

Background: The JunB transcription factor is a key mediator of proliferation, apoptosis, differentiation, and the immune response.
Results: JunB is a caspase substrate.
Conclusion: Caspase-mediated cleavage of JunB generates a C-terminal fragment, that when overexpressed, functions as an inhibitor of AP-1-dependent transcription.
Significance: We demonstrate a novel regulatory mechanism for an important transcription factor.
Keywords: AP-1 Transcription Factor, Apoptosis, Caspase, Gene Transcription, Jun Transcription Factor, Inflammasome
Abstract
The activator protein-1 (AP-1) family transcription factor, JunB, is an important regulator of proliferation, apoptosis, differentiation, and the immune response. In this report, we show that JunB is cleaved in a caspase-dependent manner in apoptotic anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma cell lines and that ectopically expressed JunB is cleaved in murine RAW 264.7 macrophage cells treated with the NALP1b inflammasome activator, anthrax lethal toxin. In both cases, we identify aspartic acid 137 as the caspase cleavage site and demonstrate that JunB can be directly cleaved in vitro by multiple caspases at this site. Cleavage of JunB at aspartic acid 137 separates the N-terminal transactivation domain from the C-terminal DNA binding and dimerization domains, and we show that the C-terminal cleavage fragment retains both DNA binding activity and the ability to interact with AP-1 family transcription factors. Furthermore, this fragment interferes with the binding of full-length JunB to AP-1 sites and inhibits AP-1-dependent transcription. In summary, we have identified and characterized a novel mechanism of JunB post-translational modification and demonstrate that the C-terminal JunB caspase cleavage product functions as a potent inhibitor of AP-1-dependent transcription.
Introduction
Activator protein-1 (AP-1)4 family transcription factors consist of homodimers and heterodimers of members of the Jun, Fos, ATF, and Maf subfamilies (1, 2). These proteins bind 12-O-tetradecanoylphorbol-13-acetate response elements (5′-TGAG/CTCA-3′), cAMP response elements (5′-TGACGTCA-3′), and variants of these sequences (1, 3, 4). Collectively, these proteins influence the expression of many genes including those associated with proliferation and survival (1, 3), differentiation (5), and inflammation (6).
JunB is a member of the AP-1 family that was first described for its ability to inhibit c-Jun transcriptional activity and block the transformation of fibroblasts by c-Jun (7, 8). JunB is broadly expressed (9, 10), and mice deficient in JunB die during embryogenesis due to defects in the generation of extraembryonic tissues (11). Additionally, JunB is able to promote important signaling events in some lymphomas. For example, JunB is highly expressed in Hodgkin lymphoma (12–14), CD30+ diffuse large B cell lymphoma (13), anaplastic lymphoma kinase-negative, anaplastic large cell lymphoma (ALK− ALCL) (13), as well as anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma (ALK+ ALCL) (12, 13). In ALK+ ALCL, JunB promotes the proliferation of tumor cells (15) and regulates the expression of several genes whose encoded proteins are either important in the pathogenesis of this lymphoma or are phenotypic characteristics of this cancer (16–20).
While investigating whether silencing of JunB sensitized ALK+ ALCL cell lines to apoptosis, we observed an alteration in the electrophoretic mobility of JunB upon treatment with apoptosis-inducing agents. In this report, we demonstrate that JunB is cleaved in a caspase-dependent manner in ALK+ ALCL cell lines treated with the apoptosis-inducing drugs staurosporine and doxorubicin. We map the site of JunB cleavage in apoptotic cells to aspartic acid 137 and show that JunB can be cleaved directly at this site in vitro by multiple caspases. Furthermore, we establish that ectopically expressed JunB is also cleaved at aspartic acid 137 in a caspase-dependent manner in murine RAW 264.7 macrophage cells treated with the inflammasome activator, anthrax lethal toxin. Importantly, cleavage of JunB at this site separates the N-terminal transcriptional activation domain from the C-terminal dimerization and DNA binding domains, and we show that the C-terminal JunB cleavage product retains the ability to bind DNA and associate with AP-1 family proteins. In this regard, overexpression of this fragment: (i) interferes with the ability of full-length JunB to bind an AP-1 target DNA sequence and (ii) inhibits transcription driven by an AP-1-dependent luciferase reporter. In conclusion, our findings reveal that JunB is cleaved by caspases in apoptotic and inflammasome-stimulated cells and that this cleavage generates a fragment that can function as an inhibitor of AP-1-dependent transcription.
EXPERIMENTAL PROCEDURES
Antibodies, cDNA Constructs, and Other Reagents
The anti-caspase 3 mouse monoclonal antibody (mAb) (3G2) and rabbit anti-cleaved caspase 3 polyclonal antibody (pAb) (9661) were purchased from Cell Signaling Technology. The mAbs for JunB (C-11 and 204C4a), c-Fos (C-10), Myc (9E10), tubulin (DM1A), and PARP-1 (C2-10) as well as the pAb for Fra2 (Q-20) were purchased from Santa Cruz Biotechnology. The mouse anti-β-actin mAb (AC-15), anti-FLAG mAb (M2), and anti-FLAG pAb were purchased from Sigma-Aldrich. The rabbit anti-pJunB (Ser-259) pAb (ab30628), and rabbit anti-caspase 1 mAb (EPR4321) were purchased from Abcam, and the rabbit anti-caspase 3 pAb (used in Fig. 1A) was a gift from Dr. Michele Barry (University of Alberta). Staurosporine and the pan-caspase inhibitor, Z-VAD-FMK, were purchased from Enzo Life Sciences. Doxorubicin was purchased from Sigma-Aldrich.
FIGURE 1.

The electrophoretic mobility of JunB is altered in a caspase-dependent manner in lysates of ALK+ ALCL cell lines treated with apoptosis-inducing drugs. A, Karpas 299 or SUP-M2 cells were left untreated or treated with staurosporine (STS) for 6 h. Lysates from these cells were then Western blotted (WB) with an anti-JunB antibody. B, cells were treated and lysates blotted as described in A with the addition of cells co-treated with staurosporine and Z-VAD-FMK. C and D, Karpas 299 cells were treated for increasing times (C) or with increasing concentrations (D) of staurosporine, and lysates from these cells were probed with the indicated antibodies. The anti-PARP blot is included to demonstrate the cleavage kinetics of a known caspase substrate. E, Karpas 299 or SUP-M2 cells were left untreated, treated with doxorubicin, or treated with doxorubicin and Z-VAD-FMK for 12 h. Anti-JunB Western blotting was then performed on the lysates. Apoptosis induction in all experiments was determined by the disappearance of pro-caspase 3 and the appearance of cleaved, active caspase 3. Anti-α-tubulin and anti-β-actin blots are included to demonstrate protein loading. The electrophoretic mobility of molecular mass standards is indicated to the sides of blots.
Myc-tagged JunB was generated by adding a double Myc tag with glycine/alanine spacer to the 5′ end of the JunB cDNA, which was then cloned into pcDNA3.1A (Invitrogen). The putative caspase cleavage site mutants were generated by site-directed mutagenesis where aspartic acid residues 31, 137, or 144 and 145 were mutated to alanine. The Myc-tagged N-terminal truncation mutant was generated by introducing a stop codon following the codon encoding for aspartic acid 137, whereas the Myc-tagged C-terminal truncation mutant consists of amino acids 138–347 of JunB. The FLAG-tagged JunB has been described previously (17). The pAP1-Luc AP-1 promoter firefly luciferase vector was purchased from BD Biosciences, and the pcDNA3.1 FLAG-tagged c-Fos (rat) (21) was obtained from Addgene (plasmid 8966).
Cells Lines and Electroporations
The Karpas 299 and SUP-M2 ALK+ ALCL cell lines were obtained from Dr. Raymond Lai (University of Alberta) and cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 1 mm sodium pyruvate, 2 mm l-glutamine, and 50 μm 2-mercaptoethanol. The RAW 264.7 murine macrophage cell line was obtained from Dr. Stefan Pukatzki (University of Alberta) and cultured in DMEM that was supplemented with 10% heat-inactivated FBS, 1 mm sodium pyruvate, and 2 mm l-glutamine. All cells were incubated at 37 °C in a 5% CO2 atmosphere.
Cells (1–2 × 107 cells/ml) were transfected by electroporation in 500 μl of RPMI 1640 medium in a 4-mm gap cuvette (VWR Canlab; Mississauga, ON, Canada). Electroporations were carried out using an ECM 830 square wave electroporator (BTX, San Diego, CA) with the following settings: 225 V, three pulses, 8-ms pulse length, 1 s between pulses. Cells were allowed to rest for 10 min following electroporation, resuspended in RPMI 1640 medium, and then incubated at 37 °C in a 5% CO2 atmosphere.
Doxorubicin and Staurosporine Treatment
Cells (5 × 105–1 × 106 cells/ml) were induced to undergo apoptosis by treatment with either 5 μm doxorubicin for 12 h or 2 μm staurosporine for 6 h at 37 °C. For time course and dose-response experiments, the treatment times and drug concentrations are indicated in the respective figures.
Anthrax Lethal Toxin Treatment
The anthrax lethal toxin components, anthrax lethal factor, and protective antigen were purchased from Cedarlane Laboratories (Burlington, ON, Canada). RAW 264.7 cells were transfected in 6-well plates with 3 μg of the indicated Myc-JunB cDNAs or vector alone using the FuGENE HD transfection reagent (Promega). Twenty h after transfection, cells were placed in fresh serum-free medium and treated with anthrax lethal factor and protective antigen at a final concentration of 1 μg/ml each for 6 h with or without Z-VAD-FMK. Staurosporine-treated cells served as a positive control.
Cell Lysis, Immunoprecipitations, and Western Blotting
Cells were collected by centrifugation, washed in PBS, and lysed in a 1% Nonidet P-40 lysis buffer (1% Nonidet P-40, 50 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 10% glycerol) containing 1 mm PMSF, 1 mm sodium orthovanadate, and protease inhibitor mixture (Sigma-Aldrich). Cell lysates were cleared by centrifugation at ∼20,000 × g for 10 min, and the protein concentration of the cleared lysates was determined using the bicinchoninic acid (BCA) Protein Assay Kit (Thermo Scientific). For immunoprecipitations, cleared lysates were incubated with 1–2 μg of antibody and protein G-Sepharose beads (Sigma-Aldrich) for 1–2 h on a nutator at 4 °C. Beads were then washed with lysis buffer, and proteins were eluted by boiling in SDS-PAGE sample buffer. For Fig. 6C, the Myc-tagged JunB proteins and rat FLAG-c-Fos were produced using the TNT T7 Quick Coupled Transcription/Translation System (Promega). 2 μl of each extract was mixed for 20 min at room temperature, and then anti-FLAG immunoprecipitations were performed as outlined above. Immunoprecipitates and cell lysates were resolved on SDS-polyacrylamide gels before being transferred to nitrocellulose membranes. Membranes were blocked in 5% nonfat milk powder in TBS before being probed overnight with primary Ab. Blots were then washed and probed with HRP-conjugated secondary antibodies, and proteins were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Reprobed blots were first stripped in 0.1% TBST, pH 2, stripping solution for 1 h before being reprobed as described above.
FIGURE 6.

The C-terminal JunB cleavage fragment can co-precipitate with c-Fos, Fra2, and full-length JunB. A, Karpas 299 cells were transfected with vector alone or cDNAs encoding for Myc-tagged JunB (WT), Myc-tagged N-terminal JunB cleavage fragment (NT), or Myc-tagged C-terminal JunB cleavage fragment (CT). Twenty-four h after transfection, cells were lysed, and lysates were immunoprecipitated (IP) with an anti-Myc antibody. Western blotting (WB) was then performed on immunoprecipitates with anti-c-Fos, anti-Fra2, or anti-Myc antibodies. B, Karpas 299 cells were transfected with the cDNAs encoding for the indicated proteins. Cells were lysed 24 h after transfection, and anti-FLAG immunoprecipitation was performed on lysates. Western blotting was then performed on the immunoprecipitates. C, the indicated in vitro transcribed/translated Myc-tagged JunB proteins were incubated with in vitro transcribed/translated FLAG-c-Fos, and anti-FLAG immunoprecipitations (IP) were performed. Western blotting (WB) was then performed on the immunoprecipitates. Cell lysates (A and B) or TNT extracts (C) were run to show the electrophoretic mobility and the presence of the respective proteins. The immunoglubulin heavy (IgH) and light chains (IgL) of the immunoprecipitating antibody are indicated. The electrophoretic mobility of molecular mass standards is indicated to the left of blots.
JunB Dephosphorylation Assays
6 × 106 Karpas 299 or SUP-M2 cells were lysed in 1 ml of 1% Nonidet P-40 lysis buffer with inhibitors. Immunoprecipitations were performed, and immunoprecipitates were washed in 1% Nonidet P-40 lysis buffer without EDTA and sodium orthovanadate followed by a wash in FastAP buffer (Fermentas). JunB was then dephosphorylated by incubating immunoprecipitates with 2 units of FastAP Thermosensitive Alkaline Phosphatase (Fermentas) in FastAP buffer for 1 h at 37 °C. Immunoprecipitations were again washed in FastAP buffer before proteins were eluted by the addition of SDS-PAGE sample buffer.
In Vitro Caspase Cleavage Assay
Assays were performed as described previously (22). Briefly, Myc-tagged JunB was produced using the TNT T7 Quick Coupled Transcription/Translation System. Two or 0.5 μl of this reaction mixture was incubated with recombinant, active caspase 3 (134 or 268 pm) (Sigma-Aldrich) in cleavage buffer (20 mm PIPES, 100 mm NaCl, 40 mm DTT, 1 mm EDTA, 0.1% CHAPS, 10% sucrose) for the indicated times. Reactions were stopped by the addition of SDS-PAGE sample buffer. For Fig. 2F, assays were performed as above with the exception that 2 units of the indicated caspases (EMD Millipore) and 0.5 μl of TNT lysate were used in reactions and reactions were allowed to incubate at 37 °C for 4 h.
FIGURE 2.

Mutation of aspartic acid 137 blocks the appearance of the lower molecular mass JunB cleavage product. A, scheme of JunB shows the location of putative caspase cleavage sites (Asp-31, Asp-137, and Asp-145) in JunB. The approximate locations of the JNK docking site, DNA binding domain, nuclear localization signal (NLS), and dimerization domain (leucine zipper; Leu. Zipper) are also shown. Also indicated is the location of the epitope recognized by the anti-JunB antibody used for Western blotting. B, Karpas 299 cells transfected with either vector alone or a Myc-JunB cDNA construct were left untreated, treated with staurosporine (STS), or treated with staurosporine and Z-VAD-FMK. Lysates of these cells were then Western blotted (WB) with the indicated antibodies. The efficacy of apoptosis was determined by the appearance of cleaved, active caspase 3. C, Karpas 299 cells transfected with the indicated JunB constructs were left untreated or treated with staurosporine (STS), and lysates were blotted (WB) with an anti-Myc antibody. The band marked with an asterisk (*) is a nonspecific band observed in some experiments. The efficacy of apoptosis induction was determined by the appearance of cleaved, active caspase 3. The anti-β-actin blot demonstrates protein loading. D and E, in vitro transcribed and translated Myc-JunB or mutant proteins were incubated with (+) or without (−) purified, active recombinant caspase 3 for the indicated times (D) or 4 h (E). Reactions were then Western blotted with an anti-Myc or anti-cleaved caspase 3 antibody. Lysates from Myc-JunB-expressing Karpas 299 cells were included in D to compare the electrophoretic mobility of cleaved Myc-JunB in cells with that in the in vitro cleavage assay. F, in vitro transcribed and translated Myc-JunB or Myc-D137A JunB was incubated with (+) or without (−) the indicated purified, active recombinant caspases. Reactions were then Western blotted (WB) with an anti-Myc antibody. Bands indicated with an asterisk (*) represent nonspecific bands, whereas the bands marked with the number sign (#) may represent additional cleavage products observed with the different caspases. In all panels, the arrows indicate the JunB cleavage product. The electrophoretic mobility of molecular mass standards is indicated to the left of blots.
Luciferase Assays
Cells were transfected with the indicated Myc-JunB cDNAs, the pAP1-Luc AP-1 promoter firefly luciferase vector, as well as a constitutively expressed Renilla luciferase construct. Twenty-four h after transfection, 1 × 106 cells were analyzed in triplicate for Renilla and firefly luciferase activity using the Dual-Glo Luciferase Assay System (Promega) in a FLUOstar OPTIMA microplate reader (BMG Labtech; Ortenberg, Germany). The firefly to Renilla luciferase activity ratio was calculated for each sample and then averaged for the triplicate measurements. Results are expressed relative to the vector alone-transfected cells.
EMSAs
For anti-Myc supershift experiments, Karpas 299 cells were transfected with 5 μg of empty vector, Myc-JunB, or Myc-JunB C-terminal fragment. For competitor experiments, Karpas 299 cells were transfected with empty vector (20 μg) or 5-, 10-, or 20-μg Myc-JunB C-terminal fragment cDNA. Empty vector was added where necessary to bring the total DNA per transfection to 20 μg. Nuclear fractions were collected 24 h after transfection using the ProteoJET cytoplasmic and nuclear protein extraction kit (Fermentas). EMSA experiments were performed using the LightShift chemiluminescent EMSA kit (Thermo Scientific). Binding reactions were performed with 20–50 fmol of biotinylated probe containing repeating AP-1 binding sites (CTC CGC TTA GTC ATT AGT CAT TAG TCA TTA GTC ATT AGT CAT AGT CAT AGT CAT TAG TCA GAT CT). This was mixed with 3.5 μg of nuclear extract and 1 μg of the indicated antibody. The nuclear extract and antibodies were preincubated on ice for 15 min prior to addition of the biotinylated probe. For Fig. 7C, rat FLAG-tagged c-Fos and the indicated JunB proteins were generated using the TNT Coupled Wheat Germ Extract System (Promega). For binding reactions, 1 μl of the JunB and/or c-Fos TNT extract was incubated with the biotinylated AP-1 probe, and EMSAs were performed as described above.
FIGURE 7.

The C-terminal JunB cleavage fragment binds DNA and competitively inhibits full-length JunB from binding DNA. A, EMSAs were performed by incubating nuclear extracts prepared from Karpas 299 cells transfected with the indicated cDNAs with a biotinylated AP-1 probe. The indicated antibodies were also included to test for supershifting of probe-protein complexes. Control IgG is an irrelevant, isotype control antibody that serves as a negative control for supershifting. In the lane labeled probe alone, no nuclear extract or antibody was included in the reaction. The Western blot below indicates the expression level of Myc-JunB and the Myc-JunB (CT) fragment in the different nuclear fractions. B, EMSAs were performed as described in A. with Karpas 299 nuclear extracts from cells expressing the indicated amounts of the Myc-JunB C-terminal (CT) fragment. Supershifts were performed using the indicated antibodies. In the lane labeled probe alone, no nuclear extract or antibody was included in the reaction. The Western blot below indicates the expression level of the Myc-JunB (CT) fragment in the different nuclear fractions. C, EMSAs were performed using the indicated in vitro transcribed/translated Myc-JunB and/or FLAG-c-Fos proteins. Bands indicated with an asterisk (*) are biotinylated proteins in the wheat germ extract, whereas the band marked with the number sign (#) is a complex between the probe and wheat germ extract protein. The Western blots to the right of the EMSA demonstrate the production of the indicated proteins using the in vitro transcription/translation system. The electrophoretic mobility of molecular mass standards is indicated to the left of Western blots.
RESULTS
The Electrophoretic Mobility of JunB Is Altered in Staurosporine- and Doxorubicin-treated ALK+ ALCL Cell Lines
In performing experiments to examine whether JunB protects ALK+ ALCL cell lines from apoptosis, we observed that treatment of Karpas 299 and SUP-M2 cells with staurosporine altered the electrophoretic mobility of the anti-JunB immunoreactive bands (Fig. 1A). The characteristic ∼43–45-kDa JunB doublet observed in untreated cells was not evident in the staurosporine-treated cells; instead we observed a single band of ∼43 kDa with the same electrophoretic mobility as the lower molecular mass band of the ∼43–45-kDa doublet. We also observed the appearance of an anti-JunB immunoreactive band of ∼28 kDa (Fig. 1A). The co-treatment of cells with staurosporine and the pan-caspase inhibitor Z-VAD-FMK (23) blocked the appearance of the ∼28-kDa anti-JunB immunoreactive band (Fig. 1B). In addition, although total JunB protein levels were reduced in co-treated cells, the ∼43–45-kDa JunB doublet was partially preserved. These changes in electrophoretic mobility in response to staurosporine treatment were also both time- and dose-dependent (Fig. 1, C and D).
We next investigated whether other apoptosis-inducing agents could similarly alter the electrophoretic mobility of JunB. Doxorubicin promotes apoptosis in ALK+ ALCL cell lines (24–26) and is one component of the CHOP (cyclophosphamide, hydroxydaunorubicin (doxorubicin), oncovin, and prednisone) chemotherapy regimen used clinically to treat ALK+ ALCL (27). Similar to staurosporine treatment, we found that doxorubicin treatment of ALK+ ALCL cell lines resulted in the caspase-dependent appearance of the ∼28-kDa anti-JunB immunoreactive band and a single ∼43-kDa band in place of the ∼43–45-kDa JunB doublet (Fig. 1E). Thus, the electrophoretic mobility of JunB is altered in a caspase-dependent manner in apoptotic cells.
JunB Is Cleaved by Caspases at Aspartic Acid Residue 137
We next examined whether JunB might be cleaved by caspases. Using Cascleave, an on-line resource to identify putative caspase cleavage sites (28), we identified three potential caspase cleavage sites in JunB at aspartic acids 31, 137, and 145 (Fig. 2A). As the JunB antibody used recognizes a C-terminal epitope in the protein, cleavage of JunB at these sites could generate C-terminal fragments consistent with the molecular masses of the JunB immunoreactive products we observed experimentally in apoptotic cells (Fig. 1). To test whether these residues were cleavage sites, we generated N-terminal double Myc epitope-tagged JunB constructs where these sites had been mutated to alanine to prevent caspase cleavage. Because aspartic acid 145 is preceded immediately by another aspartic acid residue, we generated a construct where both aspartic acids 144 and 145 were mutated to alanine (Myc-JunB D144A/D145A). We also constructed a mutant where Asp-137, Asp-144, and Asp-145 were all mutated to alanine (Myc-JunB 3A).
In staurosporine-treated Karpas 299 cells expressing Myc-JunB, we observed a loss of the ∼53–57-kDa Myc-JunB doublet and the appearance of a single band of ∼53 kDa which had the same electrophoretic mobility as the lower molecular mass band of the doublet in the untreated samples (Fig. 2B). We also observed the appearance of a ∼24-kDa anti-Myc immunoreactive band in Myc-JunB-expressing cells treated with staurosporine, which sometimes resolved as a doublet (see Fig. 2D). This likely represents the N-terminal JunB cleavage fragment and the upper band of the ∼24-kDa anti-Myc immunoreactive doublet and may represent a phosphorylated species of the N-terminal fragment. Similar to endogenous JunB, the electrophoretic mobility changes in Myc-JunB in response to staurosporine treatment were also dependent on caspase activity (Fig. 2B). With all the mutant Myc-JunB proteins we observed a collapse of the ∼53–57-kDa Myc-JunB doublet in staurosporine-treated cells (Fig. 2C). We detected the ∼24-kDa anti-Myc antibody immunoreactive fragment in cells expressing the Myc-JunB D31A and Myc-JunB D144A/D145A constructs, but not in cells expressing the Myc-JunB D137A or Myc-JunB 3A constructs (Fig. 2C). Thus, these results implicate aspartic acid 137 as a caspase cleavage site.
We next investigated whether JunB could be cleaved directly by caspases at aspartic acid 137. Using in vitro transcribed/translated Myc-JunB protein as a substrate, we observed a time-dependent increase in the ∼24-kDa anti-Myc reactive cleavage product when Myc-JunB protein was incubated with recombinant caspase 3 (Fig. 2D). Notably, the electrophoretic mobility of this cleaved product was similar in size to the lower band of the ∼24-kDa anti-Myc reactive doublet observed in Myc-JunB-expressing Karpas 299 cells treated with staurosporine (Fig. 2D). The electrophoretic mobility of the ∼53-kDa Myc-JunB generated in the in vitro transcription/translation reaction was unchanged by caspase treatment and had the same electrophoretic mobility as the lower molecular mass band of the ∼53–57-kDa Myc-JunB doublet in untreated cells and the ∼53-kDa band observed in staurosporine-treated cells (Fig. 2D). We also examined whether purified caspase 3 could cleave the putative caspase cleavage site mutants in this assay. Consistent with our results in cells, we found that Myc-JunB D31A and Myc-JunB D144A/D145A could be cleaved by recombinant caspase 3, but the Myc-JunB D137A and Myc-JunB 3A were not (Fig. 2E). We also found that purified caspases 1, 6, and 8 could cleave JunB at aspartic acid 137 in vitro, although other putative cleavage products were observed with these caspases (Fig. 2F).
JunB Is Dephosphorylated in Apoptotic Cells
Our results do not support the idea that the ∼43-kDa anti-JunB immunoreactive band in staurosporine- and doxorubicin-treated cells (Fig. 1) and ∼53-kDa Myc-JunB immunoreactive band observed in staurosporine-treated cells (Fig. 2) are caspase cleavage products of JunB. Therefore, we investigated whether this alteration in JunB electrophoretic mobility could be due to a change in another post-translational modification. JunB is phosphorylated on a number of serine and threonine residues (29–34), and phosphorylation regulates JunB levels and transcriptional activity (20, 29–32, 34). Western blotting of lysates from untreated or staurosporine-treated Karpas 299 or SUP-M2 cells with an antibody that recognizes phosphorylated serine 259 (Ser-259) of JunB demonstrated that JunB is phosphorylated on this site in untreated cells but not in staurosporine-treated cells (Fig. 3A). The phosphorylation of JunB at this residue appears to be restricted to the higher molecular mass band in the ∼43–45-kDa JunB doublet in the untreated samples (Fig. 3A). Phosphorylation of Ser-259 could be partially restored by co-treatment of staurosporine-treated cells with Z-VAD-FMK, demonstrating that dephosphorylation of Ser-259 is caspase-dependent (Fig. 3A). Similar results were observed in cells treated with doxorubicin (Fig. 3B), demonstrating that JunB dephosphorylation is not just a by-product of the kinase inhibitory activity of staurosporine. To investigate further whether collapse of the JunB doublet is due to dephosphorylation, we immunoprecipitated JunB from cell lysates and treated immunoprecipitates with alkaline phosphatase. Western blotting these immunoprecipitates with an anti-JunB antibody demonstrated a collapse in the ∼43–45-kDa JunB doublet and decreased phosphorylation of JunB on Ser-259 (Fig. 3, C and D). The shift in the electrophoretic mobility of the ∼43–45-kDa JunB doublet after phosphatase treatment was similar to the shift observed after staurosporine treatment (Fig. 3E). Furthermore, the immunoprecipitation of JunB from lysates of staurosporine-treated cells followed by phosphatase treatment did not result in a further shift in electrophoretic mobility of the ∼43-kDa anti-JunB immunoreactive species over staurosporine treatment alone (Fig. 3F). Taken together, these results argue that the ∼43-kDa JunB protein observed in apoptotic ALK+ ALCL cell lines is a dephosphorylated JunB species.
FIGURE 3.

JunB is dephosphorylated in a caspase-dependent manner in ALK+ ALCL cell lines undergoing apoptosis. A, lysates from Karpas 299 or SUP-M2 cells either untreated, treated with staurosporine, or treated with staurosporine (STS) and Z-VAD-FMK were Western blotted (WB) with antibodies that recognize JunB when phosphorylated at serine 259 (anti-pJunB (S259)) or total JunB (anti-JunB). The anti-pro-caspase 3 and anti-cleaved caspase 3 blots demonstrate the induction of apoptosis, whereas the anti-β-actin blot demonstrates protein loading. B, Karpas 299 or SUP-M2 cells were left untreated, treated with doxorubicin, or treated with doxorubicin and Z-VAD-FMK. Lysates were then Western blotted with the indicated antibodies. C and D, lysates from Karpas 299 (C) or SUP-M2 (D) cells were immunoprecipitated (IP) with an anti-JunB antibody, and immunoprecipitates were either left untreated (−) or treated with alkaline phosphatase (AP) (+). Immunoprecipitates were then Western blotted with an anti-JunB or anti-pJunB Ser-259 antibody. E, Karpas 299 or SUP-M2 lysates were immunoprecipitated, treated with or without phosphatase, and Western blotted as described in C and D. Cell lysates from untreated or staurosporine-treated cells were also included on the membranes. F, lysates from untreated or staurosporine-treated cells were immunoprecipitated, treated with or without phosphatase, and Western blotted as described in C and D. IgH indicates the heavy chain of the immunoprecipitating antibody. The arrows indicate the ∼43–45-kDa JunB proteins. The electrophoretic mobility of molecular mass standards is indicated to the left of blots.
JunB Is Cleaved at Aspartic Acid 137 in Response to Inflammasome Activation
Caspases also perform critical cellular functions independent of apoptosis. For example, caspase 1 is a component of inflammasome complexes that respond to microbial and cellular danger signals as part of the innate immune response (35). Inflammasome activation results in the activation of caspase 1 which cleaves and activates the proinflammatory cytokines pro-IL1β and pro-IL18. It also mediates a distinct type of programmed cell death termed pyroptosis (35, 36). Given that recombinant caspase 1 efficiently cleaved in vitro transcribed and translated Myc-JunB (Fig. 2F), we investigated whether JunB was cleaved in response to inflammasome activation. We used RAW 264.7 murine macrophages treated with anthrax lethal toxin as our model system because this has been shown to activate caspase 1 through engagement of the NALP1b inflammasome (37, 38). We observed the ∼24-kDa anti-Myc reactive band in lysates of Myc-JunB-expressing RAW 264.7 cells treated with anthrax lethal toxin, but not in lysates of treated cells expressing Myc-JunB D137A (Fig. 4). The appearance of the ∼24-kDa band was not evident in Myc-JunB-expressing cells co-treated with anthrax lethal toxin and Z-VAD-FMK. Moreover, the electrophoretic mobility of this ∼24-kDa band was comparable with the band observed in staurosporine-treated RAW 264.7 cells. Thus, these findings demonstrate that the caspase-dependent cleavage of JunB of aspartic acid 137 is not limited to apoptotic cells. Some collapse of the ∼53–57-kDa Myc-JunB doublet was observed in cells treated with anthrax lethal toxin, but this was less pronounced than was observed in staurosporine-treated cells (Fig. 4).
FIGURE 4.
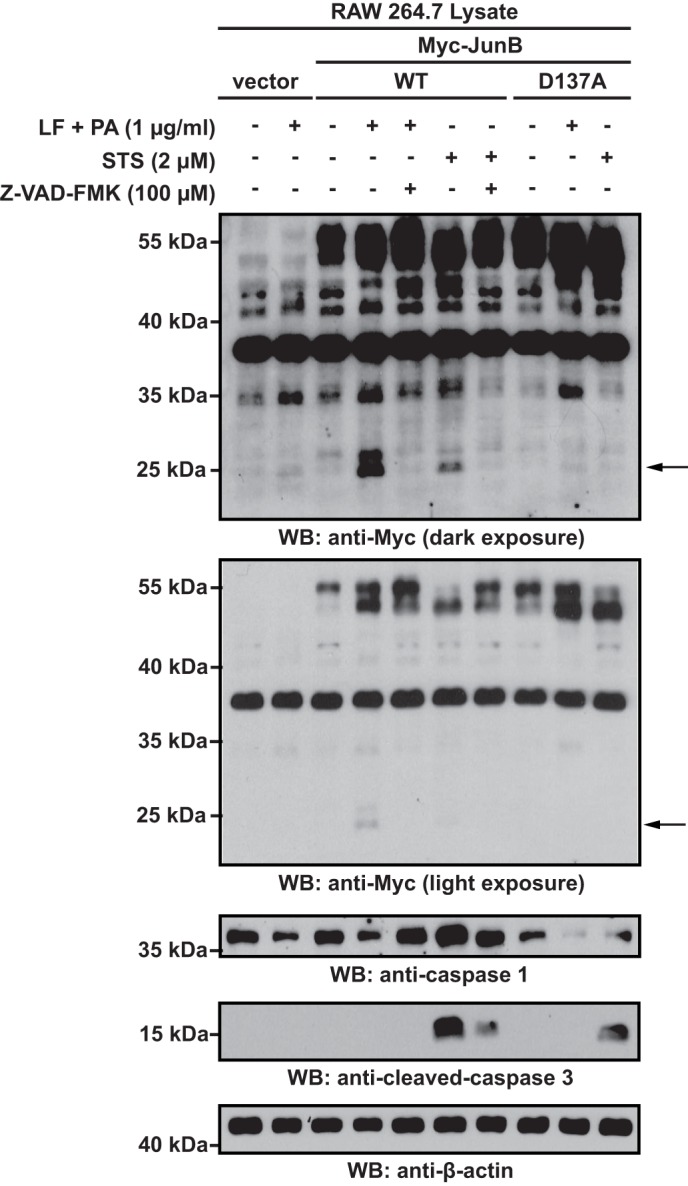
JunB is cleaved at Asp-137 in response to inflammasome activation. RAW 264.7 cells transfected with vector alone or cDNAs encoding for Myc-JunB or D137A Myc-JunB were left untreated or treated with anthrax lethal factor (LF) and protective antigen (PA), anthrax lethal factor and protective antigen and Z-VAD-FMK, staurosporine (STS), or staurosporine and Z-VAD-FMK for 6 h. Lysates of these cells were then probed with the indicated antibodies. The anti-caspase 1 blot demonstrates the cleavage of pro-caspase 1 in inflammasome-activated cells, and the anti-caspase 3 blots demonstrate apoptosis induction. The lower anti-Myc blot is a shorter exposure of the blot above and was included to demonstrate changes in electrophoretic mobility of the ∼53–57-kDa Myc-JunB doublet. The electrophoretic mobility of molecular mass standards is indicated to the left of blots.
Caspase Cleavage of JunB Generates a C-terminal Cleavage Product with Biological Activity
The cleavage of JunB at aspartic acid 137 separates the N-terminal transactivation domain from the C-terminal dimerization and DNA binding domains (see Fig. 2A). Thus, although this cleavage would be predicted to inactivate the protein, the cleavage products may retain some biological activity. To investigate this, we generated double Myc-tagged constructs of the JunB N- and C-terminal cleavage fragments and examined whether these could influence transcription from an AP-1-responsive luciferase reporter construct. Of note, both the N- and C-terminal Myc-tagged JunB cleavage fragments resolved as doublets when expressed in Karpas 299 cells (Fig. 5A), presumably due to different phosphorylated species of these proteins. We found that expression of full-length JunB in Karpas 299 cells resulted in an approximate 2.5-fold increase in luciferase activity compared with vector alone-transfected cells (Fig. 5A). In cells transfected with the N-terminal cleavage fragment, there was no significant difference in luciferase activity over cells transfected with vector alone; however, in cells transfected with the C-terminal JunB fragment, luciferase activity was significantly reduced compared with vector alone-transfected cells (Fig. 5A). We further found that the C-terminal JunB fragment functioned as a dose-dependent inhibitor of AP-1 transcriptional activity in Karpas 299 cells, whereas the N-terminal fragment had a negligible effect on AP-1 luciferase activity (Fig. 5B). Thus, these results demonstrate that the C-terminal cleavage fragment is an efficient inhibitor of AP-1-dependent transcription.
FIGURE 5.

The C-terminal JunB cleavage fragment inhibits AP-1-dependent luciferase activity. A, Karpas 299 cells were transfected with 5 μg of vector alone or cDNAs encoding for Myc-tagged JunB (WT), Myc-tagged N-terminal JunB cleavage fragment (NT), or Myc-tagged C-terminal JunB cleavage fragment (CT) along with an AP-1 firefly luciferase reporter construct and a constitutively expressed Renilla luciferase vector. Cells were lysed, and luciferase activity was measured as described under “Experimental Procedures.” The results shown represent the mean ± S.D. (error bars) of 12 independent experiments. p values were calculated by performing paired, one-tailed t tests. NS indicates no significant difference. The anti-Myc Western blot (WB; to the right) indicates the expression of the respective Myc-tagged constructs (anti-Myc blot), whereas the anti-JunB blot illustrates the level of the overexpressed protein relative to endogenous JunB. Note: the epitope recognized by the anti-JunB antibody is not present in the N-terminal JunB protein. B, Karpas 299 cells were transfected with the indicated amounts of the different Myc-JunB cDNAs, an AP-1 firefly luciferase reporter construct, and a constitutively expressed Renilla luciferase vector. Empty vector was added to some samples to ensure the total amount of transfected DNA was the same in all samples. Cells were lysed, and luciferase activity was measured as described under “Experimental Procedures.” The results shown represent the mean ± S.D. of five independent experiments. p values were calculated by performing paired, one-tailed t tests (*, p < 0.05; **, p < 0.01; ***, p < 0.001). The anti-Myc Western blot is included to illustrate the expression level of the Myc-JunB proteins. The anti-JunB blot demonstrates that endogenous JunB levels are similar in all transfected cells, and the anti-β-actin Western blot is included to demonstrate protein loading. The electrophoretic mobility of molecular mass standards is indicated to the left of Western blots.
The C-terminal JunB Cleavage Product Retains the Ability to Associate with AP-1 Family Proteins
We next explored the mechanism by which the C-terminal fragment could be inhibiting AP-1-dependent transcription. We postulated that because this fragment contains the dimerization and DNA binding domains, but lacks the transcriptional activation domain, it may bind AP-1 DNA binding sites as a homodimer or a transcriptionally inactive/impaired heterodimer. This might prevent transcriptionally competent AP-1 dimers from binding AP-1 sites and promoting transcription which is consistent with the results of our luciferase reporter experiments (Fig. 5). To test this model, we first examined whether the C-terminal fragment could associate with other AP-1 family proteins. Co-immunoprecipitation experiments demonstrated that full-length JunB and the C-terminal fragment, but not the N-terminal fragment, could co-precipitate with endogenous c-Fos and Fra2 in Karpas 299 cells (Fig. 6A). We further found that the Myc-tagged JunB C-terminal fragment could co-immunoprecipitate with FLAG-tagged JunB when expressed in Karpas 299 cells (Fig. 6B). Finally, in vitro transcribed and translated C-terminal JunB cleavage fragment was able to co-immunoprecipitate with in vitro transcribed and translated c-Fos arguing that the C-terminal fragment is able to interact directly with other AP-1 family proteins (Fig. 6C).
AP-1 Complexes Consisting of the C-terminal JunB Cleavage Product Retain the Ability to Bind DNA
We next performed EMSA to examine whether the C-terminal fragment could bind DNA. We found that a biotinylated AP-1 probe was bound by a protein(s) in Karpas 299 cell nuclear extract, and the electrophoretic mobility of these complexes was altered when the probe was incubated with extracts from cells expressing the Myc-tagged JunB C-terminal fragment (compare Fig. 7A, lanes 2 and 8). Moreover, the addition of an anti-JunB antibody to these probe-protein complexes resulted in a supershift of the complexes, demonstrating that JunB is present in these complexes (Fig. 7A, lanes 3, 6, and 9). Probe-protein complexes could also be shifted with an anti-Myc antibody when this antibody was incubated with nuclear extracts from Myc-tagged JunB (Fig. 7A, lane 7) or Myc-tagged JunB C-terminal cleavage fragment-expressing cells (Fig. 7A, lane 10), but not vector alone-transfected cells (Fig. 7A, lane 4). This last result demonstrates that the C-terminal JunB cleavage fragment can interact with an AP-1 binding site.
We also examined whether the C-terminal fragment could interfere with the binding of endogenous JunB to this AP-1 probe. We again performed EMSA, this time using an anti-JunB antibody for supershift that recognizes an epitope in the N terminus of JunB not present in the C-terminal fragment. Fig. 7B shows that with increasing expression of the C-terminal JunB fragment, a dose-dependent decrease in JunB-probe complexes super-shifted with the N-terminal anti-JunB antibody, was observed (compare lanes 6–9). However, we still observed supershifting with the C-terminal anti-JunB antibody that recognizes both endogenous JunB and the C-terminal fragment (lanes 10–13).
Finally, we examined whether in vitro transcribed/translated Myc-JunB C-terminal cleavage fragment could bind this AP-1 probe independent of other factors present in Karpas 299 nuclear extract. Intriguingly, no shifting of the AP-1 probe was observed when incubated with extracts of either in vitro transcribed/translated Myc-JunB or the C-terminal cleavage fragment (Fig. 7C, lanes 3 and 5). However, we observed shifting of this probe when incubated with extract containing FLAG-c-Fos as well as extract containing Myc-JunB (lane 8) or the C-terminal fragment (lane 10). Taken together, our EMSA results are consistent with the notion that the C-terminal JunB caspase cleavage product binds AP-1 sites, prevents endogenous JunB from binding these sites, and, at least with this synthetic AP-1 probe, binds as a heterodimer with c-Fos.
DISCUSSION
In this study, we make the novel observation that the JunB transcription factor is a caspase substrate. We show that JunB is cleaved in a caspase-dependent manner at aspartic acid 137 in both apoptotic cells and cells stimulated through the NALP1b inflammasome. Importantly, cleavage of JunB at aspartic acid 137 generates a C-terminal fragment that, when overexpressed in cells, functions as an inhibitor of AP-1-dependent transcription.
Many transcription factors are caspase substrates in apoptotic cells including the NF-κB subunits, p65/RelA, p50, and c-Rel (39–41), as well as STAT1 (42), STAT3 (43), FOXO3a (44), and GATA-1 (45). We also examined whether other members of the AP-1 family of transcription factors were cleaved in apoptotic cells. No putative FosB or c-Jun cleavage products were observed in staurosporine-treated Karpas 299 cells, but we did observe a lower molecular mass anti-Fos immunoreactive band; however, it was only evident with longer exposure times and much less prominent than was observed for JunB (results not shown). Thus, caspase-mediated cleavage in apoptotic cells is not a characteristic shared among all AP-1 family proteins and, at least with the proteins tested, is most prominent with JunB.
Similar to our observations with JunB, the caspase-mediated cleavage of several other transcription factors results in the separation of transactivation domains from DNA binding domains. These include p65/RelA (40), FOXO3a (44), Ying Yang 1 (YY1) (46), as well as PU.1 (47), and cleavage of some of these transcription factors has been demonstrated to generate a fragment that functions as an inhibitor of transcription. For example, cleavage of p65/RelA by caspases generates an N-terminal fragment where the transcriptional activation domains have been disrupted, but the DNA binding domain is still intact (40). This fragment was demonstrated to inhibit full-length p65/RelA from promoting transcription. As well, a noncleavable p65/RelA mutant was found to protect human umbilical vein endothelial cells from apoptosis induced by growth factor withdrawal, suggesting a role for the N-terminal cleavage product in apoptosis induction (40). However, we found that the noncleavable D137A JunB mutant did not affect either spontaneous or staurosporine-induced apoptosis when expressed in Karpas 299 cells (results not shown). Thus, whether cleavage of JunB is important in apoptosis or inflammasome signaling, or merely a “by-stander” whose cleavage has no role in apoptotic or NALP1b inflammasome-activated cells is an unresolved question from our study.
We were intrigued by our observation that JunB is dephosphorylated in a caspase-dependent manner in apoptotic cells. It is also likely that JunB is dephosphorylated to some extent in inflammasome-activated cells. JunB is phosphorylated at several sites, and phosphorylation at specific sites has been reported to enhance (29, 30) or impair (48) the ability of JunB to promote transcription or target JunB for ubiquitin-mediated degradation (20, 32, 34). Thus, there are many ways dephosphorylation of JunB in apoptotic cells could potentially impact JunB function. It needs to be established whether this dephosphorylation is primarily due to activation of a phosphatase(s), inactivation of a kinase(s), or both these processes. However, to begin to address these questions we first need to identify the sites of JunB phosphorylation in ALK+ ALCL and the kinases and phosphatases that regulate these sites.
The C-terminal JunB cleavage fragment we have characterized is structurally and functionally reminiscent of TAM67, a c-Jun construct generated by the Birrer group which functions as a dominant negative inhibitor of AP-1-dependent signaling (49) (Fig. 8). TAM67 lacks amino acids 3–122 of c-Jun, which comprises the majority of the transcriptional activation domain, but the protein retains the DNA binding and dimerization domains of c-Jun (49). Similar to our findings, TAM67 binds AP-1 sites and interferes with AP-1-dependent transcription (49, 50). The Birrer group proposed that TAM67 primarily inhibits transcription via a “quenching” mechanism. In their model, transcriptionally impaired TAM67/c-Fos or TAM67/c-Jun heterodimers bind AP-1 sites in place of more transcriptionally active c-Jun homodimers or c-Jun/c-Fos heterodimers (50). Our findings are consistent with the C-terminal JunB cleavage fragment functioning in a similar manner.
FIGURE 8.

Comparison of TAM67 and the JunB C-terminal cleavage fragment. The c-Jun and JunB protein sequences were collected from the NCBI protein database, and the alignment and figure were generated using the MAFFT (51) and PFAAT software (52). The deletion found in TAM67 is indicated by the line, and the Asp-137 cleavage site of JunB is indicated by the arrow.
More than 100 studies in PubMed have used the TAM67 construct to dissect AP-1-dependent signaling events. Thus, although it is unclear whether the C-terminal JunB cleavage product functions as an inhibitor of AP-1-dependent signaling in apoptotic or inflammasome-activated cells, we feel that this fragment represents an additional tool to interrogate AP-1 signaling.
Acknowledgments
We thank Drs. Troy Baldwin, Michele Barry, and Hanne Ostergaard for comments on the work and for critically reading the manuscript.
This work was supported by operating grants from the Natural Sciences and Engineering Research Council of Canada and the Alberta Cancer Foundation/Alberta Innovates Health Solutions (to R. J. I.).
- AP-1
- activator protein-1
- ALK− ALCL
- anaplastic lymphoma kinase-negative, anaplastic large cell lymphoma
- ALK+ ALCL
- anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma
- FMK
- fluoromethyl ketone
- mAb
- monoclonal antibody
- pAb
- polyclonal antibody
- PARP
- poly(ADP-ribose)polymerase
- Z
- benzyloxycarbonyl.
REFERENCES
- 1. Shaulian E., Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–136 [DOI] [PubMed] [Google Scholar]
- 2. Shaulian E. (2010) AP-1, the Jun proteins: oncogenes or tumor suppressors in disguise? Cell. Signal. 22, 894–899 [DOI] [PubMed] [Google Scholar]
- 3. Shaulian E., Karin M. (2001) AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400 [DOI] [PubMed] [Google Scholar]
- 4. Chinenov Y., Kerppola T. K. (2001) Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20, 2438–2452 [DOI] [PubMed] [Google Scholar]
- 5. Wagner E. F., Eferl R. (2005) Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 208, 126–140 [DOI] [PubMed] [Google Scholar]
- 6. Schonthaler H. B., Guinea-Viniegra J., Wagner E. F. (2011) Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. Dis. 70, i109–112 [DOI] [PubMed] [Google Scholar]
- 7. Schütte J., Viallet J., Nau M., Segal S., Fedorko J., Minna J. (1989) junB inhibits and c-fos stimulates the transforming and trans-activating activities of c-jun. Cell 59, 987–997 [DOI] [PubMed] [Google Scholar]
- 8. Chiu R., Angel P., Karin M. (1989) JunB differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 59, 979–986 [DOI] [PubMed] [Google Scholar]
- 9. Passegué E., Jochum W., Behrens A., Ricci R., Wagner E. F. (2002) JunB can substitute for Jun in mouse development and cell proliferation. Nat. Genet. 30, 158–166 [DOI] [PubMed] [Google Scholar]
- 10. Hirai S. I., Ryseck R. P., Mechta F., Bravo R., Yaniv M. (1989) Characterization of junD: a new member of the jun proto-oncogene family. EMBO J. 8, 1433–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schorpp-Kistner M., Wang Z. Q., Angel P., Wagner E. F. (1999) JunB is essential for mammalian placentation. EMBO J. 18, 934–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathas S., Hinz M., Anagnostopoulos I., Krappmann D., Lietz A., Jundt F., Bommert K., Mechta-Grigoriou F., Stein H., Dörken B., Scheidereit C. (2002) Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-κB. EMBO J. 21, 4104–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rassidakis G. Z., Thomaides A., Atwell C., Ford R., Jones D., Claret F. X., Medeiros L. J. (2005) JunB expression is a common feature of CD30+ lymphomas and lymphomatoid papulosis. Mod. Pathol. 18, 1365–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watanabe M., Ogawa Y., Ito K., Higashihara M., Kadin M. E., Abraham L. J., Watanabe T., Horie R. (2003) AP-1 mediated relief of repressive activity of the CD30 promoter microsatellite in Hodgkin and Reed-Sternberg cells. Am. J. Pathol. 163, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Staber P. B., Vesely P., Haq N., Ott R. G., Funato K., Bambach I., Fuchs C., Schauer S., Linkesch W., Hrzenjak A., Dirks W. G., Sexl V., Bergler H., Kadin M. E., Sternberg D. W., Kenner L., Hoefler G. (2007) The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood 110, 3374–3383 [DOI] [PubMed] [Google Scholar]
- 16. Watanabe M., Sasaki M., Itoh K., Higashihara M., Umezawa K., Kadin M. E., Abraham L. J., Watanabe T., Horie R. (2005) JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and Reed-Sternberg cells of Hodgkin lymphoma. Cancer Res. 65, 7628–7634 [DOI] [PubMed] [Google Scholar]
- 17. Pearson J. D., Lee J. K., Bacani J. T., Lai R., Ingham R. J. (2011) NPM-ALK and the JunB transcription factor regulate the expression of cytotoxic molecules in ALK-positive, anaplastic large cell lymphoma. Int. J. Clin. Exp. Pathol. 4, 124–133 [PMC free article] [PubMed] [Google Scholar]
- 18. Pearson J. D., Mohammed Z., Bacani J. T., Lai R., Ingham R. J. (2012) The heat shock protein-90 co-chaperone, cyclophilin 40, promotes ALK-positive, anaplastic large cell lymphoma viability and its expression is regulated by the NPM-ALK oncoprotein. BMC Cancer 12, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laimer D., Dolznig H., Kollmann K., Vesely P. W., Schlederer M., Merkel O., Schiefer A. I., Hassler M. R., Heider S., Amenitsch L., Thallinger C., Staber P. B., Simonitsch-Klupp I., Artaker M., Lagger S., Turner S. D., Pileri S., Piccaluga P. P., Valent P., Messana K., Landra I., Weichhart T., Knapp S., Shehata M., Todaro M., Sexl V., Höfler G., Piva R., Medico E., Ruggeri B. A., Cheng M., Eferl R., Egger G., Penninger J. M., Jaeger U., Moriggl R., Inghirami G., Kenner L. (2012) PDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat. Med. 18, 1699–1704 [DOI] [PubMed] [Google Scholar]
- 20. Pérez-Benavente B., Garcia J. L., Rodríguez M. S., Pineda-Lucena A., Piechaczyk M., Font de Mora J., Farràs R. (2013) GSK3-SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene 32, 2189–2199 [DOI] [PubMed] [Google Scholar]
- 21. Murphy L. O., Smith S., Chen R. H., Fingar D. C., Blenis J. (2002) Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 4, 556–564 [DOI] [PubMed] [Google Scholar]
- 22. Flygare J., Hellgren D., Wennborg A. (2000) Caspase-3-mediated cleavage of HsRad51 at an unconventional site. Eur. J. Biochem. 267, 5977–5982 [DOI] [PubMed] [Google Scholar]
- 23. Garcia-Calvo M., Peterson E. P., Leiting B., Ruel R., Nicholson D. W., Thornberry N. A. (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 273, 32608–32613 [DOI] [PubMed] [Google Scholar]
- 24. Drakos E., Rassidakis G. Z., Lai R., Herling M., O'Connor S. L., Schmitt-Graeff A., McDonnell T. J., Medeiros L. J. (2004) Caspase-3 activation in systemic anaplastic large-cell lymphoma. Mod. Pathol. 17, 109–116 [DOI] [PubMed] [Google Scholar]
- 25. Han Y., Amin H. M., Frantz C., Franko B., Lee J., Lin Q., Lai R. (2006) Restoration of shp1 expression by 5-AZA-2′-deoxycytidine is associated with down-regulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia 20, 1602–1609 [DOI] [PubMed] [Google Scholar]
- 26. Hsu F. Y., Zhao Y., Anderson W. F., Johnston P. B. (2007) Down-regulation of NPM-ALK by siRNA causes anaplastic large cell lymphoma cell growth inhibition and augments the anti cancer effects of chemotherapy in vitro. Cancer Invest. 25, 240–248 [DOI] [PubMed] [Google Scholar]
- 27. Inghirami G., Pileri S. A., and European T-Cell Lymphoma Study Group (2011) Anaplastic large-cell lymphoma. Semin. Diagn. Pathol. 28, 190–201 [DOI] [PubMed] [Google Scholar]
- 28. Song J., Tan H., Shen H., Mahmood K., Boyd S. E., Webb G. I., Akutsu T., Whisstock J. C. (2010) Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 26, 752–760 [DOI] [PubMed] [Google Scholar]
- 29. Li B., Tournier C., Davis R. J., Flavell R. A. (1999) Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 18, 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayanan K., Srinivas R., Peterson M. C., Ramachandran A., Hao J., Thimmapaya B., Scherer P. E., George A. (2004) Transcriptional regulation of dentin matrix protein 1 by JunB and p300 during osteoblast differentiation. J. Biol. Chem. 279, 44294–44302 [DOI] [PubMed] [Google Scholar]
- 31. Nikolakaki E., Coffer P. J., Hemelsoet R., Woodgett J. R., Defize L. H. (1993) Glycogen synthase kinase 3 phosphorylates Jun family members in vitro and negatively regulates their transactivating potential in intact cells. Oncogene 8, 833–840 [PubMed] [Google Scholar]
- 32. Bakiri L., Lallemand D., Bossy-Wetzel E., Yaniv M. (2000) Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 19, 2056–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farràs R., Baldin V., Gallach S., Acquaviva C., Bossis G., Jariel-Encontre I., Piechaczyk M. (2008) JunB breakdown in mid-/late G2 is required for down-regulation of cyclin A2 levels and proper mitosis. Mol. Cell. Biol. 28, 4173–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franchi L., Muñoz-Planillo R., Núñez G. (2012) Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miao E. A., Rajan J. V., Aderem A. (2011) Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243, 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cordoba-Rodriguez R., Fang H., Lankford C. S., Frucht D. M. (2004) Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1β and IL-18. J. Biol. Chem. 279, 20563–20566 [DOI] [PubMed] [Google Scholar]
- 38. Wickliffe K. E., Leppla S. H., Moayeri M. (2008) Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell. Microbiol. 10, 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravi R., Bedi A., Fuchs E. J. (1998) CD95 (Fas)-induced caspase-mediated proteolysis of NF-κB. Cancer Res. 58, 882–886 [PubMed] [Google Scholar]
- 40. Levkau B., Scatena M., Giachelli C. M., Ross R., Raines E. W. (1999) Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-κB loop. Nat. Cell Biol. 1, 227–233 [DOI] [PubMed] [Google Scholar]
- 41. Barkett M., Dooher J. E., Lemonnier L., Simmons L., Scarpati J. N., Wang Y., Gilmore T. D. (2001) Three mutations in v-Rel render it resistant to cleavage by cell-death protease caspase-3. Biochim. Biophys. Acta 1526, 25–36 [DOI] [PubMed] [Google Scholar]
- 42. King P., Goodbourn S. (1998) STAT1 is inactivated by a caspase. J. Biol. Chem. 273, 8699–8704 [DOI] [PubMed] [Google Scholar]
- 43. Darnowski J. W., Goulette F. A., Guan Y. J., Chatterjee D., Yang Z. F., Cousens L. P., Chin Y. E. (2006) Stat3 cleavage by caspases: impact on full-length Stat3 expression, fragment formation, and transcriptional activity. J. Biol. Chem. 281, 17707–17717 [DOI] [PubMed] [Google Scholar]
- 44. Charvet C., Alberti I., Luciano F., Jacquel A., Bernard A., Auberger P., Deckert M. (2003) Proteolytic regulation of Forkhead transcription factor FOXO3a by caspase-3-like proteases. Oncogene 22, 4557–4568 [DOI] [PubMed] [Google Scholar]
- 45. De Maria R., Zeuner A., Eramo A., Domenichelli C., Bonci D., Grignani F., Srinivasula S. M., Alnemri E. S., Testa U., Peschle C. (1999) Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 401, 489–493 [DOI] [PubMed] [Google Scholar]
- 46. Krippner-Heidenreich A., Walsemann G., Beyrouthy M. J., Speckgens S., Kraft R., Thole H., Talanian R. V., Hurt M. M., Lüscher B. (2005) Caspase-dependent regulation and subcellular redistribution of the transcriptional modulator YY1 during apoptosis. Mol. Cell. Biol. 25, 3704–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao M., Duan X. F., Wen D. H., Chen G. Q. (2009) PU.1, a novel caspase-3 substrate, partially contributes to chemotherapeutic agents-induced apoptosis in leukemic cells. Biochem. Biophys. Res. Commun. 382, 508–513 [DOI] [PubMed] [Google Scholar]
- 48. Boyle W. J., Smeal T., Defize L. H., Angel P., Woodgett J. R., Karin M., Hunter T. (1991) Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 64, 573–584 [DOI] [PubMed] [Google Scholar]
- 49. Brown P. H., Alani R., Preis L. H., Szabo E., Birrer M. J. (1993) Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene 8, 877–886 [PubMed] [Google Scholar]
- 50. Brown P. H., Chen T. K., Birrer M. J. (1994) Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene 9, 791–799 [PubMed] [Google Scholar]
- 51. Katoh K., Misawa K., Kuma K., Miyata T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Caffrey D. R., Dana P. H., Mathur V., Ocano M., Hong E. J., Wang Y. E., Somaroo S., Caffrey B. E., Potluri S., Huang E. S. (2007) PFAAT version 2.0: a tool for editing, annotating, and analyzing multiple sequence alignments. BMC Bioinformatics 8, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]