

Background: Mutations in SOD1 cause familial amyotrophic lateral sclerosis (ALS).
Results: SOD1 undergoes palmitoylation in the spinal cord and multiple cell types. Palmitoylation occurs predominantly on immature SOD1 and is increased for ALS-linked SOD1 mutants.
Conclusion: Palmitoylation is a reversible post-translational modification of SOD1 cysteine residues.
Significance: Palmitoylation could affect SOD1 toxicity by altering its folding, membrane targeting, and/or function.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig Disease), Neurodegenerative Diseases, Post-translational Modification, Protein Palmitoylation, Superoxide Dismutase (SOD)
Abstract
Mutations in Cu,Zn-superoxide dismutase (mtSOD1) cause familial amyotrophic lateral sclerosis (FALS), a neurodegenerative disease resulting from motor neuron degeneration. Here, we demonstrate that wild type SOD1 (wtSOD1) undergoes palmitoylation, a reversible post-translational modification that can regulate protein structure, function, and localization. SOD1 palmitoylation was confirmed by multiple techniques, including acyl-biotin exchange, click chemistry, cysteine mutagenesis, and mass spectrometry. Mass spectrometry and cysteine mutagenesis demonstrated that cysteine residue 6 was the primary site of palmitoylation. The palmitoylation of FALS-linked mtSOD1s (A4V and G93A) was significantly increased relative to that of wtSOD1 expressed in HEK cells and a motor neuron cell line. The palmitoylation of FALS-linked mtSOD1s (G93A and G85R) was also increased relative to that of wtSOD1 when assayed from transgenic mouse spinal cords. We found that the level of SOD1 palmitoylation correlated with the level of membrane-associated SOD1, suggesting a role for palmitoylation in targeting SOD1 to membranes. We further observed that palmitoylation occurred predominantly on disulfide-reduced as opposed to disulfide-bonded SOD1, suggesting that immature SOD1 is the primarily palmitoylated species. Increases in SOD1 disulfide bonding and maturation with increased copper chaperone for SOD1 expression caused a decrease in wtSOD1 palmitoylation. Copper chaperone for SOD1 overexpression decreased A4V palmitoylation less than wtSOD1 and had little effect on G93A mtSOD1 palmitoylation. These findings suggest that SOD1 palmitoylation occurs prior to disulfide bonding during SOD1 maturation and that palmitoylation is increased when disulfide bonding is delayed or decreased as observed for several mtSOD1s.
Introduction
ALS4 is a neurodegenerative disease characterized by the selective loss of motor neurons. ∼10% of ALS cases are familial (FALS), and ∼25% of FALS cases are caused by SOD1 mutations. SOD1 is a homodimeric enzyme that converts toxic superoxide free radicals into hydrogen peroxide and water. SOD1 undergoes a maturation process that involves each monomer subunit acquiring one zinc and one copper ion, dimerization, and the formation of an intramolecular disulfide bond between Cys-57 and Cys-146. Copper metallation and the intrasubunit disulfide oxidation are predominantly catalyzed by the copper chaperone for SOD1 (CCS). Because many FALS-causing mtSOD1s have full dismutase activity (1, 2) and because deletion of SOD1 does not cause an ALS-like disease in mice (3), mtSOD1-induced pathology is thought to result from a gain-of-function rather than loss-of-function. Although the toxicity of mtSOD1 is not fully understood, improper folding and aggregation of mtSOD1 are consistent features of mtSOD1-induced FALS in patients and transgenic rodent models and have been proposed to lead to motor neuron degeneration. Misfolding and aggregation of mtSOD1 could cause toxicity by sequestering critical cellular components and/or by disrupting essential cellular pathways (4). Consistent with playing a toxic role in FALS, mtSOD1 aggregation is associated with disruptions in motor neuron axonal transport (5, 6), ER stress (7–13), as well as mitochondrial dysfunction (14–16). In addition, aggregation of mtSOD1 is associated with altered SOD1 maturation and intermolecular disulfide cross-linking of SOD1 cysteine residues (17–20). Of note, wtSOD1 cysteine residues can be modified by oxidation, which alters its structure, and has been proposed to be important in the pathogenesis of sporadic ALS (21–25).
S-Palmitoylation is a dynamic and reversible post-translational modification in which the fatty acid palmitate is covalently attached to cysteine residues via a thioester bond. This post-translational modification regulates various features of a protein, including subcellular localization, trafficking, sorting, stability, and aggregation. For cytosolic proteins to be palmitoylated, they interact with palmitoyltransferases (PATs), a family of integral membrane proteins found primarily on membranes of different intracellular organelles such as the Golgi and ER. Palmitoylation provides soluble proteins with an anchor to associate with membranes and can target them to distinct compartments within cellular membranes. For instance, palmitoylation targets PSD-95 to postsynaptic densities and SNAP-25 to presynaptic termini (26). Additionally, palmitoylation is implicated in the pathogenesis of neurodegenerative diseases such as Huntington and Alzheimer (27). Here, we report that SOD1 is palmitoylated and that palmitoylation of mtSOD1s is significantly increased when expressed in mammalian cell lines and spinal cords of transgenic mice at late stages of ALS-like disease. The function of SOD1 palmitoylation is not yet known. We found that increasing SOD1 palmitoylation correlated with more SOD1 association with cellular membranes, whereas loss of SOD1 palmitoylation correlated with less membrane-associated SOD1, suggesting a role for palmitoylation in targeting SOD1 to membranes. We also observed that palmitoylation occurs predominantly on disulfide-reduced as opposed to disulfide-bonded SOD1. Increased CCS expression, which increases SOD1 disulfide bonding and other maturation steps (28, 29), caused a decrease in wtSOD1 palmitoylation, with a less significant decrease in A4V and no significant change in G93A mtSOD1 palmitoylation. These findings suggest that SOD1 palmitoylation occurs prior to the intramolecular disulfide-bonding step in SOD1 maturation and that palmitoylation is increased under conditions when there is more immature disulfide-reduced SOD1. Importantly, the immature form of SOD1 is known to be unstable (30) and is proposed to be a critical component in the misfolding and toxicity of mtSOD1 (31–34). Our findings that palmitoylation occurs on disulfide-reduced SOD1, is increased for mtSOD1, and correlates with SOD1 membrane association suggest this modification may play a role in the pathogenesis of mtSOD1-induced FALS.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following antibodies were used: mouse anti-GFP (Antibodies Inc.); mouse anti-SOD1 (Sigma); rabbit anti-SOD1 (Enzo Life Sciences); rabbit anti-CCS, rabbit anti-Tom20, and mouse anti-Na+/K+-ATPase α1 (Santa Cruz Biotechnology); and Alexa Fluor 568 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG (Invitrogen). The following reagents were used: hydroxylamine (HAM), triethanolamine, t-butanol tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, and tris(2-carboxyethyl)phosphine (TCEP) (Sigma); streptavidin conjugated to horseradish peroxidase (streptavidin-HRP) and EZ-link biotin-BMCC ((1-biotinamido-4-[4′-maleimidomethyl)cyclohexanecarboxamido] butane) (Pierce); protein G-Sepharose beads (GE Healthcare); nickel-NTA-agarose (Qiagen); N-ethylmaleimide (NEM) (Calbiochem); 2-bromopalmitate (2Br) and N-methylmaleimide (NMM) (MP Biomedicals); 17-octadecynoic acid (17-ODYA) (Cayman Chemical Co.); azide-PEG4-biotin (biotin-azide) (Click Chemistry Tools); and Alexa Fluor 647-conjugated streptavidin (Invitrogen).
DNA Constructs
Construction of plasmids, WTSOD1 (SOD1), SOD1-YFP, A4V SOD1-YFP, G93A SOD1-YFP, was previously described (35). Plasmid G85R SOD1-YFP was prepared by first generating a G85R SOD1 construct by site-directed mutagenesis of a wtSOD1 template using forward primer 5′-GTTGGAGACTTGCGCAATGTGACTGC-3′ and reverse primer 5′-GCAGTCACATTGCGCAAGTCTCCAAC-3′ followed by inserting the SOD1 coding region into pEYFP-N1 vector by PCR amplification using forward primers 5′-CCGGAATTCATGGCGACGAAGGCCGTGTGCGTG-3′ and 5′-CGCGGATCCCCTTGGGCGATCCCAATTACACC-3′. The SOD1-His6 construct was generated by PCR amplification of the SOD1 coding sequence from SOD1-YFP using forward primer 5′-CCGGAATTCATGGACTACAAGGACGACGATGACGCGACGAAGGCCGTGTGCGTG-3′ and reverse primer 5′-CCCAAGCTTTTGGGCGATCCCAATTACACC-3′. The resulting PCR product was inserted into pcDNA3.1.myc-His(−) A vector. A number of mutated WT and A4V SOD1-YFP constructs were generated with a change of cysteine to alanine at codon 6 (C6A), cysteine to serine at codon 111 (C111S), or both (C6A and C111S). SOD1-YFP C111S and A4V SOD1-YFP C111S were generated by site-directed mutagenesis of SOD1-YFP and A4V SOD1-YFP, respectively, using forward primer 5′-CAGGAGACAGGAGACCATTCCATCATTGGCCG-3′ and reverse primer 5′-CGGCCAATGATGGAATGGTCTCCTG-3′. The SOD1 coding region of SOD1-YFP and SOD1-YFP C111S was PCR-amplified to generate SOD1-YFP C6A and SOD1-YFP C6A-C111S, respectively, using forward primer 5′-CCGGAATTCATGGACTACAAGGACGACGATGACGCGACGAAGGCCGTGGCCGTG-3′ and reverse primer 5′-CGCGGATCCCCTTGGGCGATCCCAATTACACC-3′; the PCR products were inserted into pEYFP-NI vector. Similarly, the SOD1 coding region of A4V SOD1-YFP and A4V SOD1-YFP C111S was PCR-amplified to generate A4V SOD1-YFP C6A and A4V SOD1-YFP C6A-C111S, respectively, using forward primer 5′-CCGGAATTCATGGACTACAAGGACGACGATGACGCGACGAAGGTCGTGGCCGTG-3′ and reverse primer 5′-CGCGGATCCCCTTGGGCGATCCCAATTACACC-3′. All mutations were confirmed by sequencing. The construct pJP001 encoding human CCS was provided by Dr. Valeria Culotta (Johns Hopkins University) (36).
Cell Culture and Transfections
HEK 293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% calf serum and 2% penicillin/streptomycin. NSC-34 cells (a gift from Dr. Neil Cashman) were maintained in DMEM containing 10% fetal bovine serum, 1% l-glutamine, and 1% penicillin/streptomycin. For palmitoylation experiments, HEK cells in 60-mm culture dishes were transfected at 60–80% confluence with 2–5 μg of DNA using a calcium phosphate protocol for 3–5 h and then changed to normal media. To prevent protein palmitoylation, 100 μm 2-bromopalmitate was added to cells during the media change following transfection and incubated for ∼16 h prior to cell lysis. NSC-34 cells were transfected using Effectene transfection reagent (Qiagen) according to the manufacturer's instructions.
SOD1 Immunoprecipitation
HEK and NSC-34 cells were pelleted by centrifugation 24 and 48 h after transfection, respectively, washed twice with PBS, and solubilized in RIPA buffer (50 mm Tris, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) containing protease inhibitors and 50 mm NEM to block free sulfhydryls. Following 1 h to overnight solubilization at 4 °C, lysate supernatants were collected following centrifugation (14,000 rpm, 4 °C, 20 min) and incubated with either mouse anti-SOD1 or mouse anti-GFP antibodies at 1:100 or 1:250 dilutions, respectively, and were rotated for 1 h to overnight at 4 °C. To assess RIPA buffer-insoluble protein, pellets resulting from centrifugation (40,000 rpm, 4 °C, 30 min) were washed once, solubilized in 1.0% SDS, and diluted with an equal volume of 10% Triton X-100 prior to incubation with mouse anti-GFP antibody at 1:250 dilution. Protein-antibody complexes were isolated by incubation with protein G-Sepharose for 2–3 h at 4 °C. SOD1-His6 was isolated by incubation with nickel-NTA-agarose beads in NTA wash buffer lacking EDTA (50 mm NaH2PO4, 300 mm NaCl, 20 mm imidazole, pH 8.0) and rotated for 1 h at 4 °C.
Spinal cords were harvested from transgenic mice expressing either human G93A SOD1 at the end stage of disease (mean survival 161 ± 10 days), human G85R at the end stage of disease (mean survival 340.4 ± 17.9 days), or human wtSOD1 (average age 302 ± 14.6 days). Ten percent homogenates (w/v) in RIPA buffer containing protease inhibitors and 50 mm NEM were prepared from spinal cords using Kontes Microtube Pellets Pestle with motorized homogenizer (A. Daigger and Co.). Homogenates were incubated on ice for 30 min, and supernatants were collected following centrifugation (14,000 rpm, 4 °C, 10 min) and incubated with mouse anti-SOD1 antibody at 1:150 dilution. Pellets resulting from centrifugation were solubilized in 2.5% SDS and diluted 1:10 with RIPA buffer prior to incubation with mouse anti-SOD1 antibody at 1:150 dilution. Protein antibody complexes were isolated by incubation with protein G-Sepharose for 2–3 h at 4 °C.
SOD1 Palmitoylation
The acyl-biotin exchange (ABE) method was performed as described previously (37). Following immunoprecipitation, beads were washed with wash buffer (150 mm NaCl, 50 mm Tris, pH 7.4, 5 mm EDTA, pH 7.4, and 1% Triton X-100) containing 25 mm NEM, followed by treatment with 1 m HAM, pH 7.0, for 1 h at room temperature (RT). As controls, duplicate samples were incubated in buffer without HAM, preventing palmitate removal. Beads were subsequently incubated with 0.5 μm biotin-BMCC, pH 6.3, at 4 °C for 1–2 h to label reactive cysteine residues. Nickel-NTA-bound SOD1-His6 was treated identically but maintained in NTA wash buffer lacking EDTA throughout the ABE protocol. Proteins were eluted and processed for SDS-PAGE and immunoblot analysis.
Click chemistry was performed with the alkyne-containing palmitate analog, 17-ODYA, which was reacted with biotin-azide (38). 25 μm 17-ODYA was added to HEK cell medium following transfection and incubated with cells for 6–16 h at 37 °C to allow for metabolic labeling of proteins that are palmitoylated during the labeling period. SOD1 was subsequently immunoprecipitated as described above. Duplicate samples were incubated in either the absence of HAM to allow for 17-ODYA to remain bound for subsequent biotinylation or in the presence of HAM to cleave bound 17-ODYA preventing subsequent biotinylation. Next, beads were reacted with biotin azide under standard click chemistry conditions where a base solution (50 mm triethanolamine, pH 7.4, 150 mm NaCl, 0.5% Triton X-100, protease inhibitors) was mixed with 0.1 mm biotin azide, 0.1 mm t-butanol tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, 1 mm TCEP, 1 mm CuSO4. The click reaction mix was incubated with the beads in the dark at RT for 1 h. Proteins were eluted and processed for SDS-PAGE and immunoblot analysis.
Separation of Membrane-associated and Cytosolic Proteins
HEK cells were pelleted by centrifugation 24 h after transfection, washed twice with 1× PBS, and resuspended in hypotonic PBS (0.1× PBS) containing protease inhibitors. Swollen cells were incubated on ice for 10 min and subsequently homogenized with 25 passes in a motorized homogenizer (A. Daigger and Co.). 10× PBS was added to homogenates to result in 1× PBS, and nuclei were pelleted by centrifugation (2,500 rpm, 4 °C, 20 min). Total protein amounts were determined from the nucleus-free supernatants, and 25 μg of total protein was acquired for SDS-PAGE and Western blot analysis. Equal amounts of post-nuclear supernatants were centrifuged (43,000 rpm, 4 °C, 1 h) to separate cytosolic (supernatant) from integral/peripheral membrane (pellet) proteins. Membrane pellets were washed with PBS and solubilized in 100 μl of RIPA buffer containing protease inhibitors. Equal volumes of membrane lysates (25 μl) and cytosolic supernatant fractions (10 μl) were processed for SDS-PAGE and Western blot analysis.
Western Blot Analysis
ABE or click chemistry-treated proteins or proteins isolated from total, cytosolic, and membrane fractions were separated by SDS-PAGE and subsequently transferred to PVDF membranes. SDS-PAGE was performed with 7.5% polyacrylamide gels with the exception of experiments assessing untagged SOD1, G85R-YFP, and subcellular protein fractions where 12% gels were used. Total protein lysates for SOD1-YFP expressed in the presence and absence of CCS were run on a 4–15% gradient gel. SDS-PAGE was performed under reducing conditions unless specified otherwise. To detect SOD1, membranes were incubated with rabbit anti-SOD1 antibody (1:1000) for 1 h (RT) to overnight (4 °C) and subsequently with Alexa Fluor 568 goat anti-rabbit IgG (1:1000). The same membranes were also used to detect biotinylated sites of palmitoylation. To do this, membranes with SOD1-YFP were incubated with Alexa Fluor 647 streptavidin at 1:1000, and membranes with unmodified SOD1 and SOD1-His6 were incubated with streptavidin-HRP at 1:15,000 for 1 h at RT. To detect CCS, membranes were incubated with rabbit anti-CCS antibody (1:500) for 1 h (RT) to overnight (4 °C) and subsequently with Alexa Fluor 568 goat anti-rabbit IgG (1:1000). To detect Tom20, membranes were incubated with rabbit anti-Tom20 antibody (1:400) for 1 h (RT) to overnight (4 °C) and subsequently with Alexa Fluor 568 goat anti-rabbit IgG (1:1000). To detect Na+/K+-ATPase, membranes were incubated with mouse anti-Na+/K+-ATPase antibody (1:400) for 1 h (RT) to overnight (4 °C) and subsequently with Alexa Fluor 488 goat anti-mouse IgG (1:1000). Blots were imaged on a Molecular Imager Pharos-FX (Bio-Rad), whereas streptavidin-HRP signals were detected with luminol/coumaric acid/H2O2 chemiluminescence, and exposed films were developed using an SRX-101A film processor (Konica Minolta). Quantification of band intensity was done using ImageJ software (National Institutes of Health).
Statistical Analysis
Results are expressed as the means ± S.E. of n experiments. One-way analysis of variance analysis with Newman-Keuls or Bonferroni's multiple comparison tests were used to calculate statistical significance of HEK and NSC-34 cell experiments. Paired t test was performed to determine statistical significance of spinal cord experiments as well as HEK experiments with G85R SOD1-YFP.
Mass Spectrometry (MS)
HEK cultures were lysed at 24 h following transfection with SOD1, solubilized, and SOD1 immunoprecipitated with mouse anti-SOD1 antibody as described above in the presence of 50 mm NEM. Beads were next exposed to 5 μm TCEP at RT for 5 min to reduce SOD1 intramolecular disulfide bonds. Following TCEP treatment, beads were treated with 50 mm NEM at 4 °C for 15 min to allow for alkylation of newly reduced sulfhydryls. Next, the beads were divided into two portions for treatment with HAM or, as a control, without HAM for 1 h at RT and subsequently incubated with 50 mm NMM at 4 °C for 1–2 h to label reactive cysteines. Proteins were eluted with 0.1 m glycine-HCl buffer, pH 2.1, and then enzymatically digested with Glu-C to ensure that cysteine-containing peptides were of proper length (∼500–3000 Da) for optimum MS/MS by high energy collisional dissociation fragmentation in a Hybrid Orbitrap Elite MS system. Similar LC MS/MS were carried out as described previously (39). Collected MS and MS/MS data were peak-picked with MASCOT Distiller and searched against the Swiss Prot Human protein database utilizing an in-house MASCOT algorithm (version 2.2.0). The parameters used in the search included the following: glutathione, NEM, NEM + water, NMM, NMM + water, oxidation, palmitoyl, phospho, phospho, a peptide tolerance of +20 ppm, MS/MS fragment tolerance of +0.6 Da, and peptide charges of +7 or less. Normal and decoy database searches were run.
RESULTS
SOD1 Is Palmitoylated
To examine whether SOD1 is palmitoylated, we used the ABE assay for protein palmitoylation (37, 40). In the ABE assay, unmodified free cysteine thiols are first alkylated with the thiol-reactive reagent NEM, which blocks them from subsequent reactions. The thioester bond that links palmitate to its cysteine site is then cleaved by HAM. HAM cleavage exposes a free sulfhydryl group on the cysteine that was previously palmitoylated. Newly formed free sulfhydryls are then labeled with a thiol-specific biotinylation reagent (biotin-BMCC). Once the sites are labeled, palmitoylation is analyzed by SDS-PAGE and Western blotting using fluorophore or HRP-conjugated streptavidin. The same membranes were also used for Western blotting with an anti-SOD1 antibody, and streptavidin signals were normalized to the amount of SOD1 protein present on the blot.
Untagged SOD1 or SOD1 tagged with either a His6 tag (SOD1-His6) or yellow fluorescent protein (YFP; SOD1-YFP) was transiently expressed in HEK cells. The different SOD1 proteins were precipitated with a SOD1-specific monoclonal antibody (mAb), nickel-NTA-agarose, or a GFP-specific mAb, respectively, and processed with the ABE assay. As shown in Fig. 1A, only SOD1 treated with HAM was significantly biotinylated, and all three forms of SOD1 were specifically labeled, indicating that they were palmitoylated. These results also indicate that the His6 and YFP tags at the SOD1 C terminus do not interfere with its palmitoylation.
FIGURE 1.
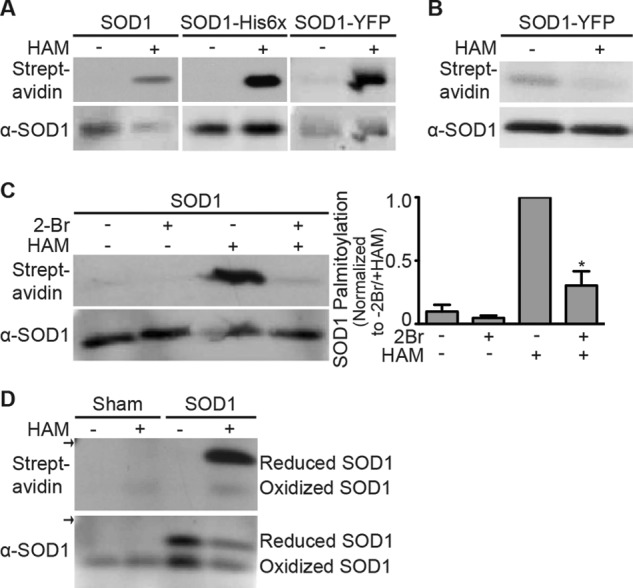
SOD1 is palmitoylated. A, unmodified SOD1, SOD1-His6, and SOD1-YFP were immunoprecipitated from transfected HEK cell lysates and subjected to the ABE protocol followed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). HAM-sensitive signals were detected for each version of SOD1. B, HEK cells transiently expressing SOD1-YFP were labeled with 17-ODYA. SOD1-YFP was immunoprecipitated and treated with biotin-azide under click chemistry reaction conditions and subsequently analyzed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Click biotin labeling of SOD1 was sensitive to HAM because treated samples had a 2-fold reduction in normalized biotin labeling (streptavidin signal divided by anti-SOD1 signal) relative to untreated samples (n = 3 experiments). C, HEK cells transiently expressing unmodified SOD1 were either untreated or treated with 2Br. SOD1 was immunoprecipitated and subjected to the ABE protocol followed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was both HAM- and 2Br-sensitive. The bar graph shows the average normalized densitometry data (streptavidin signal divided by anti-SOD1 signal) displayed as a fraction of 2Br-untreated/HAM-treated SOD1, which is set to 1. Normalized biotin labeling of 2Br-treated/HAM-treated SOD1 was reduced ∼3.3-fold relative to 2Br-untreated/HAM-treated SOD1 (n = 3 experiments, *, p < 0.001). Molecular masses for detected bands are 18 kDa (unmodified SOD1), 24 kDa (SOD1-His6), and 45 kDa (SOD1-YFP). D, palmitoylation of endogenous and overexpressed wtSOD1 in HEK cells. SOD1 was immunoprecipitated from HEK cell lysates that were either sham-transfected (endogenous SOD1) or transfected with unmodified SOD1. Immunoprecipitated SOD1 was subjected to the ABE protocol followed by SDS-PAGE under nonreducing conditions and then Western blotted with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was specific for HAM-treated samples and occurred predominantly for disulfide-reduced SOD1. Arrows indicate the 20-kDa marker. α indicates anti.
We used a second independent method, click chemistry (41), to confirm SOD1 palmitoylation. With this approach, cells are metabolically labeled with the alkyne-containing palmitic acid analog 17-ODYA. Palmitoylation occurring during the metabolic labeling period results in the incorporation of 17-ODYA. 17-ODYA labeling is then detected with the click compound, biotin-azide, which binds to the alkyne group on 17-ODYA under click chemistry reactions conditions (38, 42). HEK cells expressing SOD1-YFP were metabolically labeled with 17-ODYA. SOD1-YFP was immunoprecipitated, and duplicate samples were either incubated with HAM as a control (to cleave 17-ODYA) or left untreated (to leave 17-ODYA bound) and subsequently reacted with biotin-azide under click reaction conditions. As shown in Fig. 1B, SOD1-YFP was labeled with 17-ODYA and biotin-azide, indicating its palmitoylation. Unlike the ABE approach, click chemistry labeling is independent of palmitate cleavage by HAM. Here, palmitoylation is assayed by the incorporation of the palmitate analog, 17-ODYA, as seen with the non-HAM treated sample. As a control to test for thioester linkage of 17-ODYA to SOD1, duplicate samples were exposed to HAM prior to the click chemistry reaction. HAM exposure reduced the streptavidin signal, demonstrating that thioester cleavage of 17-ODYA prevented click biotin labeling of SOD1.
We also tested whether SOD1 palmitoylation was sensitive to the palmitoylation inhibitor 2Br. HEK cells expressing SOD1 were treated with 100 μm 2Br overnight, and the level of SOD1 palmitoylation was compared with untreated cells expressing SOD1. Exposure to the inhibitor reduced SOD1 palmitoylation by 70% relative to the level of SOD1 palmitoylation in untreated cells (Fig. 1C), further confirming the palmitoylation of SOD1.
Qualitatively, all three assays of SOD1 palmitoylation, ABE assay, click chemistry, and 2Br inhibition of cellular palmitoylation confirmed the palmitoylation of wtSOD1. However, there were quantitative differences in the results. In contrast to the robust labeling with the ABE assay, the signal from click chemistry labeling was weak, and the inhibition of ABE labeling with 2Br treatment was ∼70% despite the long periods of 17-ODYA labeling and 2Br exposure (up to 16 h for both). One difference among the three assays is that the ABE approach measures total protein palmitoylation, whereas click chemistry and 2Br inhibition measure only transient protein palmitoylation that occurs during their exposure to cells. Our finding of relatively weak labeling by click chemistry, relatively weak 2Br inhibition, and strong ABE labeling suggest that SOD1 palmitoylation does not undergo frequent cycles of depalmitoylation and repalmitoylation.
Palmitoylation Occurs Predominantly on Disulfide-reduced SOD1
An intramolecular disulfide bond forms between residues Cys-57 and Cys-146 during SOD1 maturation (43). Disulfide-bonded (mature) SOD1 can be separated from disulfide-reduced (immature) SOD1 by SDS-PAGE under nonreducing conditions following thiol protection (44). SOD1 expressed in HEK cells was solubilized and immunoprecipitated in the presence of the thiol blocker NEM and subsequently processed with the ABE assay. As observed previously, the disulfide-bonded SOD1 (lower bands) migrated faster than disulfide-reduced SOD1 (upper bands) in nonreducing SDS-PAGE and Western blot analysis (Fig. 1D, bottom). Both disulfide-reduced and disulfide-bonded forms of SOD1 were detected when SOD1 was overexpressed. However, only disulfide-bonded SOD1 was detected for endogenous SOD1 from sham-transfected cells. These results are consistent with the previous finding that SOD1 overexpression in HEK cells overwhelms the capacity of CCS to catalyze the formation of SOD1 intramolecular disulfide bonds during its maturation (36). Consequently, transfected SOD1 is increased in the disulfide-reduced form, whereas SOD1 present at endogenous levels is entirely disulfide-bonded. ABE labeling was barely detectable for disulfide-bonded SOD1, whereas disulfide-reduced SOD1 was much more highly palmitoylated (Fig. 1D, top). As a result, the palmitoylation of endogenous SOD1 was relatively low. Our results suggest that immature disulfide-reduced SOD1 is the predominantly palmitoylated form of SOD1.
SOD1 Is Palmitoylated on Cysteine 6
S-Palmitoylation occurs only on cysteine residues. SOD1 has four cysteine residues as follows: Cys-6, Cys-57, Cys-111, and Cys-146. Because Cys-57 and Cys-146 form a stable intramolecular disulfide bond (43), they are unlikely to be palmitoylated, making Cys-6 and/or Cys-111 the residues most likely to be palmitoylated. We assayed whether Cys-6 and/or Cys-111 are palmitoylated by cysteine mutagenesis substituting Cys-6 with alanine (C6A) and Cys-111 with serine (C111S) to produce two single mutants and a double mutant (C6A/C111S) of SOD1-YFP. The palmitoylation of these mutants was measured using the ABE assay as in Fig. 1. Palmitoylation of the C6A single mutant and C6A/C111S double mutant was highly reduced, almost to background levels, consistent with palmitoylation of the Cys-6 residue. However, palmitoylation of the C111S single mutant was also reduced, with ∼4-fold less of a reduction than the C6A mutant (Fig. 2A). These results suggest that Cys-111 may also be palmitoylated, but to a lesser extent than Cys-6. Another possibility is that mutation of Cys-111 changes the SOD1 conformation so that it is less palmitoylated on Cys-6.
FIGURE 2.

Palmitoylation of WT and mtSOD1 in HEK cells. wtSOD1-YFP and indicated cysteine mutants (A), wtSOD1-YFP, A4V SOD1-YFP, G93A SOD1-YFP, and G85R SOD1-YFP (B), and A4V SOD1-YFP and indicated cysteine mutants (C) were immunoprecipitated from transfected HEK cell lysates and subjected to the ABE protocol followed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was specific for HAM-treated samples. All arrows indicate the 50-kDa marker. Bar graphs on the right provide average normalized densitometry data (streptavidin signal divided by anti-SOD1 signal) after subtraction of background (−HAM) displayed as a fraction of wtSOD1-YFP (A and B) or A4V SOD1-YFP (C), with each set to 1. The normalized biotin labeling of SOD1-YFP C6A and SOD1-YFP C6A/C111S was reduced ∼16-fold, whereas labeling of SOD1-YFP C111S was reduced ∼4.5-fold relative to wtSOD1-YFP (n = 4 experiments, ***, p < 0.001) (A). The normalized biotin labeling of A4V and G93A SOD1-YFP was increased by 10.1- and 6.5-fold, respectively, relative to wtSOD1-YFP (n = 5 experiments, *, p < 0.05; **, p < 0.01). In separate experiments, the normalized biotin labeling of G85R SOD1-YFP was increased by 3.5-fold relative to wtSOD1-YFP (n = 3 experiments, *, p < 0.05) (B). Labeling of A4V SOD1-YFP C6A and A4V SOD1-YFP C6A-C111S was reduced ∼15- and 25-fold, respectively, whereas labeling of A4V SOD1-YFP C111S was reduced ∼2.3-fold relative to A4V SOD1-YFP (n = 4 experiments, ***, p < 0.001) (C). α indicates anti.
To further identify palmitoylated residue(s), we used a novel modification of ABE methodology where palmitoylated cysteines of immunopurified SOD1 were labeled with NMM, instead of the biotinylated thiol-specific reagent used in Fig. 1, and NMM-labeled cysteines were subsequently identified using high resolution tandem mass spectrometry (MS). When comparing HAM-treated and untreated (control) samples, only Cys-6 was unambiguously identified as labeled by NMM in the HAM-treated samples. The MS/MS identification of a peptide with NMM-labeled Cys-6 is displayed in Fig. 3. The labeling of the remaining three cysteines was inconclusive, consistent with our findings that Cys-6 is the primary site for palmitoylation. ABE labeling was necessary for our detection of SOD1 palmitoylation by MS because we were unable to detect the presence of palmitate on SOD1 peptides, perhaps due to the labile and hydrophobic nature of palmitate.
FIGURE 3.
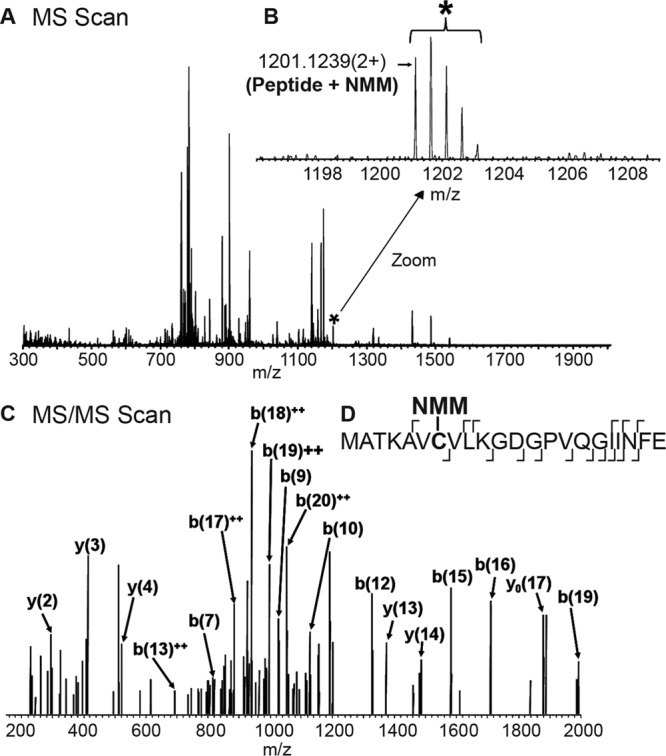
Identification of the ABE-modified cysteine (Cys-6) of SOD1 by LC-MS/MS. Immunoprecipitated and ABE-labeled SOD1 was eluted from protein beads and enzyme-digested with Glu-C, prior to injection onto a C18 RP attached to an EASY-nLC system in line with an LTQ Orbitrap ELITE mass spectrometer. Peptides were separated over a 50-min linear gradient, and MS scans (A) and MS/MS scans (C) were collected over the entire run time. B, zoomed in region of the MS scan where the molecular ion of the NMM-Cys-6-containing peptide was observed. The corresponding NMM-Cys-6 peptide ion (*) was q-isolated within the mass spectrometer and fragmented in the subsequent MS/MS scan (C). The peptide identity was based on the molecular ion of NMM-Cys-6 and the corresponding b- and y-fragment ions series labeled in D. The site of fragmentation on the NMM-Cys-6 peptide backbone is represented by the ⌈ and ⌋ for the y- and b-ion, respectively.
FALS-linked SOD1 Mutants A4V, G93A, and G85R Are More Highly Palmitoylated than wtSOD1
We next examined the palmitoylation of three FALS-linked SOD1 mutants, A4V, G93A, and G85R. Constructs expressing these SOD1s were transfected into HEK cells, and the palmitoylation of these mutants was measured using the ABE assay. Palmitoylation of A4V, G93A, and G85R SOD1-YFP was increased ∼10.1-, 6.5-, and ∼3.5-fold, respectively, relative to that of wtSOD1-YFP (Fig. 2B). The same cysteine mutagenesis performed on SOD1-YFP was also performed on A4V SOD1-YFP (Fig. 2C). Like wtSOD1, the C6A mutation and double C6A/C111S mutation blocked virtually all A4V SOD1-YFP palmitoylation, whereas the C111S mutation partially blocked palmitoylation.
To assay the palmitoylation of FALS-linked SOD1 mutants in neuronal cells, we first examined palmitoylation of G93A and A4V in the NSC-34 motor neuron cell line (45). The ABE assay demonstrated that palmitoylation of G93A and A4V SOD1-YFP was increased ∼4.8 and 6.7-fold, respectively, relative to that of wtSOD1-YFP. In addition, the C6A mutation blocked virtually all A4V SOD1-YFP palmitoylation (Fig. 4A). Next, we examined palmitoylation of SOD1 immunoprecipitated from spinal cords harvested from SOD1 transgenic mice. We assayed mice expressing human G93A SOD1 at the end stage of disease (mean survival 161 ± 10 days), human G85R SOD1 at the end stage of disease (mean survival 340.4 ± 17.9 days), and human wtSOD1 (average age 302 ± 14.6 days). Immunoprecipitated WT and G93A SOD1 protein was detected in anti-SOD1 Western blots, and ABE labeling demonstrated that palmitoylation of G93A SOD1 was increased ∼2-fold relative to that of wtSOD1 (Fig. 4B). Additionally, G93A SOD1 from RIPA buffer-insoluble pellets of spinal cord homogenates were solubilized in SDS, immunoprecipitated, and then subjected to the ABE assay. Insoluble G93A SOD1 was palmitoylated to similar levels as that seen for soluble G93A (Fig. 4, C and D). The level of palmitoylation of A4V SOD1-YFP immunoprecipitated from HEK cell RIPA buffer-insoluble pellets was also similar to that seen with RIPA buffer-soluble A4V from HEK cell lysates (data not shown).
FIGURE 4.

Palmitoylation of WT and mtSOD1 in neuronal cells. A, wtSOD1-YFP, G93A SOD1-YFP, A4V SOD1-YFP, and A4V SOD1-YFP C6A were immunoprecipitated from transfected NSC-34 cells and subjected to the ABE protocol followed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was specific for HAM-treated samples. Arrows indicate the 50-kDa marker. Bar graphs on the right provide average normalized densitometry data (streptavidin signal divided by anti-SOD1 signal) after subtraction of background (−HAM) displayed as a fraction of wtSOD1-YFP, which is set to 1. Normalized biotin labeling of G93A and A4V SOD1-YFP was increased by 4.8- and 6.7-fold, respectively, relative to wtSOD1-YFP (n = 3 experiments for wtSOD1-YFP, A4V SOD1-YFP, and G93A SOD1-YFP; n = 2 experiments for A4V SOD1-YFP C6A, *, p < 0.05). B, SOD1 was immunoprecipitated from spinal cords harvested from human G93A SOD1 transgenic mice at the end stage of disease (mean survival 161 ± 10 days), human G85R SOD1 transgenic mice at the end stage of disease (mean survival 340.4 ± 17.9 days), or human wtSOD1 transgenic mice (average age 302 ± 14.6 days) and subjected to the ABE protocol followed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was specific for HAM-treated samples. Arrows indicate the 20-kDa marker. Bar graphs on the right provide average normalized densitometry data (streptavidin signal divided by anti-SOD1 signal) after subtraction of background (−HAM) displayed as a fraction of wtSOD1, which was set to 1. Normalized biotin labeling of G93A SOD1 was increased by 1.9-fold relative to wtSOD1 (n = 3 experiments where three wtSOD1 spinal cords and four G93A SOD1 spinal cords were examined, *, p < 0.05). C, spinal cords harvested from G93A SOD1 transgenic mice at the end stage of disease and human wtSOD1 transgenic mice were solubilized in RIPA buffer, centrifuged, and then immunoprecipitated with anti-SOD1 antibody from supernatants (RIPA-soluble) and pellets (RIPA-insoluble). Immunoprecipitated SOD1 was subjected to the ABE protocol followed by SDS-PAGE under nonreducing conditions and then Western blotted with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was specific for HAM-treated samples and occurred only for reduced SOD1. Arrows indicate the 20-kDa marker. D, bar graph provides the average relative densitometry data (streptavidin signal divided by α-SOD1 signal) for G93A SOD1 processed as in C. Normalized biotin labeling was similar regardless of whether G93A SOD1 was purified from RIPA-soluble or RIPA-insoluble fractions (n = 3 experiments, four spinal cords examined). α indicates anti.
G85R SOD1 immunoprecipitated from transgenic mouse spinal cord lysates was barely detectable by Western blot. Despite the lack of detectable G85R by Western blot, ABE labeling demonstrated a biotinylated band corresponding to G85R, which, as expected, migrated characteristically faster than wtSOD1 and other mtSOD1s in SDS-PAGE (Fig. 4B) (2). The intensity of the ABE-labeled G85R SOD1 band was roughly equal to that of wtSOD1. The intensity of the G85R SOD1 protein band was too low to reliably quantify protein levels. The low protein levels may have resulted from reduced immunoprecipitation as a result of a lower affinity of the antibody for G85R or because of the relatively lower steady state of G85R SOD1 protein levels in transgenic mice (44, 46). Assuming that the low level of the G85R SOD1 Western blot signal is an accurate measure of protein levels, then G85R SOD1 palmitoylation was significantly higher than that of both wtSOD1 and G93A SOD1.
In Fig. 4D, we also tested whether palmitoylated wtSOD1 and G93A SOD1 from transgenic mouse spinal cord lysates was disulfide-bonded or disulfide-reduced. As before in Fig. 1D, we assayed SOD1 palmitoylation using ABE with subsequent SDS-PAGE carried out under nonreducing conditions allowing for the separation of disulfide-bonded from disulfide-reduced SOD1. Only disulfide-reduced SOD1 (Fig. 1D, upper band) was found to be significantly palmitoylated for WT and G93A SOD1 whether solubilized or present in the insoluble pellet. Again, the results suggest that immature disulfide-reduced SOD1 is the predominant palmitoylated species.
Co-expression of CCS Reduces SOD1 Palmitoylation Levels
To further examine the relationship between SOD1 palmitoylation and its maturation, we co-expressed CCS with wtSOD1 or mtSOD1s in HEK cells. Because CCS increases SOD1 intramolecular disulfide bonding and maturation (28, 29, 36), we wanted to test how increasing CCS levels affect SOD1 palmitoylation. As expected, we observed an increase in the relative levels of disulfide-bonded to disulfide-reduced SOD1-YFP when CCS levels were increased (Fig. 5A). The increase in disulfide bonding was most apparent for wtSOD1 and less so for A4V and G93A mtSOD1s. ABE analysis demonstrated that SOD1-YFP palmitoylation was decreased by CCS co-expression (Fig. 5, B and C). wtSOD1-YFP co-expressed with CCS had a 3-fold decrease in palmitoylation (Fig. 5C). Increasing CCS levels also significantly decreased the palmitoylation of A4V SOD1-YFP, although less strongly than with wtSOD1-YFP (2-fold; Fig. 5C). There appeared to be a decrease in palmitoylation of G93A SOD1-YFP with CCS co-expression (1.3-fold; Fig. 5C), but it was not significantly different from the palmitoylation in the absence of overexpressed CCS. These results indicate an inverse relationship between SOD1 palmitoylation and CCS-mediated disulfide bonding of SOD1. This inverse relationship appears to have a role in the increased palmitoylation of mtSOD1s relative to wtSOD1 because mtSOD1s have higher relative levels of the disulfide-reduced form (Fig. 5A).
FIGURE 5.
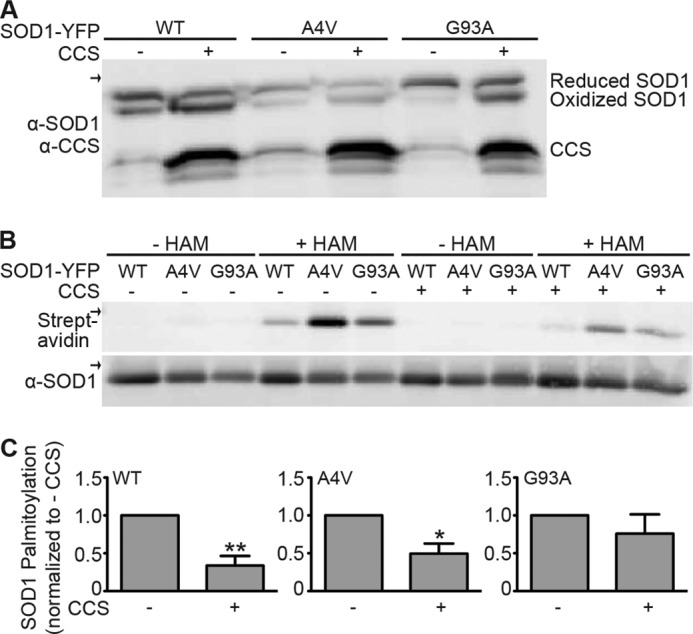
Palmitoylation of wtSOD1 and mtSOD1 in the presence or absence of overexpressed CCS. A, HEK cells were transfected with wtSOD1-YFP, A4V SOD1-YFP, or G93A SOD1-YFP as indicated and were either co-transfected or not with wtCCS. Equal amounts of total protein lysates were run on nonreducing SDS-PAGE and then Western blotted with anti-SOD1 and anti-CCS antibodies. B, wtSOD1-YFP, A4V SOD1-YFP, and G93A SOD1-YFP were immunoprecipitated from transfected cell lysates and subjected to the ABE protocol followed by Western blotting with anti-SOD1 antibody (bottom panel) and streptavidin (top panel). Biotin labeling was specific for HAM-treated samples. Arrows indicate the 50-kDa marker. C, bar graphs provide average normalized densitometry data (streptavidin signal divided by anti-SOD1 signal) after subtraction of background (−HAM) displayed as a fraction of the equivalent SOD1-YFP without coexpression of CCS, which is set to 1. Normalized biotin labeling of wtSOD1-YFP, A4V SOD1-YFP, and G93A SOD1-YFP was decreased ∼3.0-, 2.0-, and 1.3-fold respectively, relative to when CCS was present at endogenous levels (n = 3 experiments, *, p < 0.05; **, p < 0.01). α indicates anti.
Level of SOD1 Palmitoylation Correlates with Its Targeting to Membranes
To determine whether the level of SOD1 palmitoylation affects its membrane targeting, SOD1-YFPs with varying levels of palmitoylation were expressed in HEK cells, and total membrane fractions were isolated by high speed centrifugation. Total, cytoplasmic, and membrane fractions were quantitatively assessed by Western blot using an anti-SOD1 antibody to determine the relative amount of SOD1 present in each fraction. A4V SOD1-YFP, which has increased palmitoylation relative to wtSOD1-YFP (Fig. 2B), had increased membrane association relative to wtSOD1-YFP (Fig. 6B). In addition, the C6A mutation, which abolishes SOD1 palmitoylation (Fig. 2, A and C), resulted in decreased membrane association for both wtSOD1-YFP and A4V SOD1-YFP (Fig. 6).
FIGURE 6.
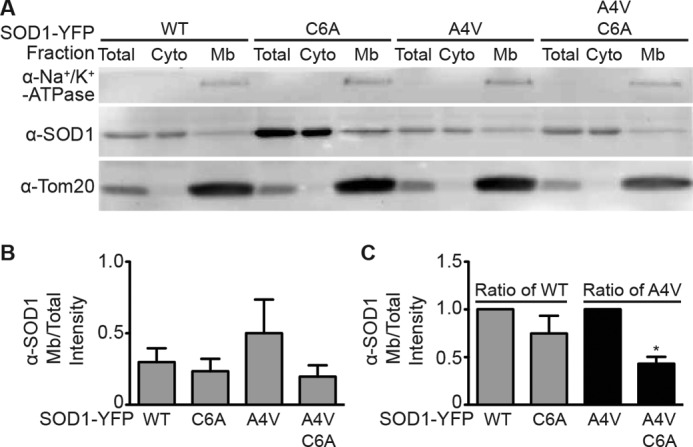
Membrane targeting of wtSOD1 compared with mtSOD1s with reduced or increased palmitoylation. A, HEK cells expressing wtSOD1-YFP, C6A SOD1-YFP (reduced palmitoylation relative to wtSOD1), A4V SOD1-YFP (increased palmitoylation relative to wtSOD1), or A4V C6A (reduced palmitoylation relative to wtSOD1 and A4V SOD1) were homogenized, and post-nuclear supernatants were subjected to high speed centrifugation to separate cytosolic (supernatant) and membrane (pellet) fractions. Membrane pellets were solubilized, and equivalent amounts of total protein from total (cytoplasmic and membrane fractions prior to high speed spin), cytosolic, and membrane fractions were run on SDS-polyacrylamide gels and Western blotted for the amount of SOD1 present in each fraction. Blots were additionally probed for the plasma membrane marker Na+/K+-ATPase and the mitochondrial marker Tom20. B, bar graph represents the densitometry values for SOD1 protein present in the membrane fraction normalized to the SOD1 protein present in the corresponding total fraction presented as raw intensity values. C, bar graph represents the normalized membrane/total SOD1 protein intensity values as a ratio of wtSOD1-YFP (for C6A SOD1-YFP) and as a ratio of A4V SOD1-YFP (for A4V C6A SOD1-YFP), with each set to one. (n = 3 experiments; *, p < 0.01). (Cyto, cytoplasmic; Mb, membrane; α, anti.)
DISCUSSION
In this study, we used five different assays (ABE, click chemistry, inhibition of palmitoylation by 2Br, cysteine mutagenesis, and MS following ABE labeling) to examine whether human wtSOD1 is palmitoylated. The results of all applied assays are consistent with wtSOD1 being palmitoylated and with Cys-6 being the primary site of palmitoylation (Figs. 2 and 3). Multiple methods were employed because palmitoylation of SOD1 had not yet been detected, despite many prior biochemical and biophysical studies of SOD1 structure (reviewed in Ref. 47). Several factors may have contributed to why SOD1 palmitoylation had gone undetected in the past. First, many studies have examined SOD1 purified from expression systems, such as E. coli, where PATs are absent and enzymatic palmitoylation is not thought to occur. Second, SOD1 palmitoylation might have gone undetected if the assay was not sensitive enough. Prior to the development of ABE and click chemistry labeling, the only assay for protein palmitoylation was metabolic labeling with radiolabeled palmitate. As discussed above, metabolic labeling only measures palmitoylation that occurs during the assay and not the total palmitoylation. The relatively weak results from click chemistry and 2Br inhibition suggest that SOD1 does not undergo cycles of depalmitoylation and repalmitoylation and therefore would not be easily assayed by metabolic labeling with radiolabeled palmitate. In addition, the large increase in mtSOD1 palmitoylation relative to that of wtSOD1 by the ABE assay suggests that a relatively small fraction of total wtSOD1 is palmitoylated. Our experiments were also conducted largely with transfected SOD1, which increased the degree to which SOD1 was palmitoylated compared with endogenous SOD1 (Fig. 1D). Thus, SOD1 palmitoylation was likely missed before more sensitive assays were developed. Consistent with this, other post-translational modifications of SOD1 have only recently been detected by MS approaches, including the report that SOD1 purified from human erythrocytes is glutathionylated at Cys-111 (48). Finally, the hydrophobic and labile nature of palmitate likely complicates detection by MS, which is why we used the less direct approach of ABE NMM labeling as the readout for palmitoylation in our MS analysis.
During the preparation of this manuscript, Marin et al. (49) published a study in which they identified endogenous SOD1 in endothelial cells as palmitoylated by an ABE and MS-based palmitoyl proteomic screen. The authors confirmed palmitoylation of SOD1 with overexpression followed by ABE analysis and tritiated palmitate incorporation, and they found that mutation of Cys-6 prevented palmitoylation (49). Our independent discovery of SOD1 palmitoylation reported here is consistent with the findings reported in Marin et al. (49). We extended these findings by demonstrating increased palmitoylation of FALS-linked mtSOD1s relative to wtSOD1 in HEK cells, NSC-34 neuronal cells, and in the spinals cords of mtSOD1 transgenic mice.
G93A SOD1 isolated from the spinal cords of G93A transgenic mice during the end stage of disease was increased 2-fold in palmitoylation relative to wtSOD1 (Fig. 4B). If the process of palmitoylation were directly causing aggregation and insolubility, then the expectation would be that insoluble mtSOD1 should be more highly palmitoylated than soluble mtSOD1. We found that G93A immunoprecipitated from RIPA buffer insoluble pellets was palmitoylated at similar levels to soluble G93A (Fig. 4, C and D). Insoluble A4V from HEK cells was also palmitoylated at a level comparable with that of soluble A4V (data not shown). Thus, mtSOD1 appears to be equally palmitoylated regardless of whether it is soluble or in insoluble aggregates.
We also examined the palmitoylation of FALS mtSOD1, G85R, from transgenic mouse spinal cords at end stage of disease. Although a biotinylated G85R band was detected at the expected molecular weight of G85R (2), indicative of palmitoylation, a corresponding band was not observed in the anti-SOD1 Western blot (Fig. 4B). This not only indicates that G85R SOD1 is palmitoylated in transgenic mouse spinal cords but suggests it is more highly palmitoylated than wtSOD1 and G93A because G85R had a similar ABE biotinylation signal to that seen with wtSOD1 and G93A despite the lower level of G85R SOD1 protein. The lack of G85R SOD1 immunolabeling could be the result of only a small amount of this protein being immunoprecipitated due to a relatively lower steady state of G85R SOD1 protein levels in transgenic mice (44, 46) and/or the high aggregation potential of this mutant (33), which could impact accessibility to immunoprecipitation. Alternatively, higher levels of G85R could have been immunoprecipitated but may be evading antibody detection by Western blot. However, we suspect the lack of G85R protein detected in the ABE Western blot is most likely due to inefficient immunoprecipitation because we were able to detect G85R in spinal cord lysates on Western blots (without immunoprecipitation) using the same antibodies (data not shown). Overall, our findings that palmitoylation of FALS-linked mtSOD1s is increased relative to wtSOD1 in multiple systems suggests that this post-translational modification of SOD1 may play a role in FALS pathogenesis.
How palmitoylation affects the biology of SOD1 is not yet clear. Palmitoylation of SOD1 is presumably not required for proper folding into a functional enzyme because SOD1 is functional when expressed in E. coli, which lack the PATs needed to palmitoylate proteins. However, SOD1 palmitoylation may still regulate the level of activity. In support of this, Marin et al. (49) report that mutation of C6S, which prevents palmitoylation, leads to a reduction in SOD1 activity in vivo and in vitro.
The cysteine residues of SOD1 have been extensively studied and are critical for several features of its biology. Cys-111, a potential site of palmitoylation, undergoes oxidation (23, 50) and glutathionylation (48) and is thought to be important for copper binding (51), all processes implicated in the structural stability of SOD1. The reactivity of Cys-111 is not surprising given its location on the surface of the protein near the dimer interface. In contrast, Cys-6 is more buried within the β-barrel of mature SOD1 (52) and becomes more solvent-accessible upon the loss of metal ions, which destabilizes the SOD1 structure (32). The inaccessibility of Cys-6 in mature SOD1 suggests that access of the PAT protein catalytic region to Cys-6 may be blocked for mature SOD1 and that palmitoylation occurs before SOD1 is in the fully mature folded state.
Consistent with palmitoylation occurring before or during its maturation, the palmitoylated SOD1 was predominantly in the disulfide-reduced state, as opposed to the disulfide-bonded state (Figs. 1D and 4C). Of note, low levels of SOD1 palmitoylation were detected for endogenous and transfected HEK cell SOD1 when disulfide-bonded (Fig. 1D). If SOD1 palmitoylation occurs prior to disulfide bonding, the low levels of palmitoylated disulfide-bonded SOD1 suggest a depalmitoylation step may occur during maturation. Alternatively, when palmitoylated, SOD1 may be less likely to undergo disulfide bonding and maturation. In support of SOD1 palmitoylation occurring before or during its maturation is the result of our experiments with increased CCS levels, which enhanced SOD1 maturation (Fig. 5A) (29, 36). The increased disulfide bonding by CCS overexpression correlated with decreased levels of both wtSOD1 and mtSOD1 palmitoylation relative to when CCS was at endogenous levels (Fig. 5). The reduction in palmitoylation by increased CCS levels was significantly less for the mtSOD1s, A4V and G93A SOD1. This differential effect of CCS on wtSOD1 versus mtSOD1 palmitoylation correlates with the ability of overexpressed CCS to facilitate SOD1 disulfide bonding. As seen in Fig. 5A and as previously reported (36), CCS causes more disulfide bonding of wtSOD1 than G93A SOD1. These results suggest that SOD1 palmitoylation increases when SOD1 folding and maturation are delayed or blocked. Indeed, FALS-causing mtSOD1s have delayed folding kinetics compared with wtSOD1, consistent with more mtSOD1 in an unfolded state relative to wtSOD1 (53). Also, a recent study found that a domain including Cys-6 of FALS-linked mtSOD1s had a different conformation than wtSOD1 (54). Thus, it is possible that delayed maturation of FALS mtSOD1s results in the observed increases in palmitoylation by increasing cysteine accessibility to PATs.
In addition to playing a role in folding, the SOD1 cysteine residues, in particular Cys-6 and Cys-111, have been implicated in several potentially pathological processes, including intermolecular disulfide bonding (17, 18, 55, 56), aggregate formation (31, 57–61), and the toxicity of mtSOD1 (60). Similar to previous studies, we found that cysteine residue mutations that block SOD1 palmitoylation (C6A and C111S) decrease the formation of aggregates in vitro (data not shown). It is unclear whether this decrease in aggregation is related to a decrease in palmitoylation of mtSOD1 or whether the cysteine itself influences aggregation for other reasons, e.g. if aggregation results from an altered protein structure or from the formation of intermolecular disulfide bonds. Palmitoylation could indirectly impact aggregation by affecting the reactivity of SOD1 cysteines, in particular Cys-6, which may be more frequently exposed in apo-SOD1 and/or misfolded FALS mtSOD1s. Mutation of all four cysteine residues of human SOD1 leads to aggregation (58), suggesting that palmitoylation is not required for aggregate formation. However, it is possible that the misfolding and aggregation resulting from mutating all four cysteines may not be equivalent to misfolding and aggregation associated with mtSOD1-induced FALS. Of note, C6S and C6F mutations cause FALS (62, 63), suggesting that palmitoylation at Cys-6 is not required for motor neuron disease or that SOD1-induced FALS can be associated with both an increase and decrease in SOD1 palmitoylation. It is possible, for example, that the C6S and C6F mutations abolish some interactions and post-translational modifications of Cys-6 that are neuroprotective. It is also possible that different mtSOD1s vary in their mechanism of toxicity. C6F SOD1 is improperly folded in vitro (53) and forms aggregates, whereas C6A, C6S, and C6G mutants do not (58, 59), suggesting that phenylalanine at position 6 is more of a toxic determinant than simply a loss of Cys-6. Understanding the roles of Cys-6 and Cys-111 in mediating SOD1 stability, aggregation, and toxicity will be critical in understanding how palmitoylation affects these processes.
A major function of cytosolic protein palmitoylation is to target proteins to the cytosolic interface of cellular membranes. In order for SOD1 to be palmitoylated, it must first be targeted to the appropriate PAT(s), which are multispanning membrane proteins located at different cellular membranes (64). Although primarily cytosolic, a fraction of SOD1 is found at membranes of different organelles, including mitochondria (65) and the ER-Golgi pathway (66, 67). Mitochondrial localization only occurs for immature SOD1 lacking copper and disulfide bonds (68), and it is dependent on CCS (69). Thus, one possibility is that SOD1 palmitoylation occurs early during its maturation and targets it from its site of palmitoylation to mitochondria. If true, increases in palmitoylation of mtSOD1 would increase targeting to mitochondria and potentially affect mitochondrial function. Consistent with this, we find that the level of SOD1 palmitoylation correlates with its membrane targeting in HEK cells. A4V SOD1, which has a 10-fold increase in palmitoylation relative to wtSOD1 (Fig. 2B), was more membrane-associated, whereas loss of SOD1 palmitoylation by introduction of the C6A mutation into both wtSOD1 and A4V SOD1 backgrounds reduced SOD1 membrane association to lower levels (Fig. 6B). Similar results have been reported in previous studies where several mtSOD1s were found to accumulate in and on mitochondria to a greater extent than wtSOD1, whereas Cys-6 and Cys-111 mutations reduced SOD1 localization to mitochondria (14, 69). Importantly, mtSOD1 accumulation on mitochondria results in mitochondrial dysfunction, which is a feature of FALS (5, 6, 14, 15, 70). It is also possible that palmitoylation targets SOD1 to the ER-Golgi pathway, a site for uptake and aggregation of mtSOD1, and could thus contribute to the pathogenic effects of ER stress (7–13) and possibly nonautonomous cell degeneration resulting from the secretion of toxic misfolded mtSOD1 (19, 66, 71, 72). Thus a role for palmitoylation in targeting SOD1 to membranes represents a means by which this modification could impact the toxicity of mtSOD1.
In summary, our results demonstrate that SOD1 is palmitoylated and that palmitoylation of mtSOD1 (A4V, G93A, and G85R) is increased. Because wtSOD1 in sporadic ALS also has aberrant structural and biochemical properties (21–25, 73, 74), altered SOD1 palmitoylation could be a pathogenic feature of sporadic disease as well as mtSOD1-induced FALS.
Acknowledgments
We thank Kathryn L. Stone, Mary LoPresti, and Kathrin Wilczak from the Yale/NIDA Neuroproteomics Center for assistance in mass spectrometry sample preparation and data collection.
This work was supported, in whole or in part, by National Institutes of Health Grants NS043782, DA019695, and MH081251 and by the Yale University/NIDA Neuroproteomics Center Grant P30DA018343 (to W. N. G.).
- ALS
- amyotrophic lateral sclerosis
- SOD1
- superoxide dismutase
- FALS
- familial ALS
- ER
- endoplasmic reticulum
- PAT
- palmitoyltransferase
- ABE
- acyl-biotin exchange
- HAM
- hydroxylamine
- NEM
- N-ethylmaleimide
- NMM
- N-methylmaleimide
- 2Br
- 2-bromopalmitate
- 17-ODYA
- 17-octadecynoic acid
- TCEP
- tris(2-carboxyethyl)phosphine
- BMCC
- 1-biotinamindo-4-[4′-maleimidomethyl)cyclohexanecarboxamido] butane
- CCS
- copper chaperone for SOD1
- NTA
- nitrilotriacetic acid.
REFERENCES
- 1. Ratovitski T., Corson L. B., Strain J., Wong P., Cleveland D. W., Culotta V. C., Borchelt D. R. (1999) Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 8, 1451–1460 [DOI] [PubMed] [Google Scholar]
- 2. Borchelt D. R., Lee M. K., Slunt H. S., Guarnieri M., Xu Z. S., Wong P. C., Brown R. H., Jr., Price D. L., Sisodia S. S., Cleveland D. W. (1994) Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. U.S.A. 91, 8292–8296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reaume A. G., Elliott J. L., Hoffman E. K., Kowall N. W., Ferrante R. J., Siwek D. F., Wilcox H. M., Flood D. G., Beal M. F., Brown R. H., Jr., Scott R. W., Snider W. D. (1996) Motor neurons in Cu,Zn-superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47 [DOI] [PubMed] [Google Scholar]
- 4. Rothstein J. D. (2009) Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65, S3–S9 [DOI] [PubMed] [Google Scholar]
- 5. De Vos K. J., Chapman A. L., Tennant M. E., Manser C., Tudor E. L., Lau K. F., Brownlees J., Ackerley S., Shaw P. J., McLoughlin D. M., Shaw C. E., Leigh P. N., Miller C. C., Grierson A. J. (2007) Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Magrané J., Sahawneh M. A., Przedborski S., Estévez Á. G., Manfredi G. (2012) Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. 32, 229–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang L., Popko B., Roos R. P. (2011) The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 20, 1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oh Y. K., Shin K. S., Yuan J., Kang S. J. (2008) Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 104, 993–1005 [DOI] [PubMed] [Google Scholar]
- 9. Atkin J. D., Farg M. A., Turner B. J., Tomas D., Lysaght J. A., Nunan J., Rembach A., Nagley P., Beart P. M., Cheema S. S., Horne M. K. (2006) Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165 [DOI] [PubMed] [Google Scholar]
- 10. Nishitoh H., Kadowaki H., Nagai A., Maruyama T., Yokota T., Fukutomi H., Noguchi T., Matsuzawa A., Takeda K., Ichijo H. (2008) ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tobisawa S., Hozumi Y., Arawaka S., Koyama S., Wada M., Nagai M., Aoki M., Itoyama Y., Goto K., Kato T. (2003) Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem. Biophys. Res. Commun. 303, 496–503 [DOI] [PubMed] [Google Scholar]
- 12. Saxena S., Cabuy E., Caroni P. (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636 [DOI] [PubMed] [Google Scholar]
- 13. Kikuchi H., Almer G., Yamashita S., Guégan C., Nagai M., Xu Z., Sosunov A. A., McKhann G. M., 2nd, Przedborski S. (2006) Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. U.S.A. 103, 6025–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferri A., Cozzolino M., Crosio C., Nencini M., Casciati A., Gralla E. B., Rotilio G., Valentine J. S., Carrì M. T. (2006) Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. U.S.A. 103, 13860–13865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Israelson A., Arbel N., Da Cruz S., Ilieva H., Yamanaka K., Shoshan-Barmatz V., Cleveland D. W. (2010) Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Q., Vande Velde C., Israelson A., Xie J., Bailey A. O., Dong M. Q., Chun S. J., Roy T., Winer L., Yates J. R., Capaldi R. A., Cleveland D. W., Miller T. M. (2010) ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. U.S.A. 107, 21146–21151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Furukawa Y., Fu R., Deng H. X., Siddique T., O'Halloran T. V. (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu,Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7148–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang L., Sharma K., Grisotti G., Roos R. P. (2009) The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 35, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chattopadhyay M., Valentine J. S. (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox Signal. 11, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ezzi S. A., Urushitani M., Julien J. P. (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 102, 170–178 [DOI] [PubMed] [Google Scholar]
- 22. Kabashi E., Valdmanis P. N., Dion P., Rouleau G. A. (2007) Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann. Neurol. 62, 553–559 [DOI] [PubMed] [Google Scholar]
- 23. Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J. P., Brady S. T., Brown R. H., Jr. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grad L. I., Guest W. C., Yanai A., Pokrishevsky E., O'Neill M. A., Gibbs E., Semenchenko V., Yousefi M., Wishart D. S., Plotkin S. S., Cashman N. R. (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. U.S.A. 108, 16398–16403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guareschi S., Cova E., Cereda C., Ceroni M., Donetti E., Bosco D. A., Trotti D., Pasinelli P. (2012) An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. U.S.A. 109, 5074–5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukata Y., Fukata M. (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175 [DOI] [PubMed] [Google Scholar]
- 27. Young F. B., Butland S. L., Sanders S. S., Sutton L. M., Hayden M. R. (2012) Putting proteins in their place: Palmitoylation in Huntington disease and other neuropsychiatric diseases. Prog. Neurobiol. 97, 220–238 [DOI] [PubMed] [Google Scholar]
- 28. Banci L., Barbieri L., Bertini I., Luchinat E., Secci E., Zhao Y., Aricescu A. R. (2013) Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 9, 297–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caruano-Yzermans A. L., Bartnikas T. B., Gitlin J. D. (2006) Mechanisms of the copper-dependent turnover of the copper chaperone for superoxide dismutase. J. Biol. Chem. 281, 13581–13587 [DOI] [PubMed] [Google Scholar]
- 30. Furukawa Y., O'Halloran T. V. (2005) Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 280, 17266–17274 [DOI] [PubMed] [Google Scholar]
- 31. Banci L., Bertini I., Durazo A., Girotto S., Gralla E. B., Martinelli M., Valentine J. S., Vieru M., Whitelegge J. P. (2007) Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. U.S.A. 104, 11263–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Banci L., Bertini I., Boca M., Calderone V., Cantini F., Girotto S., Vieru M. (2009) Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc. Natl. Acad. Sci. U.S.A. 106, 6980–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang J., Slunt H., Gonzales V., Fromholt D., Coonfield M., Copeland N. G., Jenkins N. A., Borchelt D. R. (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum. Mol. Genet. 12, 2753–2764 [DOI] [PubMed] [Google Scholar]
- 34. Lelie H. L., Liba A., Bourassa M. W., Chattopadhyay M., Chan P. K., Gralla E. B., Miller L. M., Borchelt D. R., Valentine J. S., Whitelegge J. P. (2011) Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J. Biol. Chem. 286, 2795–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghadge G. D., Wang L., Sharma K., Monti A. L., Bindokas V., Stevens F. J., Roos R. P. (2006) Truncated wild-type SOD1 and FALS-linked mutant SOD1 cause neural cell death in the chick embryo spinal cord. Neurobiol. Dis. 21, 194–205 [DOI] [PubMed] [Google Scholar]
- 36. Proescher J. B., Son M., Elliott J. L., Culotta V. C. (2008) Biological effects of CCS in the absence of SOD1 enzyme activation: implications for disease in a mouse model for ALS. Hum. Mol. Genet. 17, 1728–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drisdel R. C., Green W. N. (2004) Labeling and quantifying sites of protein palmitoylation. BioTechniques 36, 276–285 [DOI] [PubMed] [Google Scholar]
- 38. Martin B. R., Cravatt B. F. (2009) Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bianchetta M. J., Lam T. T., Jones S. N., Morabito M. A. (2011) Cyclin-dependent kinase 5 regulates PSD-95 ubiquitination in neurons. J. Neurosci. 31, 12029–12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drisdel R. C., Alexander J. K., Sayeed A., Green W. N. (2006) Assays of protein palmitoylation. Methods 40, 127–134 [DOI] [PubMed] [Google Scholar]
- 41. Speers A. E., Cravatt B. F. (2004) Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 11, 535–546 [DOI] [PubMed] [Google Scholar]
- 42. Hang H. C., Geutjes E. J., Grotenbreg G., Pollington A. M., Bijlmakers M. J., Ploegh H. L. (2007) Chemical probes for the rapid detection of fatty-acylated proteins in mammalian cells. J. Am. Chem. Soc. 129, 2744–2745 [DOI] [PubMed] [Google Scholar]
- 43. Abernethy J. L., Steinman H. M., Hill R. L. (1974) Bovine erythrocyte superoxide dismutase. Subunit structure and sequence location of the intrasubunit disulfide bond. J. Biol. Chem. 249, 7339–7347 [PubMed] [Google Scholar]
- 44. Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Lindberg M., Oliveberg M., Marklund S. L. (2006) Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129, 451–464 [DOI] [PubMed] [Google Scholar]
- 45. Cashman N. R., Durham H. D., Blusztajn J. K., Oda K., Tabira T., Shaw I. T., Dahrouge S., Antel J. P. (1992) Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 194, 209–221 [DOI] [PubMed] [Google Scholar]
- 46. Bruijn L. I., Becher M. W., Lee M. K., Anderson K. L., Jenkins N. A., Copeland N. G., Sisodia S. S., Rothstein J. D., Borchelt D. R., Price D. L., Cleveland D. W. (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 [DOI] [PubMed] [Google Scholar]
- 47. Valentine J. S., Doucette P. A., Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 48. Wilcox K. C., Zhou L., Jordon J. K., Huang Y., Yu Y., Redler R. L., Chen X., Caplow M., Dokholyan N. V. (2009) Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 284, 13940–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marin E. P., Derakhshan B., Lam T. T., Davalos A., Sessa W. C. (2012) Endothelial cell palmitoylproteomic identifies novel lipid-modified targets and potential substrates for protein acyl transferases. Circ. Res. 110, 1336–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 51. Watanabe S., Nagano S., Duce J., Kiaei M., Li Q. X., Tucker S. M., Tiwari A., Brown R. H., Jr., Beal M. F., Hayward L. J., Culotta V. C., Yoshihara S., Sakoda S., Bush A. I. (2007) Increased affinity for copper mediated by cysteine 111 in forms of mutant superoxide dismutase 1 linked to amyotrophic lateral sclerosis. Free Radic. Biol. Med. 42, 1534–1542 [DOI] [PubMed] [Google Scholar]
- 52. Parge H. E., Hallewell R. A., Tainer J. A. (1992) Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 89, 6109–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bruns C. K., Kopito R. R. (2007) Impaired post-translational folding of familial ALS-linked Cu,Zn superoxide dismutase mutants. EMBO J. 26, 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fujisawa T., Homma K., Yamaguchi N., Kadowaki H., Tsuburaya N., Naguro I., Matsuzawa A., Takeda K., Takahashi Y., Goto J., Tsuji S., Nishitoh H., Ichijo H. (2012) A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann. Neurol. 72, 739–749 [DOI] [PubMed] [Google Scholar]
- 55. Wang L., Deng H. X., Grisotti G., Zhai H., Siddique T., Roos R. P. (2009) Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum. Mol. Genet. 18, 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karch C. M., Prudencio M., Winkler D. D., Hart P. J., Borchelt D. R. (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 7774–7779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lepock J. R., Frey H. E., Hallewell R. A. (1990) Contribution of conformational stability and reversibility of unfolding to the increased thermostability of human and bovine superoxide dismutase mutated at free cysteines. J. Biol. Chem. 265, 21612–21618 [PubMed] [Google Scholar]
- 58. Karch C. M., Borchelt D. R. (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 13528–13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cozzolino M., Amori I., Pesaresi M. G., Ferri A., Nencini M., Carrì M. T. (2008) Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Niwa J., Yamada S., Ishigaki S., Sone J., Takahashi M., Katsuno M., Tanaka F., Doyu M., Sobue G. (2007) Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J. Biol. Chem. 282, 28087–28095 [DOI] [PubMed] [Google Scholar]
- 61. Prudencio M., Lelie H., Brown H. H., Whitelegge J. P., Valentine J. S., Borchelt D. R. (2012) A novel variant of human superoxide dismutase 1 harboring amyotrophic lateral sclerosis-associated and experimental mutations in metal-binding residues and free cysteines lacks toxicity in vivo. J. Neurochem. 121, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brotherton T., Polak M., Kelly C., Birve A., Andersen P., Marklund S. L., Glass J. D. (2011) A novel ALS SOD1 C6S mutation with implications for aggregation related toxicity and genetic counseling. Amyotroph. Lateral Scler. 12, 215–219 [DOI] [PubMed] [Google Scholar]
- 63. Morita M., Aoki M., Abe K., Hasegawa T., Sakuma R., Onodera Y., Ichikawa N., Nishizawa M., Itoyama Y. (1996) A novel two-base mutation in the Cu,Zn-superoxide dismutase gene associated with familial amyotrophic lateral sclerosis in Japan. Neurosci. Lett. 205, 79–82 [DOI] [PubMed] [Google Scholar]
- 64. Greaves J., Chamberlain L. H. (2011) DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends Biochem. Sci. 36, 245–253 [DOI] [PubMed] [Google Scholar]
- 65. Sturtz L. A., Diekert K., Jensen L. T., Lill R., Culotta V. C. (2001) A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084–38089 [DOI] [PubMed] [Google Scholar]
- 66. Urushitani M., Ezzi S. A., Matsuo A., Tooyama I., Julien J. P. (2008) The endoplasmic reticulum-Golgi pathway is a target for translocation and aggregation of mutant superoxide dismutase linked to ALS. FASEB J. 22, 2476–2487 [DOI] [PubMed] [Google Scholar]
- 67. Chang L. Y., Slot J. W., Geuze H. J., Crapo J. D. (1988) Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol. 107, 2169–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Field L. S., Furukawa Y., O'Halloran T. V., Culotta V. C. (2003) Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem. 278, 28052–28059 [DOI] [PubMed] [Google Scholar]
- 69. Kawamata H., Manfredi G. (2008) Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 17, 3303–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cozzolino M., Carrì M. T. (2012) Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66 [DOI] [PubMed] [Google Scholar]
- 71. Ilieva H., Polymenidou M., Cleveland D. W. (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhao W., Beers D. R., Henkel J. S., Zhang W., Urushitani M., Julien J. P., Appel S. H. (2010) Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 58, 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gruzman A., Wood W. L., Alpert E., Prasad M. D., Miller R. G., Rothstein J. D., Bowser R., Hamilton R., Wood T. D., Cleveland D. W., Lingappa V. R., Liu J. (2007) Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 104, 12524–12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haidet-Phillips A. M., Hester M. E., Miranda C. J., Meyer K., Braun L., Frakes A., Song S., Likhite S., Murtha M. J., Foust K. D., Rao M., Eagle A., Kammesheidt A., Christensen A., Mendell J. R., Burghes A. H., Kaspar B. K. (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 [DOI] [PMC free article] [PubMed] [Google Scholar]