


Abstract
The cleavage of RNA can be accelerated by a number of factors. These factors include an acidic group (Lewis acid) or a basic group that aids in the deprotonation of the attacking nucleophile, in effect enhancing the nucleophilicity of the nucleophile; an acidic group that can neutralize and stabilize the leaving group; and any environment that can stabilize the pentavalent species that is either a transition state or a short-lived intermediate. The catalytic properties of ribozymes are due to factors that are derived from the complicated and specific structure of the ribozyme–substrate complex. It was postulated initially that nature had adopted a rather narrowly defined mechanism for the cleavage of RNA. However, recent findings have clearly demonstrated the diversity of the mechanisms of ribozyme-catalyzed reactions. Such mechanisms include the metal-independent cleavage that occurs in reactions catalyzed by hairpin ribozymes and the general double-metal-ion mechanism of catalysis in reactions catalyzed by the Tetrahymena group I ribozyme. Furthermore, the architecture of the complex between the substrate and the hepatitis delta virus ribozyme allows perturbation of the pKa of ring nitrogens of cytosine and adenine. The resultant perturbed ring nitrogens appear to be directly involved in acid/base catalysis. Moreover, while high concentrations of monovalent metal ions or polyamines can facilitate cleavage by hammerhead ribozymes, divalent metal ions are the most effective acid/base catalysts under physiological conditions.
INTRODUCTION
Naturally existing catalytic RNAs include hammerhead, hairpin, hepatitis delta virus (HDV) and Varkud Satellite (VS) ribozymes; group I and II introns; and the RNA subunit of RNase P (1–6). The structures of these catalytic RNAs are shown in Figure 1. In addition, recent structural and chemical analyses strongly suggest that the ribosomal RNA is a ribozyme (7–10) and the possibility that the RNA component of the spliceosome might also be a ribozyme (11).
Figure 1.

The two-dimensional structures of various ribozymes. The ribozyme or intron portion is printed in green. The substrate or exon portion is printed in black. Arrows indicate sites of cleavage by ribozymes. (A) Left, the two-dimensional structure of a hammerhead ribozyme and its substrate. Outlined letters are conserved bases that are involved in catalysis. Right, the γ-shaped structure of the hammerhead ribozyme–substrate complex. (B–F) The two-dimensional structures of a hairpin ribozyme, the genomic HDV ribozyme, a group I ribozyme from Tetrahymena, a group II ribozyme from Saccharomyces cerevisiae (aiγ 5) and scherichiathe ribozyme of RNase P from E.coli, respectively.
Extensive efforts over the 15 years that followed the discovery of ribozymes (1,2) have revealed details of the mechanisms of the ribozyme-mediated cleavage (or ligation) of RNA. Ribozymes have been considered to be ‘fossil molecules’ that originated in a hypothetical prebiotic RNA world and it is likely that elucidation of their mechanisms of action will enhance our understanding of the life processes of primitive organisms (12–33). Since the earliest research on ribozymes, it was assumed that all ribozymes are metalloenzymes that require divalent metal ions for catalysis and that all must operate by a basically similar mechanism. However, recent advances have revealed examples of cleavage by hairpin ribozymes that are independent of divalent metal ions (34–39). Thus, the various types of ribozyme appear to exploit different cleavage mechanisms, which depend upon the architecture of the individual ribozyme. Furthermore, it was proposed recently that nucleobases in the HDV ribozyme might be candidates for participants in acid/base catalysis (40–42).
In addition, even hammerhead ribozymes, generally characterized as typical metalloenzymes, can no longer be unambiguously categorized (43,44). Recent findings indicate that the hammerhead ribozyme might operate via a variety of cleavage mechanisms, depending on the conditions of the reaction. Nevertheless, there is no doubt that RNA catalysts with groups that are poorly functional under physiological conditions do cooperate with metal ions to exert their catalytic activity and that many ribozymes can exploit divalent metal ions as cofactors and as stabilizers of their respective higher-order structures. The widespread potential utility of RNA molecules as catalysts and the events during reactions catalyzed by ribozymes, in particular the actions of catalytic functional groups such as metal ions and pKa-perturbed nucleobases, have generated considerable interest (1–49). In this article we shall review various naturally existing ribozymes that cleave RNAs, focusing mainly on the various mechanisms of catalysis.
CLEAVAGE OF THE PHOSPHODIESTER BOND
For the cleavage of RNA phosphodiester linkages, three types of large ribozyme, namely, group I and II introns and the catalytic RNA subunit of RNase P, accept external nucleophiles (the 2′-OH group of an internal adenosine in the case of the group II intron). By contrast, small ribozymes, such as hammerheads, hairpins, HDV and the VS ribozyme, use an internal nucleophile, namely, the 2′-oxygen of the ribose moiety at the cleavage site, with resultant formation of a 3′-terminal 2′,3′-cyclic phosphate. In general, ribozymes catalyze the endonucleolytic transesterification of the phosphodiester bond, requiring structural and/or catalytic divalent metal ions under physiological conditions. The reactions catalyzed by small ribozymes are considered to be roughly equivalent to the non-enzymatic hydrolysis of RNA, with inversion of the configuration at a phosphorus atom suggesting a direct in-line attack with development of a pentacoordinate transition state or intermediate. The chemical cleavage requires two events, which can occur either via a two-step mechanism or via a concerted mechanism (4,5,25,45).
In the first step of the non-enzymatic hydrolysis of RNA (25,50–52), the 2′-OH attacks the adjacent scissile phosphate, acting as an internal nucleophile (transition state 1; TS1) (Fig. 2). In the second step, the 5′-oxygen of the leaving nucleotide is released to produce a 3′-end 2′,3′-cyclic phosphate and a 5′-OH terminus (transition state 2; TS2). Of the two putative transition states, TS2 is the overall rate-limiting state [i.e., attack by the 2′-OH on the phosphorus atom is easier than cleavage of the P-O(5′) bond and, thus, TS2 always has higher energy than TS1] (25). This conclusion was confirmed in experiments with an RNA analog with a 5′-mercapto leaving group. If the formation of the intermediate were the rate-limiting step (i.e., if TS1 were a higher-energy state than TS2) in the natural RNA, the phosphorothiolate RNA (RNA with a 5′-bridging phosphorothiolate at the scissile linkage) should be hydrolyzed at a rate similar to the rate of the hydrolysis of the natural RNA because the 5′-bridging phosphorothiolate linkage would not be expected to enhance the attack by the 2′-oxygen (53). By contrast, if the decomposition of the intermediate were the rate-limiting step (i.e., if TS2 were a higher-energy state than TS1) in the natural RNA, the phosphorothiolate RNA would be expected to be hydrolyzed much more rapidly than the natural RNA because the pKa of a thiol is >5 units lower than that of the corresponding alcohol. Several groups have confirmed that the phosphorothiolate RNA is significantly more reactive than the corresponding natural RNA in non-enzymatic hydrolytic reactions (25,45,54–56) and, thus, TS2 is, indeed, always a higher-energy state than TS1.
Figure 2.

The two-step reaction scheme for the hydrolysis of a phosphodiester bond in RNA. First, the 2′-oxygen attacks the phosphorus atom, acting as an internal nucleophile, to generate the pentacoordinated intermediate or transtion state TS1. The 5′-oxygen then departs from the intermediate to complete cleavage at TS2. TS1 can be stabilized by a general base catalyst and TS2 can be stabilized by a general acid catalyst, as illustrated at the summits of the energy diagram. These transition states can also be stabilized by the direct binding of Lewis acids to the 2′-attacking oxygen and the 5′-leaving oxygen.
POSSIBLE CATALYTIC FUNCTIONS OF METAL IONS IN THE CLEAVAGE OF RNA
If ribozymes operate as metalloenzymes (4,5,15–25,27,45), the possible catalytic functions of metal ions can be summarized as follows (Fig. 3).
Figure 3.

Possible catalytic functions of metal ions in the cleavage of a phosphodiester bond. Metal ions can act as (a) a general acid catalyst, (b) a general base catalyst, (c) a Lewis acid that stabilizes the leaving group, (d) a Lewis acid that enhances the deprotonation of the attacking nucleophile and (e) an electrophilic catalyst that increases the electrophilicity of the phosphorus atom.
· A metal-coordinated hydroxide ion might act as a general base, abstracting the proton from the 2′-OH (Fig. 3b) or, alternatively, a metal ion might act as a Lewis acid to accelerate the deprotonation of 2′-OH by coordinating directly with the 2′-oxygen (Fig. 3d).
· The developing negative charge on the 5′-oxygen leaving group might be stabilized by a proton that is provided by a solvent water molecule or by a metal-bound water molecule as a general acid catalyst (Fig. 3a) or, alternatively, by direct coordination of a metal ion that acts as a Lewis acid catalyst (Fig. 3c).
· Direct coordination of a metal ion to the non-bridging oxygen might render the phosphorus center more susceptible to nucleophilic attack (electrophilic catalysis; Fig. 3e) or, alternatively, hydrogen bonding between a metal-bound water molecule and the non-bridging oxygen might stabilize the charged trigonal-bipyramidal intermediate (or transition state).
Metal ions can function in several different ways as cofactors in ribozyme-catalyzed reactions, as described above, and proposed mechanisms for the reactions catalyzed by several ribozymes have taken advantage of such functions. Moreover, it is difficult to imagine that a specific ribozyme might exploit multiple mechanisms (for example, coexistence in a reaction of the left structure and the central structure in Figure 3 at the transition state of a ribozyme-catalyzed reaction) under a single set of physiological conditions. Significant aspects of these functions of metal ions might be subsumed by nucleobases if their pKa values could be adjusted appropriately. The full details of the mechanisms of action of metalloenzymes remain to be elucidated.
LARGE RIBOZYMES
The group I intron, the group II intron and the RNA subunit of RNase P are categorized as large ribozymes. Group I and II introns are found in bacteria and in the organelles of higher plants, fungi and algae (57,58). These introns are spliced out of their primary transcripts by a two-step mechanism [Fig. 4A(i) and B(i)]. In the first step of splicing, the 5′ splice site is attacked by the 3′-OH of the external guanosine (group I). Alternatively, it is attacked by the 2′-OH of the internal adenosine residue or by a hydroxide ion, in the case of hydrolysis (group II). In the second step, the 3′-OH of the 3′-end of the upstream exon attacks the 3′ splice site to produce splicing products.
Figure 4.

A schematic representation of splicing reactions and the structures of transition states at each step. (A) The group I intron splicing reaction. (i) In the first step, the 3′-OH of the exogenous conserved G attacks the phosphorus at the 5′ splice site and generates the G-attached intron 3′–exon 2 intermediate and a free 5′ exon 1. In the second step, the 3′-OH of the 5′ exon 1 attacks the phosphorus at the 3′ splice site to produce ligated exons and the excised G-attached intron. (ii) The proposed chemical mechanism of the first step. The 3′-OH of the exogenous G is a nucleophile and the 3′-OH of the U–1 is a leaving group. One of the Mg2+ ions [site (b)] coordinates with the 3′-OH of the G to activate the attacking group. The second [site (c)] coordinates with the 2′-OH of the G. The third [site (d)] coordinates with the pro-Sp oxygen to stabilize the transition state or the intermediate. The fourth [site (a)] coordinates with the 3′-OH of the U–1 to stabilize the leaving group. The 2′-OH also protonates the 3′-leaving oxygen of the U–1. It is not known whether or not the metal ion at site (d) is the same as those at the other sites, (a), (b) and (c) (72). IGS represents the internal guide sequence. (B) The group II intron splicing reaction. (i) In the first step, the 2′-OH of an A residue that is conserved in the intron attacks the phosphorus at the 5′ splice site and generates an intron 3′–exon 2 intermediate and a free 5′ exon 1. In the second step, the free 3′-OH of the 5′ exon attacks the phosphorus at the 3′ splice site to produce ligated exons and an excised intron. SER indicates the spliced-exon reopening reaction. (ii) The proposed chemical mechanisms of the first and the second steps. In the first step, the 2′-OH of an intron A residue is the nucleophile and the 3′-OH of the 5′ splice site terminus is the leaving group. One Mg2+ ion coordinates with the 3′-OH to stabilize the leaving group. Other coordinations and/or interactions remain to be clarified. In the second step, the 3′-OH of the C, the 5′ splice site terminus, becomes the nucleophile and the 3′-OH of the U is the leaving group. One Mg2+ ion coordinates directly with the 2′-OH and the 3′-OH of the U. Other coordinations and/or interactions remain to be clarified.
RNase P is an endonuclease that generates the mature 5′-ends of tRNAs. In bacterial RNase P, the RNA subunit (RNase P ribozyme) has catalytic activity and the protein component is thought to act only to facilitate the binding of the anionic RNase P ribozyme to its substrate. However, mutations in either the gene for the RNA or the gene for the protein can inactivate RNase P in vivo, demonstrating that both components are necessary for natural enzymatic activity. In the cleavage by the RNase P ribozyme, a scissile-site phosphate is attacked by a hydroxide ion to leave a 3′-oxygen and to produce a 5′-phosphate terminus.
All of these ribozyme reactions proceed with inversion of configuration at a phosphorus atom (59–62), suggesting direct in-line attack with development of a pentacoordinate transition state or intermediate (59–72).
THE MECHANISM OF REACTIONS CATALYZED BY THE GROUP I INTRON RIBOZYME
In studies of the reactions mediated by the ribozyme from the Tetrahymena group I intron, detailed kinetic and thermodynamic analysis, combined with modifications at the atomic level, helped to define the reaction mechanism of this ribozyme at the atomic level (18,63,66,68–72). Modification at the atomic level has generally involved replacement by a sulfur atom of an oxygen atom that has the potential to interact with a catalytically important metal ion. The observed reduction in the cleavage rate in the presence of Mg2+ ions after such modification (the ‘thio effect’) and the observed restoration of a normal cleavage rate in the presence of Mn2+ ions (the ‘manganese rescue effect’) have been taken as evidence that supports the direct coordination of the atom in question with a metal ion. This phenomenon can be explained by the HSAB (Hard and Soft, Acid and Base) rule (73,74). According to this rule, a ‘hard acid’, such as a Mg2+ ion, prefers to bind to a ‘hard base’ oxygen atom rather than to a ‘soft base’ sulfur atom. By contrast, a ‘soft acid’, such as Cd2+ or Zn2+ ions, prefers to bind to a ‘soft base’ sulfur atom. A Mn2+ ion is also softer than a Mg2+ ion and, thus, the former can bind to a soft sulfur atom (as well as to a hard oxygen atom). This ability of Mn2+ ions is believed to be the origin of the manganese rescue effect.
Analysis of both the thio effect and of soft acid rescue effects, such as the rescue effects of Cd2+, Mn2+ and Zn2+ ions, has contributed significantly to our understanding of the catalytic mechanism of the first step of the reaction catalyzed by the group I intron. Such analysis has revealed the importance of three to four independent metal ions, as shown in Figure 4A(ii) (69,71). It is generally accepted that the group I intron is a metalloenzyme that operates via the general double-metal-ion mechanism of catalysis, in which a Mg2+ ion at site (b) [see Fig. 4A(ii) for locations of (a), (b), (c) and (d)] enhances the deprotonation of the 3′-OH of the guanosine nucleophile and a Mg2+ ion at site (a) stabilizes the leaving 3′-bridging oxygen of U–1 in the transition state. In this case, the divalent metal ions function as Lewis acids for activation of the nucleophile and stabilization of the leaving group by coordinating directly with them (63). This mechanism corresponds, in the reactions catalyzed by small ribozymes, to the central mechanism shown in Figure 3 with the stabilization of both TS1 and TS2 (Fig. 2) by two metal ions.
These details of coordination at the catalytic site were derived from the following observations. The substitution of the 3′-oxygen of the guanosine nucleophile with a sulfur atom reduced the rate of the reverse reaction in the presence of the hard acid, namely, Mg2+ ions, and an efficient cleavage was restored by Mn2+ ions (66). This result suggests that a Mg2+ ion at site (b) coordinates with the 3′-oxygen of the guanosine nucleophile to activate the first step. Next, the 3′-bridging phosphorothiolate substrate (3′-S substrate), in which the 3′ leaving oxygen had been replaced by a sulfur atom, had a dramatically reduced cleavage rate for the forward reaction in the presence of Mg2+ ions. An efficient cleavage was restored by Mn2+ ions (63,75). This result suggests that a Mg2+ ion at site (a) in Figure 4A(ii) coordinates with the 3′-leaving oxygen during cleavage. These observations can be explained by the double-metal-ion model, in which one Mg2+ ion coordinates with the nucleophile to activate the attacking group and the other Mg2+ ion coordinates with the 3′-leaving oxygen to stabilize the developing negative charge during RNA cleavage.
The possibility of coordination of a Mg2+ ion at site (d) in Figure 4A(ii) with the pro-Sp oxygen was suggested on the basis of the following experimental data. The RpS substrate, in which the pro-Rp oxygen at the scissile phosphate had been replaced by sulfur, was cleaved at a modestly reduced rate (76). By contrast, the SpS substrate, in which the pro-Sp oxygen at the scissile phosphate had been replaced by sulfur, had a drastically reduced cleavage rate in the presence of Mg2+ ions (71,75,77). Furthermore, the SpS/3′-S substrate, in which not only the pro-Sp oxygen but also the 3′-leaving oxygen had been replaced by sulfur atoms at the same scissile phosphate, was cleaved at a lower rate than the 3′-S substrate in the presence of Mn2+ ions on a background of Mg2+ ions. An efficient cleavage of the SpS/3′-S substrate, with the double-thio substitution, was restored by Zn2+ or Cd2+ ions, which are more thiophilic than Mn2+ ions, on a background of Mg2+ ions (75). Thus, a thio effect seemed apparent at the pro-Sp oxygen, and rescue both by Cd2+ and by Zn2+ ions was also evident. These results suggested that a Mg2+ ion(s) might coordinate with the pro-Sp oxygen, as well as with the 3′-leaving oxygen.
However, in consideration of the above results, we should note that an efficient cleavage of the SpS substrate, with the single-thio substitution, could not be restored either by Mn2+ ions or by Zn2+ or Cd2+ ions (75). This observation prevents us from ruling out the possibility that a Mg2+ ion does not coordinate with the pro-Sp oxygen in a direct manner during the first step in cleavage by the group I intron ribozyme (72). Further investigations are needed to determine whether direct coordination of a Mg2+ ion occurs at site (d).
Additional coordination has also been proposed at the catalytic site. A Mg2+ ion at site (c) in Figure 4A(ii) might interact directly with the 2′-OH of the guanosine, as suggested by experiments with a 2′-amino-2′-deoxyguanosine substrate and various metal ions in the ribozyme reaction (78–80). The cleavage rate was reduced by replacement of the 2′-OH with 2′-NH2 on a background of Mg2+ ions. An efficient cleavage was restored by addition of soft Mn2+ or Zn2+ ions (78,79). This result suggests that a metal ion at site (c) coordinates directly with the 2′-OH.
In addition to the coordination of metal ions discussed above, other interesting interactions have been proposed. Linear free-energy analysis of the cleavage of oligonucleotide substrates with a series of 2′-substituents at U–1 indicated that the effect on the rate of the 2′-OH group is larger than might be expected from simple inductive effects (81). The weaker electron-withdrawing 2′-OH enhanced the chemical cleavage step to a greater extent than did the more strongly electron-withdrawing 2′-F atom of the corresponding 2′-deoxy-2′-fluoro derivative. Therefore, the possibility was recently examined of a symmetrical transition state, in which the 2′-OH of U–1 might or might not interact with a metal ion [as observed at site (c) in Fig. 4A(ii)] (71). Despite the absence of lone-pair electrons at the 2′-NH3+ group that need to interact with a metal ion, the higher reactivity of the substrate with a 2′-deoxy-2′-NH3+ group than that of the substrate with a 2′-OH group at U–1 suggested that interaction of a metal ion with the 2′-OH of U–1 might not be important for catalysis by the group I intron ribozyme. The higher reactivity of the 2′-NH3+ derivative suggests that donation of a hydrogen bond from the 2′-group to the neighboring 3′-leaving oxygen might allow specific stabilization of the transition state relative to the ground state, thereby facilitating the chemical cleavage step.
The 2′-OH of U–1, the 2′-OH of A207 and the exocyclic amino group of G22 have been referred to as a catalytic triad (70). However, the observation that the chemical cleavage step with a 2′-NH3+ derivative is faster than that with the substrate with a 2′-OH (the natural substrate), despite the absence of lone-pair electrons at the 2′-NH3+ group that can accept a hydrogen bond from A207-OH, suggests another possibility for the arrangement of active-site groups within this network of interactions (71,72).
Even though the ribozyme-mediated chemical cleavage step with the 2′-OH group at U–1 (the natural substrate) is significantly (1000-fold) faster than that with 2′-H, with the metal-binding site (a) in Figure 4A(ii) being occupied by a Mn2+ ion, the rate constants for reactions with the 3′-S substrates are similar, irrespective of whether there is a 2′-OH or 2′-H at U–1. Moreover, in the presence of Mg2+ ions, with the metal-binding site (a) being unoccupied by a metal ion, the rate constants for reactions with the 3′-S substrates are similar with a 2′-OH or with a 2′-H at U–1, indicating that the 2′-OH at U–1 does not contribute significantly to the chemical cleavage of the phosphorus–sulfur bond with the 3′-mercapto leaving group. Since sulfur is a weaker acceptor of a hydrogen bond than is oxygen and, furthermore, since sulfur is a significantly better leaving group than oxygen, the 3′-mercapto leaving group suppresses the catalytic advantage provided by a hydrogen bond from the 2′-OH in the native transition state (71).
The second step of the splicing reaction is catalyzed within the same catalytic site as the first step (18,82,83). Moreover, in the presence of Mg2+ ions, both the reverse reaction of the first step and the forward reaction of the second step were inhibited with the RpS substrate (note that the pro-Rp oxygen at these steps corresponds to the pro-Sp oxygen in the forward reaction of the first step). These observations indicate that the stereochemical requirements are the same in both reactions (60,63,76,84). Therefore, the mechanism of the second step is considered to be analogous to the mechanism of the first step (18).
It is clear that the analysis of thio effects, rescue experiments and other experiments with derivatives have contributed significantly to our understanding of the mechanism of action of the large group I intron ribozyme of Tetrahymena. All the available data appear to support the refined double-metal-ion mechanism of catalysis (18) that is shown in Figure 4A(ii).
THE GROUP II INTRON RIBOZYME
In the splicing reactions catalyzed by the group II intron ribozyme (Fig. 4B), the first step was blocked when the RpS substrate was used (61) and a small thio effect was observed with the SpS substrate (64), suggesting that the pro-Rp oxygen makes an essential interaction in the transition state and that the pro-Sp oxygen is also involved in some kind of interaction. No cleavage of the 3′-S substrate was observed in the presence of Mg2+ ions but the reaction was rescued by Mn2+, Zn2+ or Cd2+ ions (67). This result indicates that a Mg2+ ion acted to stabilize the 3′-leaving oxygen by direct coordination.
The second step was monitored by a tripartite assay (trans-splicing), in which an oligonucleotide that corresponded to the 3′-splice site was added after the formation of the ribozyme/5′-exon RNA complex, because the second step might be masked by the rate-limiting conformational rearrangement between the first step and the second step that was observed in the bimolecular assay (cis-splicing). In contrast to the results of analysis of the first step, the cleavage of the RpS substrate was strongly inhibited but the SpS substrate had essentially no inhibitory effect (85). The actual stereospecificity for the thio substitution is reversed between the first step and the second step (both steps were inhibited in the reaction with the RpS substrate; note that the pro-Rp oxygen in the forward reaction of the first step corresponds to the pro-Sp oxygen in the reverse reaction of the first step and in the forward reaction of the second step). Thus, the second step is not the reversal of the first step, unlike results for the group I intron ribozyme. Even though the pro-Rp oxygen atom appears to make an essential interaction in the transition state, the nature of this interaction has not been defined since no rescue by thiophilic metal ions can be observed.
The group II intron ribozyme can also hydrolyze the bond between spliced exons [the spliced-exon reopening (SER) reaction, which corresponds to the reverse reaction of the second step]. This SER reaction proceeds with the RpS substrate but not with the SpS substrate (64). Since this step would be expected to be blocked with the RpS substrate if it followed the same reaction pathway as that of the first step of the splicing reaction, the observed SER reaction supports the conclusion that the second step is not the reversal of the first step, as mentioned above.
The substitution of the 2′-OH of the leaving ribonucleoside (the U residue of the intron in Fig. 4B) at the 3′-splice site with a hydrogen atom reduced the rate of the second step ∼700-fold (85). Moreover, even though substitution with a methoxy group or a fluorine atom, respectively, reduced the rate similarly to or significantly more than substitution with a hydrogen atom, substitution with an amino group resulted in a rate that was ∼10-fold higher than that with a hydrogen atom (85). These results suggest that the ability to donate a hydrogen bond from the 2′-OH group is important. The 2′-amino substrate was cleaved faster in the presence of Mn2+ ions than in the presence of Mg2+ ions at higher pH (at higher pH, the 2′-amino group exists in a neutral form, as -NH2, and not in the protonated form, -NH3+) since a Mn2+ ion binds to the 2′-NH2 group better than a Mg2+ ion (nitrogen is a softer base than oxygen and the Mn2+ ion is a softer acid than the Mg2+ ion). This interaction with the 2′-oxygen at the second step involves a single metal ion in the transition state, as indicated by the fact that the dependence on the concentration of Mn2+ ions with the 2′-amino substrate on a background of Mg2+ ions is consistent with a single-metal-ion exchange (85).
The 3′-S substrate reduced the cleavage rate of the second step by ∼100-fold in the presence of Mg2+ ions, and the reaction was rescued completely by the addition of Mn2+, Co2+ or Cd2+ ions in the tripartite assay (67,85). However, this substrate had no effect on the bimolecular cis-splicing assay in which the rate-limiting step appeared to be the conformational rearrangement (67). This result indicates that Mg2+ ions also acted in the second step to stabilize the 3′-leaving oxygen by direct coordination, as was the case in the first step. This interaction with the 3′-oxygen also involved a single Mn2+ ion in the transition state, as observed for the cleavage of the 2′-amino substrate (85). We should emphasize, however, that the preferences of the 3′-S substrate for metal ions differed between the first step (Mn2+, Zn2+ or Cd2+) and the second step (Mn2+, Co2+ or Cd2+), indicating the differences between the environments of metal ions at the two different transition states for each splicing step.
The moiety that activates the attacking nucleophile in the two independent splice steps of the reaction of the group II intron ribozyme remains to be identified but it is apparent that a Mg2+ ion binds to the leaving 3′-oxygen to stabilize the transition state at each step.
THE RNASE P RIBOZYME
In reactions catalyzed by the RNA subunit of bacterial RNase P, there is a requirement for both divalent cations (e.g., Mg2+ or Mn2+) and monovalent cations (e.g., K+ or NH4+) (62). Monovalent cations appear to be involved in the stabilization of the structure during the cleavage reaction in the absence of proteins in vitro. By contrast, divalent metal cations are required for the chemical cleavage itself, and not only for structural stabilization. During the chemical cleavage, a hydroxide ion, activated by metal ions, is thought to act as a nucleophile (86,87). Though the details of the reaction mechanism are not fully understood, it has been proposed that three Mg2+ ions participate in the transition state, because the slope of the Hill plot for the cleavage rate versus the concentration of Mg2+ ions was 3.2, and that one of the catalytic Mg2+ ions coordinates directly to the pro-Sp oxygen at the scissile phosphate. 2′-Deoxy substitution at the cleavage site reduced the apparent number of bound Mg2+ ions and decreased the apparent affinity for Mg2+ ions, suggesting that the 2′-oxygen might be one of the Mg2+-binding sites. Furthermore, 2′-methoxy substitution at the cleavage site decreased the cleavage rate, suggesting that the 2′-OH might be involved in stabilizing the 3′-leaving oxygen as the donor of a hydrogen bond (87).
According to a recent report, the cleavage rate of the RpS substrate in the presence of Mg2+ ions was at least 1000-fold lower than the cleavage rate of the natural substrate. The reduction was, however, rescued by thiophilic metal ions, such as Cd2+ and Mn2+ ions (background Mg2+ ions are needed for rescue in the case of RNase P ribozyme from Bacillus subtilis), suggesting the direct coordination of a metal ion to the pro-Rp oxygen (88,89). Since the Hill coefficient for Cd2+ rescue was 1.8, it was proposed that two metal ions coordinate to the pro-Rp oxygen in a modified model, which is consistent with the two-metal-ion model of Steitz and Steitz (18). By contrast, the cleavage reaction was also blocked with the SpS substrate (with reduction of the binding affinity of the substrate for the ribozyme in the ground state in the case of the RNase P ribozyme from Escherichia coli) and the cleavage site was shifted in the 5’ direction. The reduced rate of cleavage of the SpS substrate was not enhanced by Cd2+ and Mn2+ ions, an observation that suggests the possibility of a crucial role for the pro-Sp oxygen in stabilization of the transition state or that might be attributable to the steric exclusion of catalytic metal ions (88,89). Similar effects of RpS and SpS substrates were also observed with the eukaryotic nuclear RNase P ribozyme, in which the RNA is thought to be the catalytic component and to be evolutionarily related to the bacterial RNase P ribozyme (90). However, no thio effect was observed in the case of RNase P from plant chloroplasts, whose catalytic component appears to be a protein (91).
The 3′-S substrate also prevented cleavage at the correct site by the bacterial RNase P ribozyme and the cleavage site was moved to the next unmodified phosphodiester bond in the 5′-direction completely. The reduction in the cleavage rate was not rescued by thiophilic Cd2+ or Mn2+ ions (92). While the absence of rescue by thiophilic metal ions does not reveal the molecular nature of the inhibitory effects (the thio effect and shifting of the cleavage site), prevention of binding of a Mg2+ ion to the 3′-leaving atom as a result of the thio substitution provides one possible explanation. In addition, it is possible that several chemical and structural changes occur upon the introduction of a bulky sulfur atom.
SMALL RIBOZYMES
Hammerhead, HDV, hairpin and VS ribozymes are categorized as small ribozymes because they are smaller than the ribozymes discussed above. Each of these naturally existing ribozymes catalyzes the endonucleotic cleavage of RNA via a mechanism that involves nucleophilic attack by a 2′-OH group on the phosphorus of the neighboring phosphodiester bond, generating 5′-OH and 2′,3′-cyclic phosphate termini (for reviews, see 4,25,93). The cleavage reactions catalyzed by these ribozymes appear to proceed with inversion of the configuration at the phosphorus atom suggesting a direct in-line attack with development of a pentacoordinate transition state or intermediate (42,94–97).
The smallest of the naturally occurring catalytic RNAs that have been identified to date are the hammerhead ribozymes (Fig. 1A), which were found in several plant viral satellite RNAs, a viroid RNA and the transcript of a nuclear satellite DNA of a newt (98; for reviews, see 3,4). These ribozymes have been extensively investigated, in particular with respect to the mechanism of action of catalytic metal ions (17,25,27,45,46).
Hammerhead ribozymes have a basic requirement for divalent metal ions, such as Mg2+ ions (5,6,17–22,24,25,27,45,99–104). In studies of the hammerhead reaction, the relationship at a certain pH between the ΔpKa values of metal ions in water and the difference in the observed rates of cleavage in the presence of the corresponding metal ions suggested a single-metal-ion mechanism in which Mg2+-hydroxide acts as a general base catalyst (17). However, it was also noted that a general double-metal-ion mechanism, in which metal ions act as Lewis acids that coordinate directly to the 2′-OH and the 5′-leaving oxygen, for activation of a nucleophile and for stabilization of a developing negative charge on the leaving group, respectively, might also explain reactions catalyzed by hammerhead ribozymes (20–22,24,25).
It should also be noted that, under extreme conditions (in the presence of 1–4 M monovalent cations, such as Li+, Na+ and NH4+), hammerhead ribozymes do not require divalent metal ions for catalysis (43). On the basis of this observation, some researchers have claimed that hammerhead ribozymes are not metalloenzymes (see below).
HDV ribozymes are derived from the genomic and the antigenomic RNAs of hepatitis delta virus (105–108). In studies of reactions catalyzed by HDV ribozymes, three groups demonstrated recently that an intramolecular functional group, namely N3 at C76 in the antigenomic HDV ribozyme and N3 at C75 in the genomic HDV ribozyme, can, in fact, act as a true catalyst (40–42). However, with respect to the roles of these N3s, two different mechanisms, namely general base catalysis and general acid catalysis, were proposed. In the former scenario, it was proposed that the deprotonated N3 of C76 might be involved in cleavage as a general base that abstracts a proton from the 2′-OH to promote its nucleophilic attack on the scissile phosphate in the transition state of reactions catalyzed by the antigenomic HDV ribozyme (41). In the latter case, it was proposed that the protonated N3 of C75 in the genomic HDV ribozyme might act as a general acid to stabilize the developing negative charge at the 5′-leaving oxygen and that a metal ion might act as a general base (42). However, although further investigations are required, there remains the possibility that the catalytic mechanism of the antigenomic ribozyme is the same as that of the genomic ribozyme (109).
In discussion of the reaction catalyzed by the genomic HDV ribozyme, the importance has been emphasized of the neutralization of the substantial negative charge that develops on the 5′-leaving oxygen and the essential role of general acid catalysis in the cleavage of RNA (42). Such issues should be relevant not only in the case of reactions catalyzed by HDV ribozymes but also in the case of reactions catalyzed by other small ribozymes since cleavage of the bond between phosphorus and the 5′-oxygen is the overall rate-limiting step, as shown in Figure 2 (see below; 22,25,56). The efficient cleavage of a phosphodiester bond requires both the activation of the 2′-attacking oxygen and the stabilization of the 5′-leaving oxygen.
Hairpin ribozymes were originally derived from the minus strand of the satellite RNA of tobacco ringspot virus (sTRSV), chicory yellow mottle virus type 1 (sCYMV1) and arabis mosaic virus (sArMV) (110–113). Hairpin and hammerhead ribozymes can also catalyze the ligation of cleaved products, with the ligation efficiency being much higher for the hairpin ribozyme than the hammerhead. The ligation reaction is thought to be the reverse of the cleavage reaction since it uses the same termini as those produced upon cleavage. Hairpin ribozymes favor the ligation reaction rather than cleavage (ligation occurs 10-fold faster than cleavage). By contrast, hammerhead ribozymes favor the cleavage reaction [cleavage occurs ≥100-fold faster than ligation (47,114–116)]. The ratio of equilibrium constants (kcleavage/kligation) can be explained by the differences between entropies: the loss of entropy that occurs with ligation is smaller for the hairpin than for the hammerhead ribozyme, indicating that the more rigid hairpin structure undergoes a smaller change in dynamics on ligation than the more flexible hammerhead (117). Catalysis by hairpin ribozymes in the absence of metal ions has been reported by several groups independently (34–39). Hairpin ribozymes can be considered to be a distinct class of ribozymes that do not require metal ions as cofactors (118). The catalyst(s) seems to be a nucleobase(s).
The VS ribozyme originated from the mitochondria of certain isolates of Neurospora (119). The reaction catalyzed by the VS ribozyme requires a divalent cation such as the Mg2+ ion (120). Some regions that are important for catalysis and some interactions between the phosphate backbone and metal ions have been identified (121–123). The reaction appears to be independent of pH but the possibility exists for a conformational change prior to cleavage that might mask a dependence on pH (120,124). The catalytic group(s) in the cleavage reaction has not yet been unambiguously identified.
REACTIONS CATALYZED BY HAMMERHEAD RIBOZYMES
Hammerhead ribozymes are among the smallest catalytic RNAs. The sequence motif, with three duplex stems and a conserved core of two non-helical segments that are responsible for the self-cleavage reaction (cis-action), was first recognized in the satellite RNAs of certain viruses (3). Engineered trans-acting hammerhead ribozymes, consisting of antisense sections (stem I and stem III) and a catalytic core with a flanking stem–loop II section (Fig. 1A, left), have been used in mechanistic studies and tested as potential therapeutic agents (13,46). Hammerhead ribozymes cleave their target RNAs at specific sites, generating a 2′,3′-cyclic phosphate and a 5′-OH terminus. The NUH rule, where N can be any nucleotide and H can be A, U or C, was originally proposed to define sites of cleavage, with the most efficient cleavage occurring at GUC triplets (125–130). However, the NUH rule was reformulated into the NHH rule since other triplets, such as GAC and GCC, can also be cleaved by a hammerhead ribozyme (131).
Over the past few years, several attempts have been made to determine the overall global structure of hammerhead ribozymes (99,132–137). Although initial structural studies indicated that possible configurations of the scissile phosphate did not allow for an in-line attack mechanism, recent crystallographic studies of a ribozyme with a product or with a modification (known as a kinetic bottleneck modification) adjacent to the cleavage site succeeded in trapping an intermediate that more closely resembled the transition state (137; reviewed in 138). However, even in this case, the intermediate cannot be considered as a real transition-state intermediate. In all crystals of ribozymes examined to date, a γ-shaped configuration has been identified, with stem I forming an acute angle with stem II, and stems II and III being stacked coaxially to form a pseudo-A-form helix, in agreement with results inferred from studies of fluorescence energy transfer and from electrophoretic and chemical cross-linking studies (Fig. 1A, right) (26,139–142). Such studies indicate the involvement of two reversed-Hoogsteen G·A base pairs between G8·A13 and A9·G12, as well as a non-Watson–Crick A14·U7 base pair that is formed by a single hydrogen bond. These base pairs are followed by stem II and are stacked ‘coaxially’ onto the non-Watson–Crick A15.1·U16.1 base pair, with resultant formation of a pseudo-A-form helix by stems II and III. Four sequential nucleotides (C3U4G5A6) form a ‘uridine-turn’ motif, allowing the phosphate backbone to turn and connect with stem I. The uridine-turn forms a catalytic pocket into which the nucleobase at the cleavage site, namely C17, is inserted (133). The crystal structure of the enzyme–product complex of the hammerhead ribozyme has been determined (137). The structure suggests that the distance between C17 and G5/A6 in the transition state is smaller than previously proposed and that dramatic conformational changes, which include C17, occur in the pathway from the ground state to the transition state.
Structural metal ions in reactions catalyzed by hammerhead ribozymes
It is generally accepted that the tertiary structures of RNA molecules are stabilized by metal ions. The roles of metal ions in ribozyme-catalyzed reactions are of two distinct types: metal ions can act as catalysts during the chemical cleavage step, as shown in Figure 3; and they can also stabilize the conformation of the ribozyme–substrate complex. The ion-dependent changes in the conformation of a hammerhead ribozyme can be easily followed by monitoring the influence of metal ions on its electrophoretic mobility (26). The effects of metal ions on the formation, upon subsequent addition of Mg2+ ions, of an active complex between a hammerhead ribozyme and its substrate can also be monitored by NMR spectroscopy (143–152).
Binding sites for metal ions have been identified by capturing metal ions within the crystal structure, and such capture provides an indication of the importance of the metal ions in catalysis (99,132–136). It was proposed that a Mg2+ ion binds to the pro-Rp oxygen of the 5′-phosphate of A9 (P9 phosphate) with further hydrogen bonding associated with N7 of G10.1 (153–155). Another site for a Mg2+ ion was localized in the vicinity of the cleavage site. It was proposed that, at this cleavage site, a Mg2+ ion binds directly to the pro-Rp oxygen of the scissile phosphate. Although this possibility remains to be confirmed, the function of this second metal ion near the scissile phosphate has been proposed to be activation of the attacking 2′-OH in the transition state. The site of yet another metal ion has also been proposed. Such an ion would act as a switch that induces the conformational changes required to achieve the transition state; it would be located adjacent to G5 in the catalytic core. This last putative site was identified from an analysis of the kinetics of a Tb3+ inhibition experiment and the elucidation of the crystallographic structure of the complex (136). The coordination of a metal ion at this site in solution was also investigated by lanthanide luminescence spectroscopy (156). An additional metal ion binding site in the hammerhead ribozyme has also been identified by 31P NMR spectroscopy (149). In this case, the metal ion is associated with the A13 phosphate in the catalytic core with an apparent Kd of 250–570 µM. However, the exact role of this metal ion remains unclear. It seems likely that it might be involved in structural folding since a structural change was detected at this site upon the binding of the metal ion.
The importance of the binding of a metal ion at the P9 phosphate, not only in the ground state but also in the transition state, was demonstrated by kinetic analysis with a modified hammerhead ribozyme with a phosphorothioate modification at this site (157,158) and, in parallel, by analysis with a ribozyme with an abasic mutation at this site (159,160). The binding of a metal ion (Cd2+) to the Rp sulfur of the P9 phosphorothioate in the transition state was much stronger than the binding in the ground state, suggesting the existence of additional ligands for the metal ion in the transition state. Moreover, our own studies indicate strongly that the binding of a metal ion to N7 of G10.1 is catalytically important but not indispensable: cleavage still occurred with a minimally modified ribozyme, in which N7 of G10.1 was merely replaced by C7 (introduction of an N7-deazaguanine residue to prevent the metal ion from binding to this site) (161). Cleavage was retarded, with ∼30-fold reduction in the rate of cleavage by the modified ribozyme. By contrast, a 1000-fold reduction in the cleavage rate resulted from the introduction of an Rp-phosphorothioate at the P9 site (157).
It was proposed very recently that the first metal ion that binds to the P9 phosphate in the ground state shifts toward a non-bridging pro-Rp oxygen at the scissile phosphate during the reaction and binds to this pro-Rp oxygen in the transition state. This scenario is consistent with the prediction that additional binding to this P9 metal ion must occur in the transition state, as mentioned above. Furthermore, it was also proposed that the metal ion at P9 must be involved directly in the chemical cleavage step, acting as a base catalyst in the transition state (27), despite the fact that the P9 metal ion is located at a distance of ∼20 Å from the scissile phosphate in the ground state. However, these proposals have been questioned by other investigators (162,163).
Although it was predicted that a hydrated Mg2+ ion should participate directly in catalysis, acting as a base catalyst in deprotonation of the 2′-OH of C17, no crystal structure has been obtained with a trapped metal ion located at this cleavage position that might confirm the direct involvement of such a metal ion in deprotonation of the 2′-OH (137).
The rate-limiting departure of the 5′-oxygen in reactions catalyzed by hammerhead ribozymes
In the non-enzymatic hydrolysis of a natural RNA, the cleavage of the P-O(5′) bond is the overall rate-limiting step in hydrolysis. By contrast, attack by the 2′-OH on the phosphorus atom is the rate-limiting step with a 5′-S substrate that contains a phosphorothiolate substitution (54–56,164,165) since, as mentioned above, the latter is hydrolyzed much more rapidly than the natural substrate (Fig. 2). The same technique as that used to draw these conclusions was used to determine the rate-limiting step in reactions catalyzed by hammerhead ribozymes.
It was reported that, in the hammerhead-catalyzed cleavage reaction, the departure of the 5′-leaving group is not rate-limiting and that a metal cofactor does not interact with the leaving group (45,100,101). This conclusion was based on experiments with the 5′-S almost-DNA substrate, that was an oligodeoxynucleotide substrate that contained a 5′-bridging phosphorothiolate linkage adjacent to one ribonucleotide at the cleavage site (45). No appreciable thio effect was observed and no preference was detected for either Mg2+ or Mn2+ ions (100,101). However, the 5′-S almost-DNA substrate basically consisted of DNA and the ribozyme reaction with this substrate had an unusually limited dependence on pH. Thus, we might expect that observed rates of reaction might reflect steps other than the chemical cleavage step. In the case of a 5′-S RNA substrate consisting only of RNA, the rate of ribozyme-mediated cleavage of the 5′-S RNA substrate in the presence of Mg2+ ions was higher by almost two orders of magnitude than that of cleavage of the natural substrate (56). If TS1 were a higher energy state than TS2 in the ribozyme reaction with the natural substrate, the cleavage rate for the 5′-S RNA substrate should be similar to that for the natural substrate because the 5′-bridging phosphorothioate linkage would not be expected to enhance the attack by the 2′-OH (53). By contrast, if TS2 were a higher energy state than TS1 in the ribozyme reaction with the natural substrate, we would expect that the rate of cleavage of the 5′-S RNA substrate would be much higher than that of the natural RNA substrate because a mercapto group is a better leaving group than a hydroxyl group. On the basis of these considerations, the results indicate that TS2 is a higher energy state than TS1 in the reactions of hammerhead ribozymes with natural substrates, as indicated in Figure 2.
Catalytic metal ions in reactions catalyzed by hammerhead ribozymes: single-metal-ion mechanisms
In the case of the proteinaceous enzyme RNase A, which does not require metal ions as cofactors, the acid/base catalysts are provided by two histidine residues within the catalytic pocket (Fig. 5A). Such acid/base functionality can, in principle, be replaced by Mg2+-bound water molecules. The generally accepted mechanism of hammerhead ribozyme reactions is a single-metal-ion mechanism (27,45,99–103). In this proposed mechanism, the hydroxide ion of a hydrated Mg2+ ion acts as a general base to deprotonate the attacking 2′-OH; the Mg2+ ion coordinates directly to the pro-Rp oxygen at the scissile phosphate, acting as an electrophilic catalyst in TS1; and it is not a metal ion but a proton that acts as a general acid to stabilize TS2 (Fig. 5B).
Figure 5.
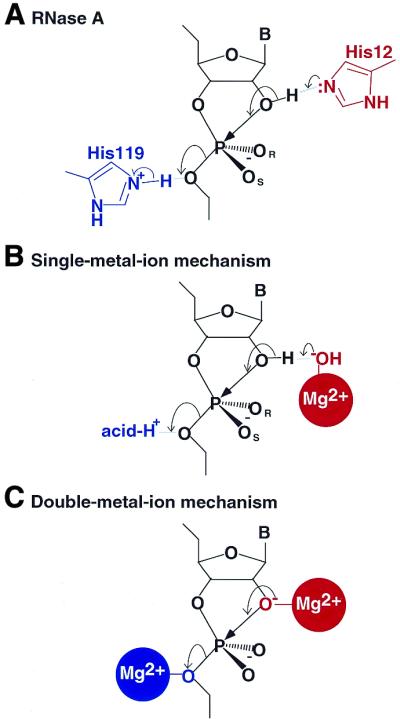
(A) The mechanism of cleavage by ribonuclease A. Two imidazole residues function as general acid–base catalysts. (B) The single-metal-ion mechanism proposed for cleavage by the hammerhead ribozyme. One metal ion binds directly to the pro-Rp oxygen and functions as a general base catalyst. (C) The double-metal-ion mechanism proposed for cleavage by the hammerhead ribozyme. Two metal ions bind directly to the 2′- and 5′-oxygens.
The single-metal-ion mechanism is supported by the fact that no metal ion was found close to the 5′-leaving oxygen in the original crystallographic structure of a freeze-trapped conformational intermediate of a hammerhead ribozyme (99). Based upon the crystal structure and a molecular dynamic simulation, a different single-metal-ion mechanism was proposed as follows (102,103). It was suggested that the involvement of just one metal ion in the transition state might be sufficient for cleavage. In this model, one Mg2+ ion coordinates simultaneously and directly to the pro-Rp oxygen and to the 2′-attacking oxygen at the cleavage site, acting as a Lewis acid to enhance the deprotonation of the 2′-OH and the subsequent attack by the nucleophile on the phosphorus atom and/or to stabilize the transition state. In addition, it was also proposed that one of the outer-sphere water molecules that surrounds the metal ion might be located at a position such that it can act as a general acid to donate a proton to the 5′-leaving group (102,103)
Another model for cleavage by hammerhead ribozymes was proposed that was based on the results of molecular dynamic studies (166). This model involves two metal ions but it is reminiscent of the single-metal-ion mechanism. In this model, two metal ions are bridged by a hydroxide ion (the µ-hydroxo-bridged Mg2+ cluster). One of the metal ions, located near the scissile phosphate, binds to the pro-Rp oxygen to act as an electrophilic catalyst. The bridging OH– between the two Mg2+ ions abstracts the proton from the Mg2+-bound water molecule. Then the activated hydroxide ion associated with the Mg2+ ion deprotonates the proximal 2′-OH at the cleavage site to activate the nucleophile, acting as a base catalyst.
As described in the previous section, formation of TS2 is the rate-limiting step in non-enzymatic reactions (25,54–56). Thus, TS2 must somehow be stabilized energetically for effective catalysis. Therefore, the hypothesis that reactions catalyzed by hammerhead ribozymes involve only a general base is insufficient.
Catalytic metal ions in reactions catalyzed by hammerhead ribozymes: double-metal-ion mechanisms
Double-metal-ion mechanisms, in which two metal ions are involved in the chemical cleavage step, have been proposed by numerous groups of investigators (5,18–22,24,25,104). From molecular orbital calculations and kinetics analysis, our group postulated that the direct coordination of Mg2+ ions with the attacking or the leaving oxygen might promote formation or cleavage of the P-O bond, with these ions acting as Lewis acids (Fig. 5C) (5,20,22,25,167). Moreover, we excluded the possible coordination of metal ions, as electrophilic catalysts, to the pro-Rp oxygen at the scissile phosphate bond (5,25,162,168,169). Studies of solvent isotope effects and kinetic analysis of a modified substrate (phosphorothiolate; 5′-S substrate), with a 5′-mercapto leaving group at the cleavage site, provided strong support for the double-metal-ion mechanism of catalysis (20,22,25).
The overall transition state structure in the hammerhead cleavage reaction is TS2, regardless of whether the reaction proceeds via a concerted one-step mechanism or via a two-step mechanism with a stable pentacoordinated intermediate. Our analysis with a 5′-S substrate demonstrated that it is important that any enzyme that catalyzes the hydrolysis of RNA should stabilize TS2 by donating a proton to the 5′-leaving oxygen or by ensuring coordination of a metal ion to the leaving oxygen. This acid catalysis by a metal ion is also supported by a recently determined crystallographic structure, in which a Co2+ ion was located close to the 5′-leaving-oxygen atom of the scissile phosphate (134).
Figure 6A shows experimentally derived profiles of pH versus rate for reactions in H2O and D2O (20,25,169). The magnitude of the apparent isotope effect (ratio of rate constants in H2O and D2O) is 4.4 and the profiles appear to support the possibility that a proton is transferred from (Mg2+-bound) water molecules. However, careful analysis led us to conclude that a metal ion binds directly to the 5′-oxygen. Since the concentration of the deprotonated 2′-oxygen in H2O should be higher than that in D2O at a fixed pH, we must take into account this difference in pKa, namely, ΔpKa (pKaD2O – pKaH2O), when we analyze the solvent isotope effect of D2O (20,25,169,170). We can estimate the pKa in D2O from the pKa in H2O using the linear relationship shown in Figure 6B (6,25,169–172). If the pKa for a Mg2+-bound water molecule in H2O is 11.4, the ΔpKa is calculated to be 0.65 (Fig. 6B, red line). Then, the pKa in D2O should be 12.0. Demonstrating the absence of an intrinsic isotope effect (kH2O/kD2O = 1), the resultant theoretical curves closely fit the experimental data, with an apparently ∼4-fold difference in observed rate constants (Fig. 6A). This result indicates that no proton transfer occurs in the hammerhead ribozyme-catalyzed reaction in the transition state and supports the hypothesis that the metal ions function as Lewis acids. In Figure 6A, the apparent plateau of rate constants above pH 8 reflects the disruption of the active hammerhead complex by the deprotonation of uridine and guanosine residues.
Figure 6.

(A) The dependence on pH of the deuterium isotope effect in the hammerhead ribozyme-catalyzed reaction. Green circles show rate vconstants in H2O; yellow circles show rate constants in D2O. Solid curves are experimentally determined. The apparent plateau of cleavage rates above pH 8 is due to disruptive effects on the deprotonation of U and G residues. Dotted lines are theoretical lines calculated from pKa values of hydrated Mg2+ ions of 11.4 in H2O and 12.0 in D2O and on the assumption that here is no intrinsic isotope effect (α = kH2O/kD2O = 1; where α is the coefficient of the intrinsic isotope effect). The following equation was used to plot the graph of pL versus log(rate): log kobs = log (kmax) – log[1 + 10(pKa(base) – pL)] – log[1 + 10(pL – pKa(acid),]. In this equation, kmax is the rate constant in the case of all acid and base catalysts in active forms: in H2O, kmax = kH2O; and in D2O, kmax = kD2O = kH2O/α. (B) The isotope effects on the acidities (pKaD2O – pKaH2O) of phenols and alcohols as a function of their acid strengths (pKa). The pKa of hydrated Mg2+ ions in H2O is 11.4, and the red arrow indicates the isotope effect of 0.65 that results in the pKa of hydrated Mg2+ ions in D2O being 12.0. The pKa of the N3 of cytosine in H2O is 6.1 and the blue arrow indicates the isotope effect of 0.53 that results in the pKa of N3 of cytosine in D2O being 6.6.
The double-metal-ion mechanism is also supported by results reported by Pontius et al. (21) and Lott et al. (24). They pointed out the minimal likelihood of base catalysis by Mg2+-bound hydroxide that deprotonates the attacking 2′-OH and the strong likelihood of Lewis acid catalysis by the direct coordination of a Mg2+ ion with the attacking 2′-OH, which enhances the deprotonation of the nucleophilic 2′-OH. Their suggestions were based on the inverse correlation between the rate of cleavage and the pKa of the added metal ions. Their argument was as follows. One of the observations used to support the single-metal-ion mechanism of catalysis (Fig. 5B) is that the lower the pKa, the higher is the cleavage rate at a given concentration of metal ions. Although metal ions with lower pKa values might be present at higher concentrations in the form of solvated metal hydroxides at a given pH, such ions should be correspondingly weaker bases and, therefore, they should be less able to remove the proton from the 2′-OH. As a result, the effect of concentration would be reduced by the effect of basicity. In other words, the dependence on pKa cannot be adequately explained by the hypothesis that the solvated metal hydroxide acts as a base in catalysis. By contrast, deprotonation of the 2′-OH can be greatly accelerated by its direct binding to a metal ion, in particular, a metal ion with a relatively low pKa, because the pKa of a 2′-OH with a bound metal ion can be reduced by several units. Such arguments are further supported by ab initio molecular orbital calculations (173).
Further evidence for a double-metal-ion mechanism was based on an analysis of the effects of changes in the concentration of La3+ ions in the presence of a fixed concentration of Mg2+ ions (24). Analysis of the effects of added La3+ ions yielded a bell-shaped curve, with activation and then inhibition of cleavage (Fig. 7A). Under the conditions of the experiment, La3+ and Mg2+ ions competed for coordination to restricted sites that are catalytically important for cleavage by the ribozyme. At lower concentrations of La3+, a La3+ ion rather than a Mg2+ ion coordinates to the 5′-leaving oxygen and absorbs the negative charge that accumulates at that position in the transition state [(b) in Fig. 7A]. The fully hydrated La3+ ion has a pKa of 9, which is >2 lower than that of Mg2+. When the leaving oxygen is directly coordinated with a trivalent La3+ ion, it is a better leaving group than when coordinated with a divalent Mg2+ ion. This difference results in a decline in the relative energy of TS2 and acceleration of cleavage. However, at higher concentrations of La3+, a La3+ ion, instead of a Mg2+ ion, coordinates not only with the 5′-leaving oxygen but also, directly, with the 2′-attacking oxygen [(c) in Fig. 7A]. Although the La3+ ion, which is more positively charged than the Mg2+ ion, might enhance the deprotonation of 2′-OH and, as a result, might increase the equilibrium concentration of 2′-O–, it would also decrease the nucleophilicity of 2′-O– toward the electropositive phosphorus. Because the coordination of a trivalent metal ion at this position reduces the nucleophilicity of the resulting 2′-O– much more dramatically than would be expected for a series of divalent ions and since this negative effect is greater than that of an induced higher concentration of 2′-O–, a higher concentration of La3+ ions decreases the overall rate of cleavage [(c) in Fig. 7A].
Figure 7.

Titration with Ln3+ ions. The hammerhead ribozyme reaction was examined on a background of Mg2+ ions. (A) Data obtained by Lott et al. (24). The proposed binding of metal ions is illustrated. (B) Data obtained by Nakamatsu et al. (161). An unmodified ribozyme (R34; red curve) and a modified ribozyme (7-deaza-R34; blue curve) were used. The rate constants were normalized by reference to the maximum rate constant ([La3+] = 3 µM). Reactions were performed under single-turnover conditions in the presence of 80 nM ribozyme and 40 nM substrate at 37°C.
However, it has been demonstrated that the binding of a metal ion to the pro-Rp oxygen of the phosphate moiety of nucleotide A9 and to N7 of nucleotide G10.1 is critical for efficient catalysis, despite the considerable distance (∼20 Å) between the P9 phosphate and the scissile phosphate in the ground state (27,99,128,132–134,157–160). In earlier discussions of the above-mentioned La3+-titration issue (24), it was difficult to completely exclude the possibility that La3+ ions might replace the metal ion at the P9 site and, as a result, might create the conditions represented by the bell-shaped curve shown in Figure 7A. In order to clarify this situation, we examined a chemically synthesized hammerhead ribozyme (7-deaza-R34) that included a minimal modification, namely, an N7-deazaguanine residue in place of G10.1 (Fig. 7B, blue curve) (161). We compared the kinetic properties of this ribozyme with those of the parental ribozyme (R34 in Fig. 7B, red curve). Kinetic analysis revealed that replacement of N7 by C7 at G10.1 reduced the catalytic activity but only to a limited extent (∼30-fold). The most important result, however, was that 7-deaza-R34 also yielded a bell-shaped curve upon addition of La3+ ions to the reaction mixture. Moreover, the apparent Kd for the replacement was the same for 7-deaza-R34 and for the parental ribozyme R34 (Fig. 7B). These results indicated that binding of a Mg2+ ion to N7 of G10.1 was catalytically important but not indispensable for hammerhead ribozyme-mediated catalysis and, furthermore, they confirmed that the La3+ titration method, as reported by von Hippel’s group, does indeed allow monitoring of the actions of catalytic metal ions at the cleavage site that are directly involved in catalysis. While such results do not, by themselves, completely exclude the possibility that other hypothetical metal-binding sites might have an allosteric effect on catalytic activity (136,156,174), the data do, at least, increase the likelihood that catalysis by hammerhead ribozymes involves a double-metal-ion mechanism since the involvement of a Mg2+ ion at N7 of nucleotide G10.1 can be ignored. The data from our experiments with 7-deaza-R34 are, obviously, completely free from potential artifacts due to a Mg2+ ion coordinated at G10.1. Thus, our results strongly support the proposal that a double-metal-ion mechanism is operative in the cleavage reaction that is catalyzed by hammerhead ribozymes (161).
Catalytic metal ions as electrophilic catalysts
The involvement of a metal ion in a strong interaction with the non-bridging pro-Rp oxygen of the scissile phosphate is generally accepted and has been supported by results from many groups (27,94–97,99,102,103,158,174–176), even though such involvement remains a matter of controversy in light of the work from our laboratory (5,6,25,104,162,168,177). It has been proposed that this metal ion acts as an electrophilic catalyst in the reaction catalyzed by hammerhead ribozymes (Fig. 3e) on the basis of results of a rescue experiment with Mn2+ ions (94–97), with further support from molecular dynamic studies (102,103,174) and spectroscopic analysis (175).
Replacement of the pro-Rp oxygen with sulfur at the cleavage site of a substrate molecule (to yield the RpS substrate for a hammerhead ribozyme) resulted in a large thio effect that was relieved by replacement of Mg2+ ions with Mn2+ ions, which have higher affinity than Mg2+ ions for sulfur. This observation led to the general conclusion that a Mg2+ ion is directly coordinated with the pro-Rp oxygen. In this arrangement, the bound metal ion can act as an electrophilic catalyst and, therefore, the proposed mechanism is very attractive as an explanation for the catalytic activity of metalloenzymes. However, as we have suggested previously, observations of the thio effect and manganese rescue do not, by themselves, prove that the direct coordination of a metal ion with the pro-Rp oxygen at the cleavage site occurs in hammerhead ribozyme-catalyzed reactions (5,25,168,177). Moreover, although results of a reinvestigation of the thio effect using another thiophilic metal ion, namely the Cd2+ ion, were used to support the proposed direct coordination of the metal ion with the pro-Rp oxygen of the scissile phosphate (27,158,176), we did not find the argument totally convincing.
In studies that we designed as part of an attempt to explain the thio effect and cadmium rescue by mechanisms that do not involve the direct coordination of a metal ion at the pro-Rp oxygen (162), we found that the rate of ribozyme-catalyzed cleavage of the RpS substrate was ∼1000-fold lower than that of the natural substrate in the absence of thiophilic Cd2+ ions and that the addition of the soft Cd2+ ions to the reaction on a background of hard Mg2+ or Ca2+ ions restored the efficient cleavage of the RpS substrate (Fig. 8A). Furthermore, at high concentrations of Cd2+ ions on a background of Ca2+ ions, the rate of cleavage of the RpS substrate was similar to that of the natural substrate.
Figure 8.

(A) Titration with Cd2+ ions. The hammerhead ribozyme reaction was examined on a background of Ca2+ ions. The substrate had a normal phosphate (natural substrate; blue) or an Rp-phosphorothioate group (RpS substrate; red) at the cleavage site. Solid curves indicate the rate constants for Cd2+-associated reactions. Dotted curves indicate the observed rate constants. (B) Thermodynamic boxes for the cleavage of the natural substrate (blue) and the RpS substrate (red) by the hammerhead ribozyme. R, ribozyme–substrate complex with all metal-binding sites occupied except the exchange site(s) examined here; M, the metal ion(s) that binds to the exchange site(s) examined here; Kd GS and Kd TS, the intrinsic dissociation constants for binding of metal ion(s) to the exchange site(s) examined here in the ground state and in the transition state, respectively; kcleave, the rate of ribozyme-catalyzed cleavage with Cd2+ ion(s) at the exchange site(s) examined here; and k, the rate of non-enzymatic cleavage. The observed rate constants can be described in terms of pseudo equilibrium constant Kd‡s for the formation of the transition state (‡) from the ground state (k = [kBT/h]Kd‡, in which kB is Boltzmann’s constant and h is Planck’s constant).
Although such results might appear to prove the direct coordination of a metal ion with the pro-Rp oxygen, we reached a completely different conclusion. It is true that Cd2+ ions at higher concentrations replaced the background Ca2+ ions, and that, above the apparent dissociation constant of the Cd2+ ions (Kd app), the RpS substrate was cleaved at a fixed rate because the Ca2+ ion(s) had been fully replaced by a catalytic Cd2+ ion(s). However, it should be noted that Cd2+ ions at higher concentrations similarly replaced the background Ca2+ ions in the reaction with the natural substrate and, moreover, that the natural substrate (PO) was cleaved at a fixed rate that was identical to the rate of cleavage of the RpS substrate (PS) above the Kd app for Cd2+ ions [kcleave(PO) = kcleave(PS)]. Clearly, Cd2+ ions enhanced the rate of cleavage not only of the RpS substrate but also of the natural substrate, an indication that Cd2+ ions replaced bound Ca2+ ions even in the case of the natural substrate. Furthermore, the Kd app for Cd2+ ions during cleavage of the RpS substrate was the same as that during cleavage of the natural substrate [Kd GS(PO) = Kd GS(PS), where GS is the ground state].
Why was the affinity for Cd2+ ions the same for both the RpS and the natural substrate? We would expect the RpS substrate to have a higher affinity for Cd2+ ions, if the metal ion were directly coordinated with the pro-Rp oxygen, because the soft Cd2+ ion is known to interact with a sulfur atom with a significantly higher affinity (two orders of magnitude higher) than with a hard oxygen atom (162). Estimates can be made of the respective kinetic and thermodynamic parameters in the thermodynamic cycle (Fig. 8B), as follows. Since the pre-existing Ca2+ ion(s) within the ribozyme–substrate complex was replaced by a Cd2+ ion(s) with the identical Kd app for both the natural and RpS substrates, the affinity of the binding of the added Cd2+ ion(s) to each respective complex in the ground state must be the same [Kd GS(PO) = Kd GS(PS)]. Moreover, the cleavage rate in the presence of saturating Cd2+ ions was the same for both the natural and RpS substrates [kcleave(PO) = kcleave(PS) = 1 min–1] and the rate of the non-enzymatic cleavage of the P-O bond in phosphate and phosphorothioate moieties is known to be similar [k(PO) = k(PS); see 178,179]. Because the thermodynamic box must be closed (Fig. 8B) and since three out of four parameters, namely, Kd GS, kcleave and k, are the same for both the natural and the RpS substrate, it is apparent that the remaining parameter (the affinity of the Cd2+ ion for the transition state complex) must also be the same for both substrates [Kd TS(PO) = Kd TS(PS)], irrespective of whether the cleavage site includes a regular phosphate or a modified Rp-phosphorothioate moiety.
In the case of a modified ribozyme in which the pro-Rp oxygen at the P9 phosphate was replaced by sulfur (RpS-P9 ribozyme), the affinity of Cd2+ ions was higher for the RpS-P9 ribozyme than for the natural ribozyme (27,157,162) [Kd app for the RpS-P9 ribozyme (25 µM) was smaller than Kd app for the natural ribozyme (220 µM) in the presence of 10 mM Mg2+ ions (157)]. Therefore, a Cd2+ ion does indeed interact with the Rp sulfur at the P9 phosphate but, contrary to the conclusion reached by other investigators, the Cd2+ ion does not interact with the sulfur atom at the Rp position of the scissile phosphate, neither in the ground state [Kd GS(PO) = Kd GS(PS)] nor in the transition state [Kd TS(PO) = Kd TS(PS)]. Thus, it is appropriate to emphasize, yet again, that observations of the thio effect and cadmium rescue by themselves are not sufficient to prove that the direct coordination of a metal ion with the pro-Rp oxygen at the cleavage site occurs in hammerhead ribozyme-catalyzed reactions.
Numerous experimental results have indicated that the substitution of only one sulfur atom for an oxygen atom has a major steric effect and that, in many cases, such substitution changes the mode of reaction (64,72,88,89,92,149,179–184). With a 3′-S or SpS substrate, the site of cleavage by the RNase P ribozyme shifted in the 5′-direction from the modified site (the expected cleavage site) to the adjacent unmodified phosphodiester linkage (88,92), as discussed above. With an SpS substrate, the cleavage site in the group II intron-catalyzed SER reaction also shifted in the 5′-direction (64). A hybridized orbital of oxygen or sulfur consists of 2s,2p atomic orbitals or 3s,3p atomic orbitals, respectively. The third periodic orbital is known to be much bigger than the second one. This difference in bulkiness might be responsible for the prevention of the correct positioning of the scissile bond at the active site. Furthermore, in the case of hammerhead ribozymes, it is possible that disruption of the symmetry of the non-bridging oxygen by the thio substitution might itself affect the structure and activity of the ribozyme (179). Introduction of a sulfur atom can also result in a significant change in the structure of the complex between the hammerhead ribozyme and its substrate (150).
Rescue experiments with Mn2+ or Cd2+ ions do not provide evidence that unequivocally supports electrophilic catalysis in hammerhead ribozyme-catalyzed reactions. Thus, Mg2+ ions that catalyze the nucleophilic attack by the 2′-oxygen or that stabilize the 5′-leaving oxygen have no kinetically detectable function as simultaneous electrophilic catalysts in hammerhead ribozyme-catalyzed reactions, as shown in Figure 5C.
HDV RIBOZYME-CATALYZED REACTIONS
Studies by three groups have revolutionized our understanding of the mechanism of HDV ribozyme-catalyzed reactions (40–42). For the cleavage of phosphodiester bonds, the nucleophile must be deprotonated and the leaving group must be protonated or stabilized by a functional group(s). As illustrated in Figure 4, the developing negative charges in the transition state in the group I and group II intron-catalyzed reactions are stabilized by direct interactions with metal ions. A similar mechanism is possible for hammerhead ribozyme-catalyzed reactions, as shown in Figure 5C. The novel finding with respect to the mechanism of catalysis by the genomic HDV ribozyme is that the pKa of cytosine 75 (C75) is perturbed to neutrality in the ribozyme–substrate complex and, more importantly, that C75 acts as a general acid catalyst in combination with a metal hydroxide which acts as a general base catalyst (Fig. 9). Discovery of this phenomenon provided the first direct demonstration that a nucleobase can act as an acid–base catalyst in an RNA. As a result, as shown by the green curve in Figure 9B, the curve that represents the dependence on pH of the self-cleavage of the precursor genomic HDV ribozyme has a slope of unity at pH values below 7 (the activity increases linearly as the pH increases, with a slope of +1). Then, at higher pH values, the observed rate constant becomes insensitive to pH.
Figure 9.


Reactions catalyzed by the genomic HDV ribozyme. (A) Fractions of the active species [AH] that acts as an acid catalyst (blue) and the active species [B:] that acts as a base catalyst (red), respectively. The pKa of the acid catalyst is 6.1 and that of the base catalyst is 11.4 in H2O. The theoretical curve for H2O in (B) was produced by the multiplication of these two curves. (B) Dependence on pH of the deuterium isotope effect in the HDV ribozyme-catalyzed reaction. Green circles, rate constants in H2O; yellow circles, rate constants in D2O; solid curves, experimental data; dotted curves, theoretical data calculated using the equation in Figure 6 and pKa values for C75 and for hydrated Mg2+ ions of 6.1 and 11.4 in H2O and 6.5 and 12.0 in D2O, respectively, assuming α = 2. The blue curve is a pH profile in 1 M NaC1 and 1 mM EDTA in the absence of divalent metal ions. (C) Energy diagram for cleavage of its substrate by an HDV ribozyme. The rate-limiting step in the reaction with the natural substrate is the cleavage of the P-(5′-O) bond. The structures of transition states TS1 and TS2 are also shown. P(V), the pentacoordinate intermediate/transition state.
The slope of unity below pH 7 is consistent with an increase, with pH, in the relative level of the metal hydroxide [B:], which acts as the general base upon deprotonation, and a constant amount of the functional protonated form of C75 [AH], which acts as the general acid. The slope of zero from pH 7 to pH 9 indicates that the relative level of the metal hydroxide [B:] increases (Fig. 9A, red curve), while the relative level of protonated C75 [AH] decreases by the same amount (Fig. 9A, blue curve) (42). In agreement with this interpretation, when C75 was replaced by uracil, the resultant C75U mutant, which was unable to assist in the transfer of a proton, did indeed lack catalytic activity. However, the activity of the C75U mutant was restored by the addition of imidazole, whose protonated form, the imidazolium ion, is known to act as a good proton donor (41,42). Another mutant, C75A, in which the ring nitrogen N1 at A75 has a slightly lower pKa than the corresponding ring nitrogen N3 of C75, supported self-cleavage activity, albeit with reduced efficiency. The observed pKa of the C75A mutant was slightly lower than that of the wild-type C75 ribozyme, supporting the interpretation in Figure 9A.
Further convincing evidence for this model comes from the observation that, in the absence of divalent metal ions (in the absence of base [B:], see Fig. 9A) and in the presence of a high concentration of monovalent cations (1 M NaCl, 1 mM EDTA), the logarithm of the observed rate constant decreased with pH with a slope of –1, as shown by the blue curve in Figure 9B. This observation is consistent with exclusively general acid catalysis, as shown by the blue curve in Figure 9A (without a metal hydroxide acting as a general base). This type of profile clearly demonstrates that the observed pKa of 6.1 for self-cleavage in the presence of divalent metal ions (Fig. 9B, green curve) reflects the pKa of a general acid rather than that of a general base. It is noteworthy that [Co(NH3)6]3+ inhibited the Mg2+-catalyzed reaction in a competitive fashion, a result that suggests that [Co(NH3)6]3+ might bind to the same site as the functional Mg2+ ion with outer-sphere coordination (note that [Co(NH3)6]3+ does not ionize to yield base catalyst [B:]) (42). The similar rate constants determined in the presence of Ca2+ ions and of Mg2+ ions are also consistent with the action of a hydrated metal ion as a Brönsted base rather than as a Lewis acid in the reaction catalyzed by the HDV ribozyme (21,42).
There was a significant, apparent D2O solvent isotope effect over the entire range of pH values, confirming that transfer of a proton occurs in the transition state [the ratio of the observed rate constants, kobs(H2O)/kobs(D2O), being as high as 10 (42)]. Moreover, since the observed pKa for self-cleavage corresponded to that for the general acid and since the overall rate-limiting step appeared to be the cleavage of the bond between the phosphorus and the 5′-leaving oxygen (Fig. 2), the transferred proton in the transition state must have been derived from C75. Under the conditions of the measurements, the pKa of C75 in H2O was 6.1 and, thus, the estimated ΔpKa was 0.53, as indicated by the blue arrow in Figure 6B. Indeed, in the pL profile (pL = pH or pD) in Figure 9B, the pKa in D2O is shifted upward by ∼0.4 ± 0.1 pH units, consistent with the estimated value (42). If we take the pKa of C75 as 6.1 in H2O and 6.5 in D2O and the respective pKa values for Mg2+-bound water to be 11.4 and 12.0; and if we assume that the value of the intrinsic D2O solvent isotope effect is 2 (kH2O/kD2O = 2), we can generate theoretical curves for HDV-catalyzed reactions in H2O (green curve) and D2O (yellow curve), as shown in Figure 9B. The good agreement of the theoretical curves with the experimentally determined curves (indicated by solid lines in Fig. 9B) (42) supports a scenario wherein transfer of a proton does indeed occur from protonated C75 in the P-O(5′) bond-cleaving transition state (TS2; Fig. 9C). This conclusion is different from the conclusion in the case of hammerhead ribozymes because, in the case of hammerhead ribozymes, the observed, apparent isotope effect (Fig. 6A) can be fully explained by the difference in relative levels of the active species in H2O and in D2O without invoking any intrinsic D2O solvent isotope effect (kH2O/kD2O = 1; Fig. 6A).
As described above, the HDV ribozyme requires a Mg2+ ion for efficient catalysis. The possibility of the coordination of the Mg2+ ion with the non-bridging oxygen during the ribozyme-mediated cleavage was examined with RpS and SpS substrates (185). Determination of the rates of the cleavage steps revealed that the cleavage rates for both substrates were almost the same as that for the natural substrate irrespective of the presence of Mg2+ ions or Mn2+ ions. These results indicate the absence of a thio effect and the possibility for manganese rescue in the HDV ribozyme-catalyzed reaction. The association constants for Mg2+ ions and the ribozyme–substrate complex were also almost the same for the two substrates. This result supports the hypothesis that no direct coordination of the Mg2+ ion with any non-bridging oxygen atoms occurs at the scissile phosphate during cleavage in HDV ribozyme-catalyzed reactions (185), and that base catalysis involves the Mg2+ ion with outer-sphere coordination (42).
All the available data support the reaction mechanism for HDV ribozyme-catalyzed reactions that is shown in Figure 9C. In this model, the donation of a proton is favored and the model involves an acid with an optimal pKa of 7 under physiological conditions (42). By contrast, the self-cleavage of the HDV ribozyme in the absence of divalent cations, but in the presence of high concentrations of monovalent cations, suggests that monovalent cations at high concentrations can replace divalent cations in the tertiary folding of the RNA. Thus, divalent cations are not absolutely essential for the folding or cleavage activity of the HDV ribozyme even though a functional Mg2+ ion does participate in cleavage under physiological conditions. These features appear to be unique among the mechanisms of action of known ribozymes. It should be emphasized that, since the overall rate-limiting step is the cleavage of the P-O(5′) bond (TS2), the acid catalysis provided by protonated C75 is mandatory in reactions catalyzed by the HDV ribozyme (Fig. 9C).
THE HAIRPIN RIBOZYME
The roles of Mg2+ ions in catalysis have been discussed for naturally existing ribozymes, such as group I and II introns, RNase P, HDV and hammerhead ribozymes. The hairpin ribozyme is exceptional in that the reaction catalyzed by the hairpin exploits a metal ion in only a passive role. Strong experimental evidence for this statement is provided by the observation that the reaction proceeds efficiently in the presence of [Co(NH3)6]3+, polyamines or aminoglycoside antibiotics, and in the absence of additional metal ions (34–39). The pKa of the coordinated NH3 groups of [Co(NH3)6]3+ is estimated to be >14 (186). Moreover, it is an exchange-inert complex (the ligand exchange rate is 10–10 s–1) (186) and it can replace a fully hydrated Mg2+ ion since both [Co(NH3)6]3+ and the fully hydrated Mg2+ ion have octahedral symmetry and similar radii. Thus, it seems unlikely that [Co(NH3)6]3+ functions as a general base or a Lewis acid to deprotonate the 2′-OH for nucleophilic attack and it is likely that [Co(NH3)6]3+ supports the structure of the RNA appropriately in the same way as the hydrated Mg2+ ions do.
Other evidence supporting the putative passive (non-catalytic) role of metal ions is that ribozyme-catalyzed cleavage in the presence of Mg2+ ions exhibits no preference for the RpS versus the SpS substrate (34). This result suggests that non-bridging oxygen atoms at the scissile phosphate do not interact with metal ions, at least in any direct manner, during cleavage. Additional evidence for a passive role comes from the observation that the ribozyme reaction can occur in films of partially hydrated RNA in the absence of divalent cations (39). This result indicates that all elements essential for catalytic function are provided by the folded RNA itself. Taken together, all the results support the hypothesis that a nucleobase(s) functions as a general acid/base catalyst. The profile of pH versus rate for hairpin ribozymes has two pKa values (5.4 and 9.8), with a flat pH-independent region at neutral pH (35). The importance of the exocyclic 2-amino group of G+1 in the substrate has been reported (187). Efforts are being made to identify a nucleobase(s) that might act as a general base/acid catalyst.
For efficient cleavage, the hairpin ribozyme seems to require not only activation of an attacking nucleophile but also stabilization of the leaving group since the cleavage mechanism is the ‘5′-leaving type’, as are the cleavage mechanisms of the HDV and hammerhead ribozymes, which involve stabilization of TS2. Hairpin ribozymes probably exploit a nucleobase(s) instead of divalent metal ions for stabilization of TS2.
REACTIONS CATALYZED BY RIBOZYMES THAT DO NOT INVOLVE DIVALENT METAL IONS
Many ribozymes are considered to be metalloenzymes, requiring divalent metal ions for efficient catalysis. However, cleavage by hairpin ribozymes occurs in the absence of divalent metal ions and some small ribozymes can catalyze cleavage in the absence of divalent metal ions but in the presence of monovalent cations, such as Na+, Li+ and NH4+. Such ribozymes have been called non-obligate metalloenzymes (43,44). It has been suggested that a dense positive charge rather than divalent metal ions is the general and fundamental requirement for catalysis by a ribozyme (43). Nucleobases are new candidates for acid/base catalysts, as described above for the HDV ribozyme. In the case of hammerhead ribozymes, typically categorized as metalloenzymes, it seems that reactions can also be catalyzed by monovalent cations but the concentrations of such cations have to be very much higher than that of divalent metal ions if cleavage is to occur at a similar rate. Monovalent cations might stabilize the transition state, in particular the overall rate-limiting formation of TS2, in the same way as divalent metal ions, by providing a dense positively charged environment at high concentrations. If this is the case, monovalent cations would be very ineffective under physiological conditions, at least in the case of reactions catalyzed by hammerhead ribozymes.
Important differences between monovalent cations and divalent metal ions include not only differences in positive-charge density but also differences in the geometry of hydration of the ions. Divalent metal ions, such as Mg2+ ions, have octahedral coordination, while monovalent metal ions, such as Na+ and Li+ ions, likely have tetrahedral coordination on the basis of the valence bond theory (186,188). These features of monovalent metal ions might not be suitable geometrically for formation of a catalytically competent structure in the transition state such as the one shown in Figure 5C (with Mg2+ ions being replaced by Na+ or Li+ ions). Therefore, it is likely that a very high concentration of such monovalent cations would be required to stabilize the overall transition state TS2. Thus, it is very difficult to envision, for example, that a hammerhead ribozyme might use monovalent cations as a catalytic cofactor under physiological conditions in vivo. Under physiological conditions, it is probable that hammerhead ribozymes are metalloenzymes that require divalent cations for catalysis.
CONCLUSIONS
The mechanisms exploited by naturally existing ribozymes appear to be more diverse than originally anticipated. It is clear, however, that at least two effective catalysts are necessary to facilitate the overall pathway for the efficient hydrolysis of RNA. In the case of the group I intron ribozyme, two metal ions function as Lewis acids by coordinating directly with the nucleophile and the leaving group. In the case of the group II intron ribozyme, a metal ion functions as a Lewis acid to stabilize the departure of the 3′-oxygen and the attacking nucleophile is activated by certain interactions with the ribozyme. In the case of the RNase P ribozyme, it has been proposed that two metal ions activate the attack of the nucleophile and stabilize the departure of the 3′-oxygen. In the genomic HDV ribozyme, a metal ion functions as a general base catalyst and the internal N3 moiety of a cytosine residue functions as a general acid. Hammerhead ribozymes also need two effective catalysts to activate the attack by the 2′-oxygen and to stabilize the departure of the 5′-oxygen. Metal ions are candidates for these catalysts: one works as a catalyst to assist in deprotonation of the nucleophile and/or the other helps in the departure of the leaving oxygen atom from the phosphorus atom (stabilization of the product). In the case of hairpin ribozymes, where catalysis is not dependent on divalent metal ions, there must be other catalysts such as a nucleobase that can assist the attack by the 2′-oxygen and the departure of the 5′-oxygen.
In conclusion, the mechanisms of hydrolysis of RNA by ribozymes in nature are becoming clearer, and the diversity of the various candidates that might serve as catalysts is becoming apparent. Catalysts that stabilize the leaving oxygen, as well as catalysts for deprotonation of the nucleophile, are required for effective catalytic reactions. It is noteworthy that the architecture of the complex between the substrate and the ribozyme allows perturbation of the pKa of nucleobases which, in addition to metal ions, can act as efficient catalysts (8,9,40–42). The importance of nucleobases with a perturbed pKa has been clearly demonstrated in the case of the genomic HDV ribozyme (42) and more examples are being presented that involve ribosomes (7–10). The recent advances in this exciting field have revealed fundamental aspects of the catalysis that results in the cleavage of phosphodiester bonds.
Acknowledgments
ACKNOWLEDGEMENTS
W.J.S. would like to thank the Ministry of Economy, Trade and Industry, METI (formerly MITI) for the financial support that paid for his 3 month stay at the National Institute of Advanced Industrial Science and Technology (AIST) in Tsukuba.
References
- 1.Cech T.R., Zaug,A.J. and Grabowski,P.J. (1981) In vitro splicing of the ribosomal RNA precursor of Tetrahymena: involvement of a guanosine nucleotide in the excision of the intervening sequence. Cell, 27, 487–496. [DOI] [PubMed] [Google Scholar]
- 2.Guerrier-Takada C., Gardiner,K., Marsh,T., Pace,N. and Altman,S. (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell, 35, 849–857. [DOI] [PubMed] [Google Scholar]
- 3.Symons R.H. (1992) Small catalytic RNAs. Annu. Rev. Biochem., 61, 641–671. [DOI] [PubMed] [Google Scholar]
- 4.Carola C. and Eckstein,F. (1999) Nucleic acid enzymes. Curr. Opin. Chem. Biol., 3, 274–283. [DOI] [PubMed] [Google Scholar]
- 5.Warashina M., Zhou,D.M., Kuwabara,T. and Taira,K. (1999) Ribozyme structure and function. In Söll,D., Nishimura,S. and Moore,P.B. (eds), Comprehensive Natural Products Chemistry. Elsevier Science Ltd, Oxford, UK, Vol. 6, pp. 235–268.
- 6.Warashina M., Takagi,Y., Stec,W.J. and Taira,K. (2000) Differences among mechanisms of ribozyme-catalyzed reactions. Curr. Opin. Biotechnol., 11, 354–362. [DOI] [PubMed] [Google Scholar]
- 7.Noller H.F., Hoffarth,V. and Zimniak,L. (1992) Unusual resistance of peptidyl transferase to protein extraction procedures. Science, 256, 1416–1419. [DOI] [PubMed] [Google Scholar]
- 8.Nissen P., Hansen,J., Ban,N., Moor,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 9.Muth G.W., Ortoleva-Donnelly,L. and Strobel,S.A. (2000) A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science, 289, 947–950. [DOI] [PubMed] [Google Scholar]
- 10.Cech T.R. (2000) Structural biology. The ribosome is a ribozyme. Science, 289, 878–879. [DOI] [PubMed] [Google Scholar]
- 11.Collins C.A. and Guthrie,C. (2000) The question remains: is the spliceosome a ribozyme? Nat. Struct. Biol., 10, 850–854. [DOI] [PubMed] [Google Scholar]
- 12.Uhlenbeck O.C. (1987) A small catalytic oligoribonucleotide. Nature, 328, 596–600. [DOI] [PubMed] [Google Scholar]
- 13.Haseloff J. and Gerlach,W.L. (1988) Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature, 334, 585–591. [DOI] [PubMed] [Google Scholar]
- 14.Bratty J., Chartrand,P., Ferbeyre,G. and Cedergren,R. (1993) The hammerhead RNA domain, a model ribozyme. Biochim. Biophys. Acta, 1216, 345–359. [DOI] [PubMed] [Google Scholar]
- 15.Pyle A.M. (1993) Ribozymes: a distinct class of metalloenzymes. Science, 261, 709–714. [DOI] [PubMed] [Google Scholar]
- 16.Yarus M. (1993) How many catalytic RNAs? Ions and the Cheshire cat conjecture. FASEB J., 7, 31–39. [DOI] [PubMed] [Google Scholar]
- 17.Dahm S.C., Derrick,W.B. and Uhlenbeck,O.C. (1993) Evidence for the role of solvated metal hydroxide in the hammerhead cleavage mechanism. Biochemistry, 32, 13040–13045. [DOI] [PubMed] [Google Scholar]
- 18.Steitz T.A. and Steitz,J.A. (1993) A general two-metal-ion mechanism for catalytic RNA. Proc. Natl Acad. Sci. USA, 90, 6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uebayashi M., Uchimaru,T., Koguma,T., Sawata,S., Shimayama,T. and Taira,K. (1994) Theoretical and experimental consideration on the hammerhead ribozyme reactions – divalent magnesium-ion mediated cleavage of phosphorus–oxygen bonds. J. Org. Chem., 59, 7414–7420. [Google Scholar]
- 20.Sawata S., Komiyama,M. and Taira,K. (1995) Kinetic evidence based on solvent isotope effects for the nonexistence of a proton-transfer process in reactions catalyzed by a hammerhead ribozyme – implication to the double-metal-ion mechanism of catalysis. J. Am. Chem. Soc., 117, 2357–2358. [Google Scholar]
- 21.Pontius B.W., Lott,W.B. and von Hippel,P.H. (1997) Observations on catalysis by hammerhead ribozymes are consistent with a two-divalent-metal-ion mechanism. Proc. Natl Acad. Sci. USA, 94, 2290–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou D.M., Zhang,L.H. and Taira,K. (1997) Explanation by the double-metal-ion mechanism of catalysis for the differential metal ion effects on the cleavage rates of 5′-oxy and 5′-thio substrates by a hammerhead ribozyme. Proc. Natl Acad. Sci. USA, 94, 14343–14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birikh K.R., Heaton,P.A. and Eckstein,F. (1997) The structure, function and application of the hammerhead ribozyme. Eur. J. Biochem., 245, 1–16. [DOI] [PubMed] [Google Scholar]
- 24.Lott W.B., Pontius,B.W. and von Hippel,P.H. (1998) A two-metal ion mechanism operates in the hammerhead ribozyme-mediated cleavage of an RNA substrate. Proc. Natl Acad. Sci. USA, 95, 542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou D.M. and Taira,K. (1998) The hydrolysis of RNA: from theoretical calculations to the hammerhead ribozyme-mediated cleavage of RNA. Chem. Rev., 98, 991–1026. [DOI] [PubMed]
- 26.Lilley D.M.J. (1999) Structure, folding and catalysis of the small nucleolytic ribozymes. Curr. Opin. Struct. Biol., 9, 330–338. [DOI] [PubMed] [Google Scholar]
- 27.Wang S., Karbstein,K., Peracchi,A., Beigelman,L. and Herschlag,D. (1999) Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry, 38, 14363–14378. [DOI] [PubMed] [Google Scholar]
- 28.Yarus M. (1999) Boundaries for an RNA world. Curr. Opin. Chem. Biol., 3, 260–267. [DOI] [PubMed] [Google Scholar]
- 29.Walter N.G. and Burke,J.M. (1998) The hairpin ribozyme: structure, assembly and catalysis. Curr. Opin. Chem. Biol., 2, 24–30. [DOI] [PubMed] [Google Scholar]
- 30.Li Y. and Breaker,R.R. (1999) Deoxyribozymes: new players in the ancient game of biocatalysis. Curr. Opin. Struct. Biol., 9, 315–323. [DOI] [PubMed] [Google Scholar]
- 31.Scott W.G (1999) RNA structure, metal ions and catalysis. Curr. Opin. Chem. Biol., 3, 705–709. [DOI] [PubMed] [Google Scholar]
- 32.Strobel S.A. (1999) A chemogenetic approach to RNA function/structure analysis. Curr. Opin. Struct. Biol., 9, 346–352. [DOI] [PubMed] [Google Scholar]
- 33.Westhof E. (1999) Chemical diversity in RNA cleavage. Science, 286, 61–62. [DOI] [PubMed] [Google Scholar]
- 34.Hampel A. and Cowan,J.A. (1997) A unique mechanism for RNA catalysis: the role of metal cofactors in hairpin ribozyme cleavage. Chem. Biol., 4, 513–517. [DOI] [PubMed] [Google Scholar]
- 35.Nesbitt S., Hegg,L.A. and Fedor,M.J. (1997) An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem. Biol., 4, 619–630. [DOI] [PubMed] [Google Scholar]
- 36.Young K.J., Gill,F. and Grasby,J.A. (1997) Metal ions play a passive role in the hairpin ribozyme catalysed reaction. Nucleic Acids Res., 25, 3760–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chowrira B.M., Berzal-Herranz,A. and Burke,J.M. (1993) Ionic requirements for RNA binding, cleavage and ligation by the hairpin ribozyme. Biochemistry, 32, 1088–1095. [DOI] [PubMed] [Google Scholar]
- 38.Earnshaw D.J. and Gait,M.J. (1998) Hairpin ribozyme cleavage catalyzed by aminoglycoside antibiotics and the polyamine spermine in the absence of metal ions. Nucleic Acids Res., 26, 5551–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seyhan A.A. and Burke,J.M. (2000) Mg2+-independent hairpin ribozyme catalysis in hydrated RNA films. RNA, 6, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferré-D’Amaré A.R., Zhou,K. and Doudna,J.A. (1998) Crystal structure of a hepatitis delta virus ribozyme. Nature, 395, 567–574. [DOI] [PubMed] [Google Scholar]
- 41.Perrotta A.T., Shih,I. and Been,M.D. (1999) Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science, 286, 123–126. [DOI] [PubMed] [Google Scholar]
- 42.Nakano S., Chadalavada,D.M. and Bevilacqua,P.C. (2000) General acid–base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science, 287, 1493–1497. [DOI] [PubMed] [Google Scholar]
- 43.Murray J.B., Seyhan,A.A., Walter,N.G., Burke,J.M. and Scott,W.G. (1998) The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol., 5, 587–595. [DOI] [PubMed] [Google Scholar]
- 44.Hanna R. and Doudna,J.A. (2000) Metal ions in ribozyme folding and catalysis. Curr. Opin. Chem. Biol., 4, 166–170. [DOI] [PubMed] [Google Scholar]
- 45.Kuimelis R.G. and McLaughlin,L.W. (1998) Mechanisms of ribozyme-mediated RNA cleavage. Chem. Rev., 98, 1027–1044. [DOI] [PubMed] [Google Scholar]
- 46.Stage-Zimmermann T.K. and Uhlenbeck,O.C. (1998) Hammerhead ribozyme kinetics. RNA, 4, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedor M.J. (2000) Structure and function of the hairpin ribozyme. J. Mol. Biol., 297, 269–291. [DOI] [PubMed] [Google Scholar]
- 48.Komatsu Y., Kanzaki,I., Shirai,M., Kumagai,I., Yamashita,S. and Ohtsuka,E. (2000) Functional domain-assembly in hairpin ribozymes. J. Biochem., 127, 531–536. [DOI] [PubMed] [Google Scholar]
- 49.Westhof E. and Fritsch,V. (2000) RNA folding: beyond Watson–Crick pairs. Structure Fold. Des., 8, R55–R65. [DOI] [PubMed] [Google Scholar]
- 50.Kuusela S. and Lönnberg,H. (1997) Metal ion dependent hydrolysis of RNA. Curr. Top. Soln Chem., 2, 29–47. [Google Scholar]
- 51.Oivanen M., Kuusela,S. and Lönnberg,H. (1998) Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by Brönsted acids and bases. Chem. Rev., 98, 961–990. [DOI] [PubMed] [Google Scholar]
- 52.Komiyama M., Takeda,N. and Shigekawa,H. (1999) Hydrolysis of DNA and RNA by lanthanide ions: mechanistic studies leading to new applications. Chem. Commun., 1443–1451. [Google Scholar]
- 53.Komiyama M. and Bender,M.L. (1980) The cyclodextrin-accelerated cleavage of thiocarboxylic S-esters. Bull. Chem. Soc. Jpn, 53, 1073–1076. [Google Scholar]
- 54.Liu X. and Reese,C.B. (1995) Uridylyl-(3′-5′)-(5′-thiouridine)—an exceptionally base-labile di-ribonucleoside phosphate analog. Tetrahedron Lett., 36, 3413–3416. [Google Scholar]
- 55.Thomson J.B., Patel,B.K., Jiménez,V., Eckart,K. and Eckstein,F. (1996) Synthesis and properties of diuridine phosphate analogues containing thio and amino modifications. J. Org. Chem., 61, 6273–6281. [DOI] [PubMed] [Google Scholar]
- 56.Zhou D.M., Usman,N., Wincott,F.E., Matulic-Adamic,J., Orita,M., Zhang,L.H., Komiyama,M., Kumar,P.K.R. and Taira,K. (1996) Evidence for the rate-limiting departure of the 5′-oxygen in nonenzymatic and hammerhead ribozyme-catalyzed reactions. J. Am. Chem. Soc., 118, 5862–5866. [Google Scholar]
- 57.Cech T.R. and Herschlag,D. (1996) Group I ribozyme: substrate recognition, catalytic strategies and comparative mechanistic analysis. In Eckstein,F. and Lilley,D.M.J. (eds), Nucleic Acids and Molecular Biology. Catalytic RNA, Vol. 10. Springer, New York, NY, pp. 1–17.
- 58.Michel F. Umesono,K. and Ozeki,H. (1989) Comparative and functional anatomy of group II catalytic introns – a review. Gene, 82, 5–30. [DOI] [PubMed] [Google Scholar]
- 59.McSwiggen J.A. and Cech,T.R. (1989) Stereochemistry of RNA cleavage by the Tetrahymena ribozyme and evidence that the chemical step is not rate-limiting. Science, 244, 679–683. [DOI] [PubMed] [Google Scholar]
- 60.Rajagopal J., Doudna,J.A. and Szostak,J.W. (1989) Stereochemical course of catalysis by the Tetrahymena ribozyme. Science, 244, 692–694. [DOI] [PubMed] [Google Scholar]
- 61.Padgett R.A., Podar,M., Boulanger,S.C. and Perlman,P.S. (1994) The stereochemical course of group II intron self-splicing. Science, 266, 1685–1688. [DOI] [PubMed] [Google Scholar]
- 62.Chen Y., Li,X. and Gegenheimer,P. (1997) Ribonuclease P catalysis requires Mg2+ coordinated to the pro-Rp oxygen of the scissile bond. Biochemistry, 36, 2425–2438. [DOI] [PubMed] [Google Scholar]
- 63.Piccirilli J.A., Vyle,J.S., Caruthers,M.H. and Cech,T.R. (1993) Metal ion catalysis in the Tetrahymena ribozyme reaction. Nature, 361, 85–88. [DOI] [PubMed] [Google Scholar]
- 64.Podar M., Perlman,P.S. and Padgett,R.A. (1995) Stereochemical selectivity of group II intron splicing, reverse splicing and hydrolysis reactions. Mol. Cell. Biol., 15, 4466–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sontheimer E.J., Sun,S. and Piccirilli,J.A. (1997) Metal ion catalysis during splicing of premessenger RNA. Nature, 388, 801–805. [DOI] [PubMed] [Google Scholar]
- 66.Weinstein L.B., Jones,B.C., Cosstick,R. and Cech,T.R. (1997) A second catalytic metal ion in group I ribozyme. Nature, 388, 805–808. [DOI] [PubMed] [Google Scholar]
- 67.Sontheimer E.J., Gordon,P.M. and Piccirilli,J.A. (1999) Metal ion catalysis during group II intron self-splicing: parallels with the spliceosome. Genes Dev., 13, 1729–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cech T.R. and Golden,B.L. (1999) Building a catalytic active site using only RNA. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 69.Shan S., Yoshida,A., Sun,S., Piccirilli,J.A. and Herschlag,D. (1999) Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc. Natl Acad. Soc. USA, 96, 12299–12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strobel S.A. and Ortoleva-Donnelly,L. (1999) A hydrogen-bonding triad stabilizes the chemical transition state of a group I ribozyme. Chem. Biol., 6, 153–165. [DOI] [PubMed] [Google Scholar]
- 71.Yoshida A., Shan,S., Herschlag,D. and Piccirilli,J.A. (2000) The role of the cleavage site 2′-hydroxyl in the Tetrahymena group I ribozyme reaction. Chem. Biol., 7, 85–96. [DOI] [PubMed] [Google Scholar]
- 72.Shan S. and Herschlag,D. (2000) An unconventional origin of metal-ion rescue and inhibition in the Tetrahymena group I ribozyme reaction. RNA, 6, 795–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pearson R.G. (1968) Hard and soft acids and bases, HSAB, part I: fundamental principles. J. Chem. Educ., 45, 581–587. [Google Scholar]
- 74.Pearson R.G. (1968) Hard and soft acids and bases, HSAB, part II: underlying theories. J. Chem. Educ., 45, 643–648. [Google Scholar]
- 75.Yoshida A., Sun,S. and Piccirilli,J.A. (1999) A new metal ion interaction in the Tetrahymena ribozyme reaction revealed by double sulfur substitution. Nat. Struct. Biol., 6, 318–321. [DOI] [PubMed] [Google Scholar]
- 76.Herschlag D., Piccirilli,J.A. and Cech,T.R. (1991) Ribozyme-catalyzed and nonenzymatic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry, 30, 4844–4854. [DOI] [PubMed] [Google Scholar]
- 77.McConnell T.S. and Cech,T.R (1995) A positive entropy change for guanosine binding and for the chemical step in the Tetrahymena ribozyme reaction. Biochemistry, 34, 4056–4067. [DOI] [PubMed] [Google Scholar]
- 78.Sjögren A.-S., Pettersson,E., Sjöberg,B.M. and Strömberg,R. (1997) Metal ion interaction with cosubstrate in self-splicing of group I introns. Nucleic Acids Res., 25, 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shan S. and Herschlag,D. (1999) Probing the role of metal ions in RNA catalysis: kinetic and thermodynamic characterization of a metal ion interaction with the 2′-moiety of the guanosine nucleophile in the Tetrahymena group I ribozyme. Biochemistry, 38, 10958–10975. [DOI] [PubMed] [Google Scholar]
- 80.Shan S., Narlikar,G.J. and Herschlag,D. (1999) Protonated 2′-aminoguanosine as a probe of the electrostatic environment of the active site of the Tetrahymena group I ribozyme. Biochemistry, 38, 10976–10988. [DOI] [PubMed] [Google Scholar]
- 81.Herschlag D., Eckstein,F. and Cech,T.R. (1993) The importance of being ribose at the cleavage site in the Tetrahymena ribozyme reaction. Biochemistry, 32, 8312–8321. [DOI] [PubMed] [Google Scholar]
- 82.Inoue T., Sullivan,F.X. and Cech,T.R. (1986) New reactions of the ribosomal RNA precursor of Tetrahymena and the mechanism of self-splicing. J. Mol. Biol., 189, 143–165. [DOI] [PubMed] [Google Scholar]
- 83.Been M.D. and Perrotta,A.T. (1991) Group I intron self-splicing with adenosine: evidence for a single nucleoside-binding site. Science, 252, 434–437. [DOI] [PubMed] [Google Scholar]
- 84.Suh E. and Waring,R.B. (1992) A phosphorothioate at the 3′ splice-site inhibits the second splicing step in a group I intron. Nucleic Acids Res., 20, 6303–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gordon P.M., Sontheimer,E.J. and Piccirilli,J.A. (2000) Kinetic characterization of the second step of group II intron splicing: role of metal ions and the cleavage site 2′-OH in catalysis. Biochemistry, 39, 12939–12952. [DOI] [PubMed] [Google Scholar]
- 86.Guerrier-Takada C., Haydock,K., Allen,L. and Altman,S. (1986) Metal ion requirements and other aspects of the reaction catalyzed by M1 RNA, the RNA subunit of ribonuclease P from Escherichia coli. Biochemistry, 25, 1509–1515. [DOI] [PubMed] [Google Scholar]
- 87.Smith D. and Pace,N.R. (1993) Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry, 32, 5273–5281. [DOI] [PubMed] [Google Scholar]
- 88.Warnecke J.M., Fürste,J.P., Hardt,W.D., Erdmann,V.A. and Hartmann,R.K. (1996) Ribonuclease P (RNase P) RNA is converted to a Cd2+-ribozyme by a single Rp-phosphorothioate modification in the precursor tRNA at the RNase P cleavage site. Proc. Natl Acad. Sci. USA, 93, 8924–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Warnecke J.M., Held,R., Busch,S. and Hartmann,R.K. (1999) Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis. J. Mol. Biol., 290, 433–445. [DOI] [PubMed] [Google Scholar]
- 90.Thomas B.C., Chamberlain,J., Engelke,D.R. and Gegenheimer,P. (2000) Evidence for an RNA-based catalytic mechanism in eukaryotic nuclear ribonuclease P. RNA, 6, 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thomas B.C., Li,X. and Gegenheimer,P. (2000) Chloroplast ribonuclease P does not utilize the ribozyme-type pre-tRNA cleavage mechanism. RNA, 6, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Warnecke J.M., Sontheimer,E.J., Piccirilli,J.A. and Hartmann,R.K. (2000) Active site constraints in the hydrolysis reaction catalyzed by bacterial RNase P: analysis of precursor tRNAs with a single 3′-S-phosphorothiolate internucleotide linkage. Nucleic Acids Res., 28, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saville B.J. and Collins,R.A. (1990) A site-specific self-cleavage reaction performed by a novel RNA in Neurospora mitochondria. Cell, 61, 685–696. [DOI] [PubMed] [Google Scholar]
- 94.van Tol H., Buzayan,J.M., Feldstein,P.A., Eckstein,F. and Bruening,G. (1990) Two autolytic processing reactions of a satellite RNA proceed with inversion of configuration. Nucleic Acids Res., 18, 1971–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dahm S.C. and Uhlenbeck,O.C. (1991) Role of divalent metal ions in the hammerhead RNA cleavage reaction. Biochemistry, 30, 9464–9469. [DOI] [PubMed] [Google Scholar]
- 96.Slim G. and Gait,M.J. (1991) Configurationally defined phosphorothioate-containing oligoribonucleotides in the study of the mechanism of cleavage of hammerhead ribozymes. Nucleic Acids Res., 19, 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koizumi M. and Ohtsuka,E. (1991) Effects of phosphorothioate and 2-amino groups in hammerhead ribozymes on cleavage rates and Mg2+ binding. Biochemistry, 30, 5145–5150. [DOI] [PubMed] [Google Scholar]
- 98.Foster A.C. and Symons,R.H. (1987) Self-cleavage of virusoid RNA is performed by the proposed 55-nucleotide active site. Cell, 50, 9–16. [DOI] [PubMed] [Google Scholar]
- 99.Scott W.G., Murray,J.B., Arnold,J.R.P., Stoddard,B.L. and Klug,A. (1996) Capturing the structure of a catalytic RNA intermediate: the hammerhead ribozyme. Science, 274, 2065–2069. [DOI] [PubMed] [Google Scholar]
- 100.Kuimelis R.G. and McLaughlin,L.W. (1996) Ribozyme-mediated cleavage of a substrate analogue containing an internucleotide-bridging 5′-phosphorothioate: evidence for the single-metal model. Biochemistry, 35, 5308–5317. [DOI] [PubMed] [Google Scholar]
- 101.Kuimelis R.G. and McLaughlin,L.W. (1997) Application of a 5′-bridging phosphorothioate to probe divalent metal and hammerhead ribozyme mediated RNA cleavage. Bioorg. Med. Chem., 5, 1051–1061. [DOI] [PubMed] [Google Scholar]
- 102.Torres R.A. and Bruice,T.C. (1998) Molecular dynamics study displays near in-line attack conformations in the hammerhead ribozyme self-cleavage reaction. Proc. Natl Acad. Sci. USA, 95, 11077–11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Torres R.A. and Bruice,T.C. (2000) The mechanism of phosphodiester hydrolysis: near in-line attack conformation in the hammerhead ribozyme. J. Am. Chem. Soc., 122, 781–791. [Google Scholar]
- 104.Uchimaru T., Uebayasi,M., Tanabe,K. and Taira,K. (1993) Theoretical analyses on the role of Mg2+ ions in ribozyme reactions. FASEB J., 7, 137–142. [DOI] [PubMed] [Google Scholar]
- 105.Sharmeen L., Kuo,M.Y.-P., Dinter-Gottlieb,G. and Taylor,J. (1988) Antigenomic RNA of human hepatitis delta virus can undergo self-cleavage. J. Virol., 62, 2674–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kuo M.Y.-P., Sharmeen,L., Dinter-Gottlieb,G. and Taylor,J. (1988) Characterization of self-cleaving RNA sequences on the genome and antigenome of human hepatitis delta virus. J. Virol., 62, 4439–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perrotta A.T. and Been,M.D. (1991) A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature, 350, 434–436. [DOI] [PubMed] [Google Scholar]
- 108.Lai M.M. (1995) The molecular biology of hepatitis delta virus. Annu. Rev. Biochem., 64, 259–286. [DOI] [PubMed] [Google Scholar]
- 109.Shih I. and Been,M.D. (2000) Kinetic scheme for intermolecular RNA cleavage by a ribozyme derived from hepatitis delta virus RNA. Biochemistry, 39, 9055–9066. [DOI] [PubMed] [Google Scholar]
- 110.Buzayan J.M., Gerlach,W.L. and Bruening,G. (1986) Non-enzymatic cleavage and ligation of RNAs complementary to a plant virus satellite RNA. Nature, 323, 349–353. [Google Scholar]
- 111.Feldstein P.A., Buzayan,J.M., van Tol,H., de Bear,J., Gough,G.R., Gilham,P.T. and Bruening,G. (1990) Specific association between an endoribonucleolytic sequence from a satellite RNA and a substrate analogue containing a 2′-5′ phosphodiester. Proc. Natl Acad. Sci. USA, 87, 2623–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hampel A., Tritz,R., Hicks,M. and Cruz,P. (1990) ‘Hairpin’ catalytic RNA model: evidence for helices and sequence requirement for substrate RNA. Nucleic Acids Res., 18, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Feldstein P.A. and Bruening,G. (1993) Catalytically active geometry in the reversible circularization of ‘mini-monomer’ RNAs derived from the complementary strand of tobacco ringspot virus satellite RNA. Nucleic Acids Res., 21, 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hertel K.J., Herschlag,D. and Uhlenbeck,O.C. (1994) A kinetic and thermodynamic framework for the hammerhead ribozyme reaction. Biochemistry, 33, 3374–3385. [DOI] [PubMed] [Google Scholar]
- 115.Hegg L.A. and Fedor,M.J. (1995) Kinetics and thermodynamics of intermolecular catalysis by hairpin ribozymes. Biochemistry, 34, 15813–15828. [DOI] [PubMed] [Google Scholar]
- 116.Shippy R., Lockner,R., Farnsworth,M. and Hampel,A. (1999) The hairpin ribozyme. Discovery, mechanism and development for gene therapy. Mol. Biotechnol., 12, 117–129. [DOI] [PubMed] [Google Scholar]
- 117.Nesbitt S.M., Erlacher,H.A. and Fedor,M.J. (1999) The internal equilibrium of the hairpin ribozyme: temperature, ion and pH effects. J. Mol. Biol., 286, 1009–1024. [DOI] [PubMed] [Google Scholar]
- 118.Sargueil B., McKenna,J. and Burke,J.M. (2000) Analysis of the functional role of a G•A sheared base pair by in vitro genetics. J. Biol. Chem., 41, 32157–32166. [DOI] [PubMed] [Google Scholar]
- 119.Collins R.A. and Saville,B.J. (1990) Independent transfer of mitochondrial chromosomes and plasmids during unstable vegetative fusion in Neurospora. Nature, 345, 177–179. [DOI] [PubMed] [Google Scholar]
- 120.Collins R.A. and Olive,J.E. (1993) Reaction conditions and kinetics of self-cleavage of a ribozyme derived from Neurospora VS RNA. Biochemistry, 32, 2795–2799. [DOI] [PubMed] [Google Scholar]
- 121.Rastogi T., Beattie,T.L., Olive,J.E. and Collins,R.A. (1996) A long-range pseudoknot is required for activity of the Neurospora VS ribozyme. EMBO J., 15, 2820–2825. [PMC free article] [PubMed] [Google Scholar]
- 122.Sood V.D., Beattie,T.L. and Collins,R.A. (1998) Identification of phosphate groups involved in metal binding and tertiary interactions in the core of the Neurospora VS ribozyme. J. Mol. Biol., 282, 741–750. [DOI] [PubMed] [Google Scholar]
- 123.Rastogi T. and Collins,R.A. (1998) Smaller, faster ribozymes reveal the catalytic core of Neurospora VS RNA. J. Mol. Biol., 277, 215–224. [DOI] [PubMed] [Google Scholar]
- 124.Guo H.C.T. and Collins,R.A. (1995) Efficient trans-cleavage of a stem–loop RNA substrate by a ribozyme derived from Neurospora VS RNA. EMBO J., 14, 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koizumi M., Iwai,S. and Ohtsuka,E. (1988) Construction of a series of several self-cleaving RNA duplexes using synthetic 21-mers. FEBS Lett., 228, 228–230. [DOI] [PubMed] [Google Scholar]
- 126.Jeffries A.C. and Symons,R.H. (1989) A catalytic 13-mer ribozyme. Nucleic Acids Res., 17, 1371–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ruffner D.E., Stormo,G.D. and Uhlenbeck,O.C. (1990) Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry, 29, 10695–10702. [DOI] [PubMed] [Google Scholar]
- 128.Perriman R., Delves,A. and Gerlach,W.L. (1992) Extended target-site specificity for a hammerhead ribozyme. Gene, 113, 157–163. [DOI] [PubMed] [Google Scholar]
- 129.Zoumadakis M. and Tabler,M. (1995) Comparative analysis of cleavage rates after systematic permutation of the NUX consensus target motif for hammerhead ribozymes. Nucleic Acids Res., 23, 1192–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shimayama T., Nishikawa,S. and Taira,K. (1995) Generality of the NUX rule: kinetic analysis of the results of systematic mutations in the trinucleotide at the cleavage site of hammerhead ribozymes. Biochemistry, 34, 3649–3654. [DOI] [PubMed] [Google Scholar]
- 131.Kore A.R., Vaish,N.K., Kutzke,U. and Eckstein,F. (1998) Sequence specificity of the hammerhead ribozyme revisited; the NHH rule. Nucleic Acids Res., 26, 4116–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pley H.W., Flaherty,K.M. and McKay,D.B. (1994) Three-dimensional structure of a hammerhead ribozyme. Nature, 372, 68–74. [DOI] [PubMed] [Google Scholar]
- 133.Scott W.G., Finch,J.T. and Klug,A. (1995) The crystal structure of an all-RNA hammerhead ribozyme: a proposed mechanism for RNA catalytic cleavage. Cell, 81, 991–1002. [DOI] [PubMed] [Google Scholar]
- 134.Murray J.B., Terwey,D.P., Maloney,L., Karpeisky,A., Usman,N., Beigelman,L. and Scott,W.G. (1998) The structural basis of hammerhead ribozyme self-cleavage. Cell, 92, 665–673. [DOI] [PubMed] [Google Scholar]
- 135.Wedekind J.E. and McKay,D.B. (1998) Crystallographic structures of the hammerhead ribozyme: relationship to ribozyme folding and catalysis. Annu. Rev. Biophys. Biomol. Struct., 27, 475–502. [DOI] [PubMed] [Google Scholar]
- 136.Feig A.L., Scott,W.G. and Uhlenbeck,O.C. (1998) Inhibition of the hammerhead ribozyme cleavage reaction by site-specific binding of Tb(III). Science, 279, 81–84. [DOI] [PubMed] [Google Scholar]
- 137.Murray J.B., Szöke,H., Szöke,A. and Scott,W.G. (2000) Capture and visualization of a catalytic RNA enzyme–product complex using crystal lattice trapping and X-ray holographic reconstruction. Mol. Cell, 5, 279–287. [DOI] [PubMed] [Google Scholar]
- 138.Doudna J.A. (1998) Ribozymes: the hammerhead swings into action. Curr. Biol., 8, R495–R497. [DOI] [PubMed] [Google Scholar]
- 139.Tuschl T., Gohlke,C., Jovin,T.M., Westhof,E. and Eckstein,F. (1994) A three-dimensional model for the hammerhead ribozyme based on fluorescence measurements. Science, 266, 785–789. [DOI] [PubMed] [Google Scholar]
- 140.Perkins T.A., Wolf,D.E. and Goodchild,J. (1996) Fluorescence resonance energy transfer analysis of ribozyme kinetics reveals the mode of action of a facilitator oligonucleotide. Biochemistry, 35, 16370–16377. [DOI] [PubMed] [Google Scholar]
- 141.Bassi G.S., Mollegaard,N.E., Murchie,A.I. and Lilley,D.M. (1999) RNA folding and misfolding of the hammerhead ribozyme. Biochemistry, 38, 3345–3354. [DOI] [PubMed] [Google Scholar]
- 142.Singh K.K., Parwaresch,R. and Krupp,G. (1999) Rapid kinetic characterization of hammerhead ribozymes by real-time monitoring of fluorescence resonance energy transfer (FRET). RNA, 5, 1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Orita M., Vinayak,R., Andrus,A., Warashina,M., Chiba,A., Kaniwa,H., Nishikawa,F., Nishikawa,S. and Taira,K. (1996) Magnesium-mediated conversion of an inactive form of a hammerhead ribozyme to an active complex with its substrate. An investigation by NMR spectroscopy. J. Biol. Chem., 271, 9447–9454. [DOI] [PubMed] [Google Scholar]
- 144.Sakamoto T., Kim,M.H., Kurihara,Y., Sasaki,N., Noguchi,T., Katahira,M. and Uesugi,S. (1997) Properties of a hammerhead ribozyme with deletion of stem II. J. Biochem., 121, 288–294. [DOI] [PubMed] [Google Scholar]
- 145.Ojha R.P., Dhingra,M.M., Sarma,M.H., Myer,Y.P., Setlik,R.F., Shibata,M., Kazim,A.L., Ornstein,R.L., Rein,R., Turner,C.J. and Sarma,R.H. (1997) Structure of an anti-HIV-1 hammerhead ribozyme complex with a 17-mer DNA substrate analog of HIV-1 gag RNA and a mechanism for the cleavage reaction: 750 MHz NMR and computer experiments. J. Biomol. Struct. Dyn., 15, 185–215. [DOI] [PubMed] [Google Scholar]
- 146.Seela F., Debelak,H., Usman,N., Burgin,A. and Beigelman,L. (1998) 1-Deazaadenosine: synthesis and activity of base-modified hammerhead ribozymes. Nucleic Acids Res., 26, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kuwabara T., Warashina,M., Orita,M., Koseki,S., Ohkawa,J. and Taira,K. (1998) Formation of a catalytically active dimer by tRNAVal-driven short ribozymes. Nat. Biotechnol., 16, 961–965. [DOI] [PubMed] [Google Scholar]
- 148.Scott W.G. (1998) RNA catalysis. Curr. Opin. Struct. Biol., 8, 720–726. [DOI] [PubMed] [Google Scholar]
- 149.Hansen M.R., Simorre,J.P., Hanson,P., Mokler,V., Bellon,L., Beigelman,L. and Pardi,A. (1999) Identification and characterization of a novel high affinity metal-binding site in the hammerhead ribozyme. RNA, 5, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Suzumura K., Warashina,M., Yoshinari,K., Tanaka,Y., Kuwabara,T., Orita,M. and Taira,K. (2000) Significant change in the structure of a ribozyme upon introduction of a phosphorothioate linkage at P9: NMR reveals a conformational fluctuation in the core region of a hammerhead ribozyme. FEBS Lett., 473, 106–112. [DOI] [PubMed] [Google Scholar]
- 151.Maderia M., Hunsicker,L.M. and DeRose,V.J. (2000) Metal-phosphate interactions in the hammerhead ribozyme observed by 31P NMR and phosphorothioate substitutions. Biochemistry, 39, 12113–12120. [DOI] [PubMed] [Google Scholar]
- 152.Tanaka Y., Morita,E.H., Hayashi,H., Kasai,Y., Tanaka,T. and Taira,K. (2000) Well-conserved tandem G·A pairs and the flanking C·G pair in hammerhead ribozymes are sufficient for capture of structurally and catalytically important metal ions. J. Am. Chem. Soc., 122, 11303–11310. [Google Scholar]
- 153.Ruffner D.E. and Uhlenbeck,O.C. (1990) Thiophosphate interference experiments locate phosphates important for the hammerhead RNA self-cleavage reaction. Nucleic Acids Res., 18, 6025–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ruffner D.E., Stormo,G.D. and Uhlenbeck,O.C. (1990) Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry, 29, 10695–10702. [DOI] [PubMed] [Google Scholar]
- 155.Tuschl T. and Eckstein,F. (1993) Hammerhead ribozymes: importance of stem–loop II for activity. Proc. Natl Acad. Sci. USA, 90, 6991–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Feig A.L., Panek,M., Horrocks,W.D.,Jr and Uhlenbeck,O.C. (1999) Probing the binding of Tb(III) and Eu(III) to the hammerhead ribozyme using luminescence spectroscopy. Chem. Biol., 6, 801–810. [DOI] [PubMed] [Google Scholar]
- 157.Peracchi A., Beigelman,L., Scott,E.C., Uhlenbeck,O.C. and Herschlag,D. (1997) Involvement of a specific metal ion in the transition of the hammerhead ribozyme to its catalytic conformation. J. Biol. Chem., 272, 26822–26826. [DOI] [PubMed] [Google Scholar]
- 158.Knöll R., Bald,R. and Fürste,J.P. (1997) Complete identification of nonbridging phosphate oxygens involved in hammerhead cleavage. RNA, 3, 132–140. [PMC free article] [PubMed] [Google Scholar]
- 159.Peracchi A., Beigelman,L., Usman,N. and Herschlag,D. (1996) Rescue of abasic hammerhead ribozymes by exogenous addition of specific bases. Proc. Natl Acad. Sci. USA, 93, 11522–11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peracchi A., Karpeisky,A., Maloney,L., Beigelman,L. and Herschlag,D. (1998) A core folding model for catalysis by the hammerhead ribozyme accounts for its extraordinary sensitivity to abasic mutations. Biochemistry, 37, 14765–14775. [DOI] [PubMed] [Google Scholar]
- 161.Nakamatsu Y., Kuwabara,T., Warashina,M., Tanaka,Y., Yoshinari,K. and Taira,K. (2000) Significant activity of a modified ribozyme with N7-deazaguanine at G10.1: the double-metal-ion mechanism of catalysis in reactions catalysed by hammerhead ribozymes. Genes Cells, 5, 603–612. [DOI] [PubMed] [Google Scholar]
- 162.Yoshinari K. and Taira,K. (2000) A further investigation and reappraisal of the thio effect in the cleavage reaction catalyzed by a hammerhead ribozyme. Nucleic Acids Res., 28, 1730–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Murray J.B. and Scott,W.G. (2000) Does a single metal ion bridge the A-9 and scissile phosphate groups in the catalytically active hammerhead ribozyme structure? J. Mol. Biol., 296, 33–41. [DOI] [PubMed] [Google Scholar]
- 164.Uchimaru T., Stec,W.J., Tsuzuki,S., Hirose,T., Tanabe,K. and Taira,K. (1996) Ab initio investigation on nucleophilic ring opening of 1,3,2-oxathiaphospholane: nucleophilic substitution at phosphorus coupled with pseudorotation. Chem. Phys. Lett., 263, 691–696. [Google Scholar]
- 165.Uchimaru T., Stec,W.J. and Taira,K. (1997) Mechanism of the chemoselective and stereoselective ring opening of oxathiaphospholanes: an ab initio study. J. Org. Chem., 62, 5793–5800. [Google Scholar]
- 166.Hermann T., Auffinger,P., Scott,W.G. and Westhof,E. (1997) Evidence for a hydroxide ion bridging two magnesium ions at the active site of the hammerhead ribozyme. Nucleic Acids Res., 25, 3421–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Taira K., Uebayasi,M., Maeda,H. and Furukawa,K. (1990) Energetics of RNA cleavage: implications for the mechanism of action of ribozymes. Protein Eng., 3, 691–701. [DOI] [PubMed] [Google Scholar]
- 168.Zhou D.M., Kumar,P.K.R., Zhang,L.H. and Taira,K. (1996) Ribozyme mechanism revisited: evidence against direct coordination of a Mg2+ ion with the pro-R oxygen of the scissile phosphate in the transition state of a hammerhead ribozyme-catalyzed reaction. J. Am. Chem. Soc., 118, 8969–8970. [Google Scholar]
- 169.Warashina M., Takagi,Y., Sawata,S., Zhou,D.M., Kuwabara,T. and Taira,K. (1997) Entropically driven enhancement of cleavage activity of a DNA-armed hammerhead ribozyme: mechanism of action of hammerhead ribozymes. J. Org. Chem., 62, 9138–9147. [Google Scholar]
- 170.Kumar P.K.R., Zhou,D.M., Yoshinari,K. and Taira,K. (1996) Mechanistic studies on hammerhead ribozymes. In Eckstein,F. and Lilley,D.M.J. (eds), Nucleic Acids and Molecular Biology. Catalytic RNA, Vol. 10. Springer, New York, NY, pp. 217–230.
- 171.Bell R.P. and Kuhn,A.T. (1963) Dissociation constants of some acids in deuterium oxide. Trans. Faraday Soc., 59, 1789–1793. [Google Scholar]
- 172.Jenckes W.P. (1969) Catalysis in Chemistry and Enzymology. McGraw-Hill, New York, NY, pp. 250–253.
- 173.Lyne P.D. and Karplus,M. (2000) Determination of the pKa of the 2′-hydroxyl group of a phosphorylated ribose: implications for the mechanism of hammerhead ribozyme catalysis. J. Am. Chem. Soc., 122, 166–167. [Google Scholar]
- 174.Grasby J.A., Jonathan,P., Butler,G. and Gait,M.J. (1993) The synthesis of oligoribonucleotides containing O6-methylguanosine: the role of conserved guanosine residues in hammerhead ribozyme cleavage. Nucleic Acids Res., 21, 4444–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cunningham L.A., Li,J. and Li,Y. (1998) Spectroscopic evidence for inner-sphere coordination of metal ions to the active site of a hammerhead ribozyme. J. Am. Chem. Soc., 120, 4518–4519. [Google Scholar]
- 176.Scott E.C. and Uhlenbeck,O.C. (1999) A re-investigation of the thio effect at the hammerhead cleavage site. Nucleic Acids Res., 27, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhou D.M., He,Q.C., Zhou,J.M. and Taira,K. (1998) Explanation by a putative triester-like mechanism for the thio effects and Mn2+ rescues in reactions catalyzed by a hammerhead ribozyme. FEBS Lett., 431, 154–160. [DOI] [PubMed] [Google Scholar]
- 178.Ora M., Peltomäki,M., Oivanen,M. and Lönnberg,H. (1998) Metal-ion-promoted cleavage, isomerization and desulfurization of the diastereomeric phosphoromonothioate analogues of uridylyl(3′,5′)uridine. J. Org. Chem., 63, 2939–2947. [Google Scholar]
- 179.Derrick W.B., Greef,C.H., Caruthers,M.H. and Uhlenbeck,O.C. (2000) Hammerhead cleavage of the phosphorodithioate linkage. Biochemistry, 39, 4947–4954. [DOI] [PubMed] [Google Scholar]
- 180.Warnecke J.M., Green,C.J. and Hartmann,R.K. (1997) Role of metal ions in the cleavage mechanism by the E.coli RNase P holoenzyme. Nucl. Nucl., 16, 721–725. [Google Scholar]
- 181.Brautigam C.A. and Steitz,T.A. (1998) Structural principles for the inhibition of the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I by phosphorothioates. J. Mol. Biol., 277, 363–377. [DOI] [PubMed] [Google Scholar]
- 182.Hamm M.L., Schwans,J.P. and Piccirilli,J.A. (2000) The hammerhead ribozyme catalyzes the deglycosylation of 2′-mercaptocytidine. J. Am. Chem. Soc., 122, 4223–4224. [Google Scholar]
- 183.Hamm M.L., Nikolic,D., van Breemen,R.B. and Piccirilli,J.A. (2000) Unconventional origin of metal ion rescue in the hammerhead ribozyme reaction: Mn2+-assisted redox conversion of 2′-mercaptocytidine to cytidine. J. Am. Chem. Soc., 122, 12069–12078. [Google Scholar]
- 184.Smith J.S. and Nikonowicz,E.P. (2000) Phosphorothioate substitution can substantially alter RNA conformation. Biochemistry, 39, 5642–5652. [DOI] [PubMed] [Google Scholar]
- 185.Hamid F., Kawakami,J., Nishikawa,F. and Nishikawa,S. (1997) Analysis of the cleavage reaction of a trans-acting human hepatitis delta virus ribozyme. Nucleic Acids Res., 25, 3124–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Basolo F. and Pearson,R.G. (1967) Mechanism of Inorganic Reactions. A Study of Metal Complexes in Solution. John Wiley and Sons, Inc, New York, NY.
- 187.Chowrira B.M., Berzal-Herranz,A. and Burke,J.M. (1991) Novel guanosine requirement for catalysis by the hairpin ribozyme. Nature, 354, 320–322. [DOI] [PubMed] [Google Scholar]
- 188.Matsushima Y. and Takashima,Y. (1999) Seimei no Mukikagaku. Hirokawa Publishing Co, Tokyo, Japan. [Google Scholar]