds to the classical genetic observation that dominant-negative (“antimorphic”) mutations confer a more severe loss-of-function phenotype than null (“amorphic”) mutations in the same gene (Muller, 1932). Another defining characteristic of dominant-negative mutations is that their inhibitory potency is directly related to the mutant allele dosage (Muller, 1932; Herskowitz, 1987). Consistent with this property, we found that the extent of trans-dominant inhibition of APP processing by WT PS1 progressively increased as a function of increasing mutant gene dosage. Collectively, these findings demonstrate that PSEN1 mutations do not cause a simple loss of the mutant protein's function, but rather confer a gain of negative function that impairs the activity of both mutant and coexpressed WT proteins. The demonstration of a dominant-negative effect of PSEN1 mutations on WT PS1 bolsters the hypothesis that loss of presenilin function plays an important role in FAD pathogenesis (Shen and Kelleher, 2007; Kelleher and Shen, 2010).
The dominant-negative mechanism that we report here is fundamentally distinct from the reported ability of overexpressed mutant PS1 to inhibit γ-secretase activity by replacing endogenous PS1 (Thinakaran et al., 1996, 1997). Notably, our activity assays were conducted within a linear dose–response range for PS1 expression and activity, thereby avoiding competition between mutant and WT PS1 for limiting cofactors. Indeed, mutant PS1 could exert dominant-negative effects when expressed at equal gene dosage with WT PS1 (i.e., in the absence of overexpression), without compromising the expression or endoproteolysis of coexpressed WT PS1 (i.e., in the absence of replacement). Moreover, PSEN1 mutations acted in trans to enhance Aβ42 production by WT PS1, whereas replacement of WT PS1 by mutant PS1 would be expected to decrease all PS1 activities. Collectively, these observations provide strong evidence that mutant PS1 exerts dominant-negative effects by interfering with the catalytic activity rather than the expression or maturation of WT PS1.
To account for the ability of pathogenic PSEN1 mutations to inactivate Aβ production by the mutant protein and yet engender cerebral Aβ deposition, we previously proposed that mutant PS1 might stimulate Aβ42 production by WT PS1 (Shen and Kelleher, 2007; Heilig et al., 2010; Kelleher and Shen, 2010). Our results provide strong empirical support for this proposal, as evidenced by the synergistic effect on Aβ42 production observed when mutant PS1 was coexpressed with WT PS1. Remarkably, coexpression of L435F mutant PS1 with WT PS1 elevated Aβ42 production by ∼40% relative to the level produced by WT alone, even though the L435F mutation rendered the mutant protein itself incapable of producing Aβ42. By contrast, mutant PS1 with a simple loss of function would not be expected to increase either Aβ42 production or the Aβ42/Aβ40 ratio when coexpressed with WT PS1, consistent with observations in Psen1+/− mice (Qian et al., 1998). Thus, our findings strongly imply that the excess Aβ42 observed under these conditions is produced by WT PS1 under the trans-dominant influence of proteolytically inactive mutant PS1. This phenomenon may help to explain the seemingly paradoxical ability of PSEN1 mutations that abrogate γ-secretase activity to promote Aβ deposition in the FAD brain (Heilig et al., 2010). Our observations suggest that PSEN1 mutations may further contribute to elevation of the Aβ42/Aβ40 ratio and Aβ deposition through dominant-negative inhibition of Aβ40 production by WT PS1.
We suggest that the capacity of PSEN1 mutations to stimulate Aβ42 production by WT PS1 is a manifestation of dominant-negative impairment of PS1 function. Consistent with this view, we found that PSEN1 mutations could stimulate Aβ42 production in trans while inhibiting all other PS1 activities analyzed both in cis and in trans. Further support for this interpretation comes from the widely reported yet enigmatic ability of transition-state analog inhibitors of γ-secretase to stimulate Aβ42 production while inhibiting Aβ40 production (Wolfe et al., 1999; Li et al., 2000; Zhang et al., 2001). This effect occurs under conditions of submaximal inhibition when the PS1 population is partially occupied by inhibitor, suggesting that PS1 molecules with active site-bound inhibitor stimulate Aβ42 production by PS1 molecules without bound inhibitor. Thus, the ability of both PSEN1 mutations and active site-directed γ-secretase inhibitors to stimulate Aβ42 production may be similarly explained by a trans-dominant effect of catalytically impaired PS1 on catalytically intact PS1. Notably, our observation that PS1 bearing FAD mutations that abolish its catalytic activity can nevertheless stimulate Aβ42 production by WT PS1 argues against a model in which mutant PS1 itself produces a relative or absolute excess of Aβ42 owing to purely cis-acting effects (Wolfe, 2007).
Results from mouse genetic studies support the view that elevation of the Aβ42/Aβ40 ratio and brain Aβ deposition are manifestations of dominant-negative action of PSEN1 mutations. In APP transgenic mice heterozygous for a Psen1 M146V allele, deletion of the WT Psen1 allele markedly enhanced the Aβ42/Aβ40 ratio and Aβ deposition (Wang et al., 2006). Among the classes of dominant gain-of-function mutations, only antimorphic mutations cause phenotypic exaggeration upon deletion of the WT allele from heterozygous individuals; by contrast, deletion of the WT allele from heterozygotes ameliorates the phenotype of hypermorphic mutations and has no effect on the phenotype of neomorphic mutations (Muller, 1932). Thus, the effect of the M146V mutation on Aβ production and deposition in the mouse brain is most consistent with a dominant-negative mechanism.
Dominant-negative inhibition is commonly mediated by physical interaction between mutant and WT proteins (Herskowitz, 1987). Our demonstration of reciprocal coimmunoprecipitation of differentially tagged mutant and WT PS1 proteins suggests that mutant PS1 can stably interact with WT PS1 to interfere with its proteolytic activity. Consistent with these results, some prior studies have suggested that γ-secretase may form a stable higher-order assembly (Li et al., 2000; Edbauer et al., 2002; Steiner et al., 2002; Farmery et al., 2003; Li et al., 2003; Nyabi et al., 2003). Moreover, PS1 dimerization has been detected in cross-linking studies (Schroeter et al., 2003), and recent crystallographic analysis of an archael presenilin homolog revealed a tetrameric structure (Li et al., 2013). Our results provide independent evidence that PS1 functions as a multimer within the γ-secretase complex, and further imply that multimerization of mutant and WT PS1 could have strongly deleterious consequences for γ-secretase activity. For example, if mutant and WT PS1 form homodimers and heterodimers according to a standard binomial distribution, our results indicate that the L435F mutation renders mutant-WT heterodimers completely inactive for Notch processing.
The ability of mutant PS1 to interact with WT PS1 and thereby influence its catalytic activity implies that PS1 can adopt multiple conformations with distinct functional properties. Although mutant PS1 could in principle directly block WT catalysis through an orthosteric interaction (e.g., formation of a dimeric active site) (Schroeter et al., 2003), we suggest that an allosteric interaction is most compatible with the observation that mutant PS1 can inhibit WT PS1 activity while enhancing its production of Aβ42. Specifically, we propose a model in which FAD mutations cause PS1 to adopt an altered conformation with impaired catalytic activity; interaction of mutant PS1 with WT PS1 in turn induces an allosteric conformational change in the WT protein that impairs its catalytic activity, in some cases altering its substrate specificity to favor production of Aβ42 (Fig. 7). Recent structural analysis of related GXGD proteases supports the idea that PS1 can assume multiple conformations with differing catalytic properties (Hu et al., 2011). Both PSEN1 mutations and transition-state analog inhibitors appear to induce conformational changes within PS1 that can affect catalytic and specificity subsites (Takagi et al., 2010). The prediction that such conformational distortions can be transmitted allosterically from mutant or inhibitor-bound PS1 to intact PS1 awaits experimental confirmation.
Figure 7.

Dominant-negative model for inhibition of PS1 activity and stimulation of Aβ42 production by PSEN1 mutations and γ-secretase inhibitors. Schematic representation of possible interactions between WT and mutant or inhibitor-bound PS1 is shown. If PSEN1 mutations caused a simple loss of mutant protein function (i.e., a hypomorphic or amorphic mechanism), coexpression of WT and mutant PS1 as in FAD patients would be expected to result in additive summation of their individual activities, yielding total PS1 activity ≥ 100% of the level contributed by WT PS1 alone (left). In contrast, a reduction in total PS1 activity below the level displayed by WT PS1 alone would indicate dominant-negative inhibition of WT PS1 by mutant PS1 (i.e., an antimorphic mechanism) (middle). Our data suggest that mutant PS1 antagonizes WT PS1 activity through an allosteric physical interaction and that dominant-negative inhibition of WT PS1 can alter its substrate specificity to enhance its production of Aβ42. At low to moderate concentrations, active site-directed γ-secretase inhibitors can decrease Aβ40 production yet increase Aβ42 production, analogous to the effect of dominant-negative PSEN1 mutations (right). We suggest that inhibitor-bound (and thus catalytically inactive) PS1 can interact allosterically with unbound PS1, altering its substrate specificity to favor production of Aβ42.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke Grant R01NS041783 to J.S., National Institute of Neurological Disorders and Stroke Grant R01NS075346 to R.J.K., and the Alzheimer's Association (to J.S. and R.J.K.). E.A.H. was supported by National Institutes of Health Training Grant T32 AG000222-18. R.J.K. was supported by Pew Scholar and John Merck Scholar awards. We thank Dr. Alison Goate for the APP C99 construct, Dr. Raphael Kopan for the NotchΔE construct, and members of the J.S. and R.J.K. laboratories for helpful discussions.
The authors declare no competing financial interests.
References
- Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML, Sisodia SS, Li YM. Activation and intrinsic γ-secretase activity of presenilin 1. Proc Natl Acad Sci U S A. 2010;107:21435–21440. doi: 10.1073/pnas.1013246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/S0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Cai XD, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Haass C, Steiner H. Presenilin and nicastrin regulate each other and determine amyloid β-peptide production via complex formation. Proc Natl Acad Sci U S A. 2002;99:8666–8671. doi: 10.1073/pnas.132277899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmery MR, Tjernberg LO, Pursglove SE, Bergman A, Winblad B, Näslund J. Partial purification and characterization of γ-secretase from post-mortem human brain. J Biol Chem. 2003;278:24277–24284. doi: 10.1074/jbc.M211992200. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heilig EA, Xia W, Shen J, Kelleher RJ., 3rd A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of γ-secretase activity. J Biol Chem. 2010;285:22350–22359. doi: 10.1074/jbc.M110.116962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Van Gassen G, Bentahir M, Nyabi O, Craessaerts K, Mueller U, Annaert W, De Strooper B. γ-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J Cell Sci. 2003;116:1127–1136. doi: 10.1242/jcs.00292. [DOI] [PubMed] [Google Scholar]
- Herskowitz I. Functional inactivation of genes by dominant negative mutations. Nature. 1987;329:219–222. doi: 10.1038/329219a0. [DOI] [PubMed] [Google Scholar]
- Hu J, Xue Y, Lee S, Ha Y. The crystal structure of GXGD membrane protease FlaK. Nature. 2011;475:528–531. doi: 10.1038/nature10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Shen J. Genetics: γ-secretase and human disease. Science. 2010;330:1055–1056. doi: 10.1126/science.1198668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan D, Lee J, Song L, Manning R, Wong G, Parker E, Zhang L. PS1 N- and C-terminal fragments form a complex that functions in APP processing and Notch signaling. Proc Natl Acad Sci U S A. 2001;98:12186–12190. doi: 10.1073/pnas.211321898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ma G, Cai H, Price DL, Wong PC. Nicastrin is required for assembly of presenilin/gamma-secretase complexes to mediate Notch signaling and for processing and trafficking of β-amyloid precursor protein in mammals. J Neurosci. 2003;23:3272–3277. doi: 10.1523/JNEUROSCI.23-08-03272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Presenilin 1 is linked with γ-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem. 2003;278:7850–7854. doi: 10.1074/jbc.C200648200. [DOI] [PubMed] [Google Scholar]
- Muller HJ. Further studies on the nature and causes of gene mutations. Proceedings of the 6th International Congress of Genetics; 1932. pp. 213–255. [Google Scholar]
- Nyabi O, Bentahir M, Horré K, Herreman A, Gottardi-Littell N, Van Broeckhoven C, Merchiers P, Spittaels K, Annaert W, De Strooper B. Presenilins mutated at Asp-257 or Asp-385 restore Pen-2 expression and Nicastrin glycosylation but remain catalytically inactive in the absence of wild type Presenilin. J Biol Chem. 2003;278:43430–43436. doi: 10.1074/jbc.M306957200. [DOI] [PubMed] [Google Scholar]
- Qian S, Jiang P, Guan XM, Singh G, Trumbauer ME, Yu H, Chen HY, Van de Ploeg LH, Zheng H. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Abeta1–42/43 expression. Neuron. 1998;20:611–617. doi: 10.1016/S0896-6273(00)80999-X. [DOI] [PubMed] [Google Scholar]
- Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, Lewis HD, Saxena MT, De Strooper B, Coonrod A, Tomita T, Iwatsubo T, Moore CL, Goate A, Wolfe MS, Shearman M, Kopan R. A presenilin dimer at the core of the γ-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci U S A. 2003;100:13075–13080. doi: 10.1073/pnas.1735338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J Neurochem. 2007;101:674–681. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- Steiner H, Winkler E, Edbauer D, Prokop S, Basset G, Yamasaki A, Kostka M, Haass C. PEN-2 is an integral component of the γ-secretase complex required for coordinated expression of presenilin and nicastrin. J Biol Chem. 2002;277:39062–39065. doi: 10.1074/jbc.C200469200. [DOI] [PubMed] [Google Scholar]
- Sun X, Beglopoulos V, Mattson MP, Shen J. Hippocampal spatial memory impairments caused by the familial Alzheimer's disease-linked presenilin 1 M146V mutation. Neurodegener Dis. 2005;2:6–15. doi: 10.1159/000086426. [DOI] [PubMed] [Google Scholar]
- Takagi S, Tominaga A, Sato C, Tomita T, Iwatsubo T. Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the gamma-secretase. J Neurosci. 2010;30:15943–15950. doi: 10.1523/JNEUROSCI.3318-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid β precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–339. doi: 10.1016/S0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/S0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wang J, Brunkan AL, Hecimovic S, Walker E, Goate A. Conserved “PAL” sequence in presenilins is essential for γ-secretase activity, but not required for formation or stabilization of γ-secretase complexes. Neurobiol Dis. 2004;15:654–666. doi: 10.1016/j.nbd.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem. 2006;281:15330–15336. doi: 10.1074/jbc.M512574200. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40: talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:136–140. doi: 10.1038/sj.embor.7400896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Rahmati T, Donkor IO, Selkoe DJ. Peptidomimetic probes and molecular modeling suggest that Alzheimer's γ-secretase is an intramembrane-cleaving aspartyl protease. Biochemistry. 1999;38:4720–4727. doi: 10.1021/bi982562p. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Zhang L, Song L, Terracina G, Liu Y, Pramanik B, Parker E. Biochemical characterization of the γ-secretase activity that produces β-amyloid peptides. Biochemistry. 2001;40:5049–5055. doi: 10.1021/bi0028800. [DOI] [PubMed] [Google Scholar]