

Background: The role of intracellular lipid metabolism as it relates to nutrient-coupled glucose-stimulated insulin secretion has not been fully elucidated.
Results: Knockdown of acyl-CoA synthetase isoform 4 impairs glucose-stimulated insulin secretion caused by decreased cellular epoxyeicosatrienoic acids.
Conclusion: Acyl-CoA synthetase 4 is required for optimal glucose-stimulated insulin secretion.
Significance: Understanding the role of beta-cell lipid metabolism is needed to develop novel strategies to enhance insulin secretion.
Keywords: Arachidonic Acid, Beta-Cell, Eicosanoid, Insulin Secretion, Polyunsaturated Fatty Acids, Acyl-CoA Synthetase, Epoxyeicosatrienoic Acids
Abstract
Glucose-stimulated insulin secretion (GSIS) in pancreatic beta-cells is potentiated by fatty acids (FA). The initial step in the metabolism of intracellular FA is the conversion to acyl-CoA by long chain acyl-CoA synthetases (Acsls). Because the predominantly expressed Acsl isoforms in INS 832/13 cells are Acsl4 and -5, we characterized the role of these Acsls in beta-cell function by using siRNA to knock down Acsl4 or Acsl5. Compared with control cells, an 80% suppression of Acsl4 decreased GSIS and FA-potentiated GSIS by 32 and 54%, respectively. Knockdown of Acsl5 did not alter GSIS. Acsl4 knockdown did not alter FA oxidation or long chain acyl-CoA levels. With Acsl4 knockdown, incubation with 17 mm glucose increased media epoxyeicosatrienoic acids (EETs) and reduced cell membrane levels of EETs. Further, exogenous EETs reduced GSIS in INS 832/13 cells, and in Acsl4 knockdown cells, an EET receptor antagonist partially rescued GSIS. These results strongly suggest that Acsl4 activates EETs to form EET-CoAs that are incorporated into glycerophospholipids, thereby sequestering EETs. Exposing INS 832/13 cells to arachidonate or linoleate reduced Acsl4 mRNA and protein expression and reduced GSIS. These data indicate that Acsl4 modulates GSIS by regulating the levels of unesterified EETs and that arachidonate controls the expression of its activator Acsl4.
Introduction
Insulin secretion from beta-cells in the islets of Langerhans is a complex process initiated by nutrient secretagogues, including glucose, amino acids, and fatty acids (FA).2 McGarry and co-workers (1, 2), in classic experiments, showed that FA are essential for insulin secretion, because glucose-stimulated insulin secretion (GSIS) was diminished when nicotinic acid infusion deprived rats of circulating free FA. Conversely, when plasma-free FA are elevated by an infusion of lard oil plus heparin, insulin secretion is supranormal. Other studies designed to evaluate the role of long chain acyl-CoAs in INS-1 cells and rat islets also support the concept that lipids acutely stimulate GSIS (3–5). FA appear to augment GSIS by at least two separate mechanisms: 1) activation of the G-protein-coupled receptor, GPR40, resulting in increased intracellular calcium (6, 7), and 2) increasing intracellular levels of FA metabolites (8), whose mechanism of action has remained uncertain.
Arachidonate (AA; 20:4ω6) is normally esterified to glycerophospholipids and represents ∼30% of the fatty acid mass in pancreatic islet glycerophospholipids (9). Based on studies that show that glucose stimulates the release and metabolism of AA, it has been hypothesized that AA and/or its metabolites play a significant role in insulin secretion (10–12). The initial hydrolysis of AA from glycerophospholipids seems to be critical for insulin secretion, because knockdown or knock-out of phospholipase A2 in INS-1 cells (13) or mouse islets (14) diminishes GSIS. Free AA is metabolized by one of several different enzyme systems that yield potent bioactive compounds with critical signaling roles. Isolated pancreatic islets produce prostaglandins, leukotrienes, thromboxanes, hydroxyeicosatetraenoic acids (HETEs), and epoxyeicosatrienoic acids (EETs) (10, 15, 16). Glucose stimulates the synthesis of 12-HETE, and inhibiting 12-HETE synthesis in isolated rat islets suppresses GSIS. Similarly, different EET species also alter beta-cell function. Thus, 5,6-EET, but not 8,9-, 11,12-, or 14,15-EET, stimulates insulin secretion in isolated rat islets (17), and 8,9-, 11,12-, and 14,15-EET stimulate glucagon release from the same islets. Although AA and its metabolites play a role in GSIS, it is not understood how these metabolites are regulated in the beta-cell or how they might stimulate or impair insulin secretion.
Central to the intracellular metabolism of fatty acids is their activation by one of a family of five long chain acyl-CoA synthetases (Acsl1, 3–6). Each of the long chain acyl-CoA synthetases is encoded by a separate gene and differs in substrate preference, enzyme kinetics, subcellular location, and tissue-specific expression (18). Activation to an acyl-CoA is required before FA can be oxidized for ATP production, esterified for the synthesis of glycerolipids or cholesterol esters, or used in a signaling pathway. Both loss of function and gain of function studies suggest that each Acsl isoform is distinct in directing acyl-CoAs to one or more specific pathways. For example, Acsl1 deficiency in adipose or heart does not alter glycerolipid synthesis but profoundly diminishes FA oxidation (19, 20).
A stimulatory role for acyl-CoAs in GSIS has been hypothesized (3), but the Acsl isoforms that synthesize acyl-CoAs and their role in regulating FA flux and FA metabolites in beta-cells have not been directly examined. Using INS 832/13 insulinoma cells, we show that knocking down Acsl4 impairs GSIS by increasing free EETs, that direct incubation of INS 832/13 cells with EETs reduces insulin secretion, and that Acsl4 activates EETs to produce EET-CoAs that may then be re-esterified into glycerophospholipids.
EXPERIMENTAL PROCEDURES
Materials
[1-14C]Palmitic acid and [1-14C]oleic acid were purchased from PerkinElmer Life Sciences (Waltham, MA). [1-14C]Arachidonic acid was purchased from American Radiolabeled Chemicals (St. Louis, MO). (±)-5,6-EET, (±)-8,9-EET, (±)-11,12-EET, (±)-14,15-EET, and 14,15-EEZE were purchased from Cayman Chemical (Ann Arbor, MI). All other chemicals and reagents were purchased from Sigma unless otherwise specified. Silica-gel G plates were from Whatman (Springfield Mill, UK).
Cell Culture
INS 832/13 rat insulinoma cells were cultured (21) and used at passage numbers between 43 and 60.
siRNA Transfection
Duplex siRNAs were designed and synthesized by Integrated DNA Technologies (Coralville, IA). Targets against rat Acsl4 (GenBankTM accession number NM_053623.1) were nucleotide positions 283–307 and 3377–3401 for siAcsl4-1 and siAcsl4-2, respectively. The target sequence against rat Acsl5 (GenBankTM accession number NM_053607.1) was nucleotide positions 784–808. Nonspecific siRNA, siControl (AAGTCGGTTAAACGTTGGCAT), served as control. The cells were transfected with DharmaFect reagent 1 (Dharmacon Products, Lafayette, CO) per the manufacturer's specifications with a final duplex siRNA concentration of 25 nm. Briefly, 24 h after cells were plated at the appropriate densities, they were treated with siRNAs for 48 h, after which the medium was changed, and experiments were performed on the treated cells 24 h later.
Quantitative Real Time RT-PCR
Total RNA was extracted from INS 832/13 cells using TRIzol (Invitrogen) and reverse transcribed with a high capacity cDNA archive kit (Applied Biosystems, Foster City, CA). cDNA was amplified by real time PCR in a total reaction volume of 25 μl using Absolute QPCR Sybr Green Fluorescein Mix (ThermoScientific, Waltham, MA). Primer sets are identified in Table 1. PCR amplifications were performed in an iCycler detection system (Bio-Rad). Target gene expression was normalized to endogenous beta-actin and expressed as 2−ΔΔCt.
TABLE 1.
| Gene | Forward primer | Reverse primer | GenBankTM accession number |
|---|---|---|---|
| β-Actin | GGCTCCTAGCACCATGAAGA | GAAAGGGTGTAAAACGCAGC | NM_031144 |
| Acsl1 | GTTCCGAAGAATGACCTGGA | TTCTGAAGACATGCTGTGCC | NM_012820.1 |
| Acsl3 | CTCCACTTTCTGCAACGACA | TCTGCCGGTATTGTAGTCCC | NM_057107.1 |
| Acsl4 | GAGGCTGAATTGCTCCTTTG | TCAGCAACAGCAAACAGACC | NM_053623.1 |
| Acsl5 | TGACACACTGGGAGCAGAAG | CAGGTCATCATCGAAAGGGT | NM_053607.1 |
| Acsl6 | TGGGGAAAGAAAGTGGACTG | AAACTTTGCTGGGCTGAGAA | NM_130739.1 |
| Cyp2j4 | CGCACCGCATCTGTGCTGTC | GCAAAGGCACGCCACTGAGC | NM_023025.2 |
| Ephx2 | GGCGCTGCCCAGAGACTTCC | GCCTGAAGCATGGGGCGGTT | NM_022936.1 |
Western Blot Analysis
The cells were homogenized in lysis buffer (20 mm Tris, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 50 mm NaF, 2.5 mm Na4P2O7·10 H2O, 1% Triton X-100, and Complete Mini Protease Inhibitor Mixture; Roche Applied Science, Indianapolis, IN). Equal amounts of protein (40 μg) were loaded and resolved on 8% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were probed with antibodies against Acsl4 (Santa Cruz Biotechnology, Dallas, TX) or rabbit anti-human ACSL4 (22), a generous gift from S. M. Prescott (University of Utah, Salt Lake, UT), Acsl5 (Novus Biologics, Littleton, CO), and Gapdh for the loading control (Pierce). Densitometry was performed using ImageJ, where the protein of interest was normalized to Gapdh loading control.
ACS Activity Assay
ACS activity was measured in INS 832/13 cell total membrane fractions (4 to 10 μg) at 25 °C using an isotopic method with either 50 μm [1-14C]palmitic acid, [1-14C]oleic acid, or [1-14C]arachidonic acid, 10 mm ATP, and 0.25 mm CoA (23). An indirect ACS activity assay using synthesized and purified recombinant rat FLAG-Acsl4 (24) with different substrates was performed spectrophotometrically (25).
Flux Studies
Incorporation of oleate or AA into cellular lipids was measured in INS 832/13 cells after siAcsl4 knockdown. Using 6-well plates, transfected cells were incubated for 4 h with 0.25 μCi of [1-14C]oleic acid or [1-14C]arachidonic acid in the presence of 100 μm unlabeled oleate or AA, respectively. The lipids were extracted and separated by thin layer chromatography (26). Labeled lipid products were visualized and quantified using a BioScan Image 200 System (Washington, DC).
Insulin Secretion Studies
To assay insulin secretion, INS 832/13 cells were transfected as described above at 3.5 × 105 cells/well in 12-well tissue culture dishes. After 72 h, the cells were washed in Hanks' balanced salt solution with 0.2% bovine serum albumin and 3 mm glucose followed by preincubation in 1 ml of the same buffer for 1 h. Insulin secretion was measured by static incubation of the cells for 2 h in 1 ml of Hanks' balanced salt solution containing glucose, oleate/palmitate (2:1 molar ratio), oleate, palmitate, AA, KCl, or 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE; Cayman Chemicals, Ann Arbor, MI) at the concentrations indicated in the figure legends. In experiments with EET exposure, INS 832/13 cells were grown to confluence in 12-well plates and exposed to 1 μm EET species in both the preincubation buffer and the stimulation buffer. Insulin released into the stimulation buffer was measured using an enzyme-linked immunosorbent assay (rat/mouse insulin ELISA; Millipore, Billerica, MA, catalog number EZRMI-13K) and normalized to total cellular protein.
Measurement of Palmitate Oxidation
Fatty acid oxidation was determined by measuring 14CO2 and acid-soluble metabolites produced from [1-14C]palmitate in siControl and siAcsl4 cells (19).
Extraction and Analysis of Media and Cellular Eicosanoids
Following a 48-h siRNA treatment, the cell medium was changed. Twenty-four hours later the medium was collected and stored on ice until extraction. Cell lipids were saponified (27). Briefly, the cells were scraped from plates with 0.2 n methanolic NaOH and heated to 50 °C for 2 h. After cooling to room temperature, PBS was added to bring the pH to ∼8.0. The saponified lipid products and media lipids were extracted twice with ethyl ether after adding internal standards (30 ng of prostaglandin E2-d4, 10,11-dihydroxynonadecanoic acid, and 10,11-epoxyheptadecanoic acid (Cayman Chemicals)) and butylated hydroxytoluene. Media and cellular eicosanoid levels were quantified by liquid chromatography with an Agilent 1200 Series capillary HPLC (Agilent Technologies, Santa Clara, CA). The samples were analyzed in triplicate. Negative ion electrospray ionization tandem mass spectrometry was used for detection. Analyses were performed on an MDS Sciex API 3000 equipped with a TurboIonSpray source (Applied Biosystems, Foster City, CA) (28).
Extraction and Quantification of Long Chain Acyl-CoAs
Cellular long chain acyl-CoAs were extracted, purified, and analyzed as described (29–31). The acyl-CoAs were analyzed by flow injection analysis using positive electrospray ionization on Quattro micro, triple quadrupole mass spectrometer (Waters, Milford, MA). Heptadecanoyl CoA was employed as an internal standard.
Statistical Analysis
The data are expressed as means ± S.E. The significance of data was declared at p < 0.05 by Student's t test.
RESULTS
Acsl5 siRNA Suppresses Acsl5 mRNA and Protein but Does Not Affect GSIS
Profiling of Acsl mRNA expression by quantitative PCR in INS-1 832/13 revealed that Acsl4 and -5 are the predominantly expressed Acsl isoforms; Acsl1, -3, and -6 were expressed at lower levels (Fig. 1A). To determine the role of the major Acsls in INS 832/13 cell lipid metabolism and to determine whether their activity plays a role in insulin secretion, we used siRNAs to knock down either Acsl4 or Acsl5. A siRNA targeted against Acsl5 reduced the level of Acsl5 mRNA by more than 80% and reduced Acsl5 protein expression by ∼30% relative to siControl (Fig. 1, B and C), but the expression of Acsl4 mRNA did not significantly change, and glucose-stimulated insulin secretion was not significantly altered (Fig. 1D). These data indicate that, despite being expressed at a level higher than any of the other Acsl isoforms, Acsl5 in INS 832/13 cells does not play a role in GSIS.
FIGURE 1.

Acsl4 and Acsl5 are the predominant Acsl isoforms in INS 832/13 cells. Glucose-stimulated insulin secretion was not reduced by Acsl5 knockdown. A, relative mRNA expression of Acsl isoforms in INS 832/13 cells. B, a siRNA targeted against Acsl5 reduced mRNA expression by >90%. C, corresponding reduction of Acsl5 protein expression by ∼30% as quantified by densitometry relative to Gapdh loading control. A.U., arbitrary units. D, siRNA knockdown of Acsl5 had no significant effect on glucose stimulated insulin secretion with or without KCl. The results represent means ± S.E. of three separate experiments carried out at least in triplicate. *, p < 0.05 relative to siControl.
Acsl4 siRNA Effectively Suppressed Acsl4 Expression and Cellular Acsl Activity
To determine the role of Acsl4 in INS 832/13 cell lipid metabolism and insulin secretion, two siRNAs targeted against Acsl4 reduced the level of Acsl4 mRNA by more than 75% (Fig. 2A). This knockdown of Acsl4 did not significantly change the expression of Acsl5 mRNA, indicating that Acsl5 did not compensate for the Acsl4 deficiency. Likewise, the other Acsl isoforms (1, 3, and 6) and fatty acid transport proteins (Fatp4 and Fatp5) mRNAs were unaffected (data not shown). Both of the Acsl4 siRNAs reduced the level of Acsl4 protein expression after transfection by greater than 80% (Fig. 2B). Acsl specific activity in total cell membranes was then measured with three different FA substrates, AA, palmitate, and oleate (32). Knockdown of Acsl4 significantly decreased Acsl specific activity by 57% for AA, 46% for palmitate, and 53% for oleate (Fig. 2C). Residual Acsl activity results from the other Acsl isoforms that are still expressed and remain functional. Interestingly, compared with both palmitate and oleate, AA gave the highest Acsl specific activity, as suggested previously (32), and its activity was diminished the most by siAcsl4. These data suggest that in INS 832/13 cells, Acsl4 contributes approximately half of the total cellular Acsl activity.
FIGURE 2.

Acsl4 siRNA knockdown in INS 832/13 cells reduced Acsl4 mRNA expression, Acsl4 protein expression, acyl-CoA synthetase activity, glucose-stimulated insulin secretion, and fatty acid augmented insulin secretion. A, two separate siRNAs targeted against Acsl4 reduced mRNA expression by more than 75%. B, corresponding reduction of Acsl4 protein expression with Acsl4 siRNA knockdown by two independent Acsl4 siRNAs. Duplicate samples were examined with knockdown of >80% for each Acsl4 siRNA as quantified by densitometry relative to Gapdh loading control. A.U., arbitrary units. C, Acsl specific activity from siControl and siAcsl4-treated INS 832/13 cells with AA, palmitate (PA), and oleate (OA) as substrates. D, glucose-stimulated insulin secretion with or without KCl and fatty acid augmented insulin secretion with 2:1 oleate/palmitate (OA/PA) at the concentrations indicated. The results represent means ± S.E. of three separate experiments carried out at least in triplicate. *, p < 0.05 relative to siControl.
Acsl4 Knockdown Reduced Both GSIS and FA-augmented GSIS
Given the substrate preference and tissue distribution of Acsl4 (32, 33), we evaluated the effect of Acsl4 knockdown on insulin secretion. In the presence of various secretagogues, including glucose, specific fatty acids, and KCl, knockdown of Acsl4 expression significantly reduced insulin secretion (Fig. 2D). In siAcsl4 knockdown cells stimulated solely with 17 mm glucose, GSIS was reduced by 32%. Further, the addition of a combination of oleate and palmitate in a physiologic 2:1 unsaturated:saturated molar ratio increased insulin secretion relative to the effect of glucose alone. However, in all treatment groups compared with the siControl cells, the siAcsl4 cells secreted less insulin. In both siControl and siAcsl4 cells, the differential rise in insulin secretion with exogenous FA was likely due to the known effects of GPR40 on FA-augmented insulin secretion (7). Likewise, in siAcsl4 cells relative to siControl, the reduction in insulin secretion was probably due to generation of FA metabolites with signaling properties formed from AA. These results indicate that a single specific Acsl isoform, Acsl4, mediates the majority of intracellular Acsl activity and that normal GSIS requires Acsl4 activity.
Knockdown of Acsl4 Altered the Incorporation of Arachidonate into Neutral Lipids and Increased Palmitate Oxidation
To further characterize the consequences of Acsl4 knockdown, we evaluated the incorporation of oleate and AA into neutral lipids and phospholipids. Incorporation of oleate into total glycerolipids was not affected by the knockdown of Acsl4 (Fig. 3A), and no changes were observed in the incorporation of oleate into neutral lipids (triacylglycerol, diacylglycerol, or cholesterol esters) (Fig. 3C), or phospholipids (Fig. 3E). However, Acsl4 knockdown resulted in 20% lower incorporation of AA into total lipids (Fig. 3B) with a 51% reduction in its incorporation into neutral lipids (Fig. 3D), without altering the incorporation of AA into phospholipids (Fig. 3F). These results suggest that Acsl4 preferentially channels arachidonate toward the synthesis of specific intracellular glycerolipids.
FIGURE 3.

Incorporation of oleate into INS 832/13 cellular lipids was not affected by Acsl4 knockdown; however, Acsl4 knockdown reduced incorporation of AA into neutral lipid while preserving phospholipid levels. Acsl4 knockdown did not affect palmitate (PA) oxidation. A, C, and E, oleate (OA) incorporation into total cellular lipids (A), neutral lipids (C), and phospholipids (E). B, D, and F, AA incorporation into total cellular lipids (B), neutral lipids (D), and phospholipids (F). G and H, PA incorporation into CO2 (G) and PA incorporation into acid-soluble metabolites (ASM) (H). CE, cholesterol esters; TAG, triacylglycerol; DAG, diacylglycerol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PI/S, combination of phosphatidylinositol/phosphatidylserine. The values are reported as means ± S.E. from three separate experiments carried out at least in triplicate. *, p < 0.05 relative to siControl.
We evaluated the effect of Acsl4 knockdown on the oxidation of palmitate at basal and stimulatory glucose concentrations (3 or 17 mm glucose) to determine whether a defect in FA oxidation might have reduced insulin secretion. Consistent with previous studies (34), in siControl and siAcsl4 cells, the oxidation of palmitate was greater at low glucose concentrations than at higher glucose concentrations (Fig. 3, G and H). Surprisingly, however, at both 3 and 17 mm glucose, knockdown of Acsl4 increased the oxidation of palmitate. Thus, a defect in FA oxidation did not cause the reduced GSIS of the Acsl4 knockdown. Additionally, these studies suggest that knocking down Acsl4 diverted palmitate toward oxidative metabolism.
Knockdown of Acsl4 Did Not Alter Content of Total CoA or Long Chain Acyl-CoA Species
To understand whether lack of Acsl4 would alter the fatty acid composition of INS 832/13 cells, we measured both total CoA and long chain acyl-CoA species by LC-MS/MS, expecting that the knockdown of Acsl4 would result in a decrease in long chain acyl-CoA species, especially arachidonyl-CoA. However, siControl and siAcsl4 cells did not significantly differ in their content of either total CoA or long chain acyl-CoA species (Fig. 4, A and B) compared with control vehicle-treated cells that were devoid of supplemental FA. To ensure that the knockdown of Acsl4 was specific for the reduction in arachidonyl-CoA, siControl and siAcsl4 cells were incubated for 4 h with either 50 μm oleate or AA. The addition of exogenous FA increased the total acyl-CoA content of both siControl and siAcsl4-treated cells (Fig. 4A). The addition of exogenous oleate to siAcsl4 cells had no effect on any of the individual acyl-CoAs, relative to siControl cells (Fig. 4C). However, adding exogenous AA to siAcsl4 cells specifically reduced arachidonyl-CoA, relative to siControl cells (Fig. 4D), confirming the known AA substrate preference of Acsl4 (32). Because it was necessary to add exogenous AA to observe the reduced arachidonyl-CoA in the siAcsl4 cells and because the content of long chain acyl-CoAs did not change, especially in the vehicle-treated cells, these findings suggest that long chain acyl-CoAs do not regulate insulin release directly. Instead, insulin secretion appeared to be mediated by a lipid intermediate whose levels were altered by Acsl4.
FIGURE 4.

Knockdown of Acsl4 did not change cellular total acyl-CoA levels except with the addition of exogenous AA where Acsl4 knockdown reduced arachidonoyl-CoA levels. A, effect of Acsl4 knockdown in INS 832/13 cells on total CoA species after a 4-h incubation with either vehicle or 50 μm oleate (OA) or 50 μm AA (final concentration). B, long chain acyl-CoA species from vehicle control. C, long chain acyl-CoA species from 50 μm oleate treated cells. D, long chain acyl-CoA species from 50 μm arachidonate-treated cells. 16:1-CoA, palmitoleoyl-CoA; 16:0-CoA, palmitoyl-CoA; 18:3-CoA, linolenoyl-CoA; 18:2-CoA, linoleoyl-CoA; 18:1-CoA, oleoyl-CoA; 18:0-CoA, stearoyl-CoA; 20:4-CoA, arachidonoyl-CoA; 20:1-CoA, eicosanoyl-CoA; 20-CoA, arachidoyl-CoA. The values are reported as means ± S.E. from three separate experiments carried out in triplicate. *, p < 0.05 relative to siControl.
Acsl4 Modulates Epoxyeicosatrienoic Acid Incorporation into Cell Membranes
To determine the lipid intermediates responsible for reduced GSIS, we profiled AA metabolites in the media from Acsl4 knockdown cells. The media from siControl or siAcsl4-treated cells did not contain significantly different levels of prostaglandins, thromboxanes, or HETEs (Fig. 5, A and B). However, compared with media from the siControl cells, media from the siAcsl4 cells contained 81% more total EETs (Fig. 5C). Likewise, compared with the siControl cells, the cell membranes from siAcsl4 cells contained 69% less total EETs (Fig. 5D). The Acsl4 knockdown did not alter the mRNA expression of soluble epoxide hydrolase (Ephx2) or cytochrome P-450 2j4 (Cyp2j4), the enzymes responsible for the degradation and synthesis of EETs, respectively (Fig. 5E). Additionally, the amounts of dihydroxyeicosatrienoic acids (DHETs), the breakdown products of EETs by soluble epoxide hydrolase (sEH), were elevated in the siAcsl4 cells to the same extent as the free EETs (Fig. 5F), suggesting that Acsl4 knockdown did not alter sEH activity.
FIGURE 5.

Acsl4 knockdown did not alter the formation of thromboxanes, prostaglandins, or HETEs. However, knockdown of Acsl4 increased media EET levels, whereas cellular membrane EET levels were reduced. Acsl4 knockdown increased the formation of DHETs, indicating that sEH activity is intact. Purified rat Acsl4 is able to use EETs as a substrate. A, thromboxane B2 (TXB2), prostaglandin D2 (PGD2), and prostaglandin B2 (PGB2) levels in media surrounding INS 832/13 cells after knockdown of Acsl4. B, HETE levels in media surrounding INS 832/13 cells after knockdown of Acsl4. C, EET levels in media surrounding INS 832/13 cells after knockdown of Acsl4. D, cellular membrane content of EET levels in INS 832/13 cells after knockdown of Acsl4. E, effect of Acsl4 knockdown on the mRNA expression of Ephx2 and Cyp2j4. F, DHET levels in media surrounding INS 832/13 cells after knockdown of Acsl4. G, purified rat Acsl4 enzyme specific activity using AA and EETs as substrates. The values are reported as means ± S.E. from two separate experiments carried out at least in triplicate. *, p < 0.05 relative to siControl. Because the ability to detect 11,12-EET was not sufficiently sensitive, these data are not shown in C and D.
EETs are incorporated into the sn-2 position of glycerophospholipids through a CoA-dependent process (35, 36). Our data suggest that Acsl4 is the isoform responsible for activating EETs to form EET-CoAs, thereby facilitating their esterification into glycerophospholipids by an as yet unknown acyltransferase. To confirm the ability of Acsl4 to use EETs as substrates, we measured the activity of purified rat Acsl4 with each of the four EET species. All EETs were substrates for Acsl4 (Fig. 5G). Although the specific activity of rat Acsl4 with EETs was ∼50% lower than with AA, distinct EET preferences were observed. Rat Acsl4 preferred 8,9-EET > 14,15-EET > 5,6-EET > 11,12-EET. Taken together, these results suggest that Acsl4 activates EETs before they are esterified into glycerophospholipids.
EETs Inhibit GSIS and Inhibiting EET Actions Rescue the Reduced GSIS of Acsl4 Knockdown
Because knockdown of Acsl4 increased free EETs in the media, we asked whether the unesterified EETs provided a mechanism for the reduced insulin secretion observed when Acsl4 activity was decreased. We used the EET concentrations (1 μm) that were previously used in rat islet experiments (17). At basal glucose concentrations, the EET species did not alter insulin secretion (Fig. 6A). However compared with vehicle-exposed cells at 17 mm glucose, 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET each diminished insulin secretion by as much as 30%. Apart from 8,9-EET, the effect was lost when 40 mm KCl was added. It is likely that exogenous application of EETs did not diminish insulin secretion as strongly as observed with the Acsl4 knockdown (Fig. 2D) because the exogenous addition did not directly target the EETs to the insulin secretion machinery within the cell.
FIGURE 6.

INS 832/13 cells exposed to EETs reduced GSIS and an inhibitor of EET action partially rescued the impaired GSIS in Acsl4 knockdown cells. A, Reduced GSIS in INS 832/13 cells exposed to EETs (±) 5,6-, 8,9-, 11,12-, or 14,15-EET at 1 μm final concentration with or without 40 mm KCl. B, reduced GSIS by Acsl4 knockdown was partially rescued with the addition of 10 μm 14,15-EEZE, an antagonist to the putative EET receptor. The values are reported as means ± S.E. from three separate experiments carried out at least in triplicate. *, p < 0.05 relative to siControl. The bar indicates difference between siAcsl4-treated cells exposed to EEZE relative to vehicle.
To further confirm the role of EETs on GSIS, we exposed siControl and siAcsl4-treated cells to 14,15-EEZE, an antagonist to the putative EET receptor (37). 14,15-EEZE increased insulin secretion 28 and 57% at 3 and 17 mm glucose, respectively, in the siAcsl4 cells (Fig. 6B), supporting the idea that EETs suppress insulin secretion. Taken together, these results suggest that 5,6-, 8,9-, 11,12-, and 14,15-EETs inhibit GSIS and that inhibiting EET action rescues the ability of siAcsl4 cells to secrete insulin in the presence of a stimulatory concentration of glucose.
Incubating INS 832/13 Cells with Polyunsaturated Fatty Acid (PUFA) Reduced Both Acsl4 Expression and GSIS
Chronic exposure of INS 832/13 cells to exogenous FA reduces GSIS (28). To determine the effect of FA on Acsl4 expression, we incubated INS 832/13 cells with 0.5–1 mm concentrations of either oleate:palmitate (2:1 unsaturated:saturated molar ratio) or oleate:palmitate:AA (6:3:1 molar ratio) for 72 h. AA was added to the oleate:palmitate incubation because PUFAs are present in the typical Western diet and associated with metabolic disease (38). Although the oleate:palmitate combination had no effect on Acsl4 protein expression, the combination of oleate:palmitate:AA resulted in >50% reduction in Acsl4 protein expression (data not shown). Thus, at plausible physiologic concentrations of AA (50 μm) (38), Acsl4 protein expression was reduced. When INS 832/13 cells were incubated for 72 h with 10, 25, or 50 μm palmitate, oleate, AA, or linoleate, the AA and linoleate reduced Acsl4 mRNA expression (Fig. 7A) and Acsl4 protein expression (Fig. 7B) in a dose-dependent manner. This reduction in Acsl4 expression corresponded to a ∼20% reduction in GSIS at 3 mm glucose and ∼40% reduction at 17 mm glucose (Fig. 7C). These experiments indicate that PUFAs decrease Acsl4 expression, which then reduces GSIS similar to the reduction observed with selective knockdown of Acsl4.
FIGURE 7.

Incubation of INS 832/13 cells with AA or linoleate reduced, in a dose-dependent manner, the expression of Acsl4 mRNA and protein that caused a reduction in glucose-stimulated insulin secretion. A, Acsl4 mRNA expression in INS 832/13 cells exposed to 10, 25, or 50 μm oleate (OA), AA, or linoleate (LA) for 72 h. B, representative Western blot of corresponding Acsl4 protein expression in INS 832/13 cells exposed to 10, 25, or 50 μm palmitate (PA), oleate, arachidonate, or linoleate for 72 h. Acsl4 protein expression in the 50 μm AA- and LA-treated cells is reduced by >70% relative to vehicle-treated cells as quantified by densitometry relative to Gapdh loading control. A.U., arbitrary units. C, glucose-stimulated insulin secretion in 832/13 cells exposed to 10, 25, or 50 μm oleate, arachidonate, or linoleate for 72 h with or without 40 mm KCl. The values are reported as means ± S.E. from three separate experiments carried out at least in triplicate. *, p < 0.05 relative to vehicle.
DISCUSSION
The major finding of this study is that unesterified EETs inhibit GSIS. Acsl4 promotes GSIS by producing EET-CoAs that are then esterified into glycerolipids. Previous studies to determine the role of long chain acyl-CoAs in GSIS used triacsin C, an inhibitor of acyl-CoA synthetase activity, but yielded conflicting results (39, 40). Because inhibiting acyl-CoA synthetase activity with triacsin C reduced insulin secretion in one of these studies, it was thought that long chain acyl-CoAs themselves were important mechanistically in coupling FA metabolism and augmented insulin secretion (40). Our studies suggest, instead, that an eicosanoid substrate of Acsl4 is an inhibitor.
Despite previous studies that concluded that acyl-CoAs were important for insulin secretion, no studies have directly evaluated the role of acyl-CoA synthetases in insulin secretion. Acyl-CoA synthetases have different substrate preferences, enzyme kinetics, subcellular locations, and tissue-specific expression (18). Using a beta-cell model, INS 832/13 cells, we found that, of the five Acsl isoforms, the mRNAs for Acsl4 and Acsl5 were the most prominent. To determine the role of acyl-CoA synthetases in INS 832/13 cells, we selectively knocked down either Acsl4 or Acsl5 using siRNAs. Knockdown of Acsl5 did not result in changes in GSIS, indicating that Acsl5 and its resultant acyl-CoA products do not play a direct role in GSIS. However, the knockdown of Acsl4 resulted in reduced GSIS without changes in acyl-CoA content, indicating that the mechanism for the reduced GSIS was not via a lack of either a specific long chain acyl-CoA or a pool of acyl-CoAs synthesized by Acsl4.
GSIS is a complicated process involving the metabolism of glucose leading to a rise in ATP, depolarization events, insulin granule translocation, and insulin exocytosis. Our data indicate that the role of Acsl4 and EETs in the GSIS cascade is distal to the depolarization event because GSIS in siAcsl4-treated cells was reduced with 40 mm KCl treatment. With a rise in intracellular calcium, cytosolic phospholipase A2 is activated leading to hydrolysis of lipids from the sn-2 position of glycerophospholipids. Under normal conditions, the AA that is metabolized by the cytochrome P450 (CYP) pathway to produce EETs, or the EETs that are released from glycerophospholipids by lipases serve to dampen insulin secretion, thus preventing untoward insulin release and hypoglycemia. The levels of free EETs are modulated either by rapid inactivation by sEH or by re-esterification by Acsl4. If, however, Acsl4 is absent, the concentration of free EETs increases and diminishes GSIS (Fig. 8). Thus, in siAcsl4 cells, the reduction in GSIS appears to result from increased levels of unesterified EETs. Our studies strongly suggest that Acsl4 normally synthesizes EET-CoAs that are incorporated into glycerophospholipids (35, 36), thereby moderating any rise in EETs. Our data suggest that the sequestration of EETs into glycerophospholipids is a major route of metabolism of EETs in INS 832/13 cells.
FIGURE 8.
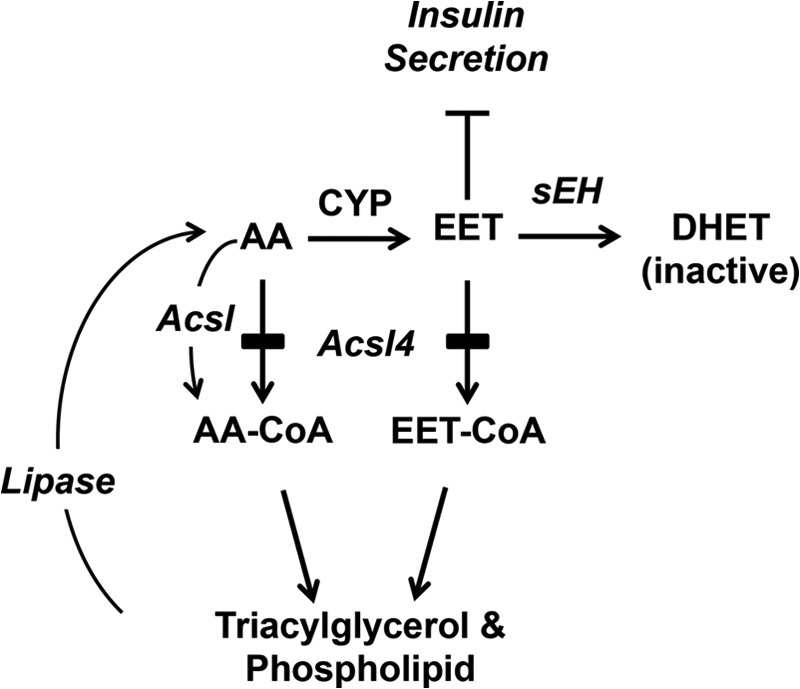
Pathway showing the proposed critical role of Acsl4 in diverting AA away from cytochrome P450 oxidation (CYP) to prevent the over production of EETs that reduce glucose-stimulated insulin secretion. Acsl4 normally generates EET-CoAs, allowing rapid and efficient re-esterification of EETs into glycerophospholipids. The remaining free EETs are further metabolized by sEH to inactive DHETs. Selective Acsl4 knockdown shunts EETs away from re-esterification, thereby increasing free EETs, which reduce insulin secretion.
Our finding that EETs inhibit insulin secretion contrasts with a previous study showing that 5,6-EET increases insulin secretion from rat islets (17). In this study, however, 8,9-EET, 11,12-EET, and 14,15-EET increased glucagon secretion, but not insulin secretion. Additionally, the insulin stimulatory effect seen with 5,6-EET may not be due to EET-like effects but rather to a prostaglandin-like effect because 5,6-EET is converted to 5,6-epoxy-prostaglandin E1 (41), and exogenous prostaglandins stimulate GSIS (42–44). The differences in these studies are most likely due to the presence of glucagon-secreting alpha-cells in the rat islets, in contrast to our studies with a pure beta-cell line. Another study in mice lacking soluble epoxide hydrolase (45), the enzyme that converts EET to inactive DHET, suggested that EETs might stimulate insulin secretion; membrane and free EETs, however, were not measured in this study. Our data in a beta-cell model suggest that, despite normal sEH activity as measured by DHET production, the absence of Acsl4 allowed EETs to accumulate and reduce insulin secretion.
The uptake and storage of FA metabolites has been implicated as a cause of beta-cell dysfunction. However, most experiments have used supraphysiologic levels of FA, usually palmitate, which is toxic when given alone to cells (46, 47). We show here that chronic exposure (72 h) at a physiologic level of the omega-6 PUFAs AA or linoleate to INS 832/13 cells reduces Acsl4 expression in a dose-dependent manner together with a reduction in GSIS. These findings open the possibility that, in addition to other environmental factors, a specific class of FA contributes to the development of diabetes by reducing Acsl4 expression, whose activity moderates the content of specific oxidized lipid metabolites (EETs), thereby leading to decreased insulin secretion.
Our study shows that Acsl isoform 4, which activates AA and EETs, enhances GSIS. The fact that EETs diminish GSIS in INS 832/13 cells suggests that this effect must be regulated by converting EETs into EET-CoAs that can then be sequestered in glycerophospholipids and that optimal GSIS is maintained by this cycle of EET metabolism. The data presented here shift the paradigm that lipid intermediates stimulate insulin secretion to one identifying specific lipid intermediates that act as repressors of insulin secretion.
This work was supported, in whole or in part, by National Institutes of Health Grants K08DK090141 (to E. L. K.), DK59935 (to R. A. C.), and DK058398 (to C. B. N.). This work was also supported by University of North Carolina Nutrition Obesity Research Center (NORC) Grant DK056350 and by Grant Z01 ES025034 from the Intramural Research Program of the National Institutes of Health, NIEHS (to D. C. Z.).
- FA
- fatty acids
- GSIS
- glucose-stimulated insulin secretion
- Acsl
- long chain acyl-CoA synthetase
- EETs
- epoxyeicosatrienoic acids
- AA
- arachidonate
- HETE
- hydroxyeicosatetraenoic acid
- 14,15-EEZE
- 14,15-epoxyeicosa-5(Z)-enoic acid
- PUFA
- polyunsaturated fatty acid
- DHET
- dihydroxyeicosatrienoic acid
- sEH
- soluble epoxide hydrolase.
REFERENCES
- 1. McGarry J. D., Dobbins R. L. (1999) Fatty acids, lipotoxicity and insulin secretion. Diabetologia 42, 128–138 [DOI] [PubMed] [Google Scholar]
- 2. Stein D. T., Esser V., Stevenson B. E., Lane K. E., Whiteside J. H., Daniels M. B., Chen S., McGarry J. D. (1996) Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J. Clin. Invest. 97, 2728–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corkey B. E., Deeney J. T., Yaney G. C., Tornheim K., Prentki M. (2000) The role of long chain fatty acyl-CoA esters in beta-cell signal transduction. J. Nutr. 130, 299S-304S [DOI] [PubMed] [Google Scholar]
- 4. Corkey B. E., Glennon M. C., Chen K. S., Deeney J. T., Matschinsky F. M., Prentki M. (1989) A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic beta-cells. J. Biol. Chem. 264, 21608–21612 [PubMed] [Google Scholar]
- 5. Prentki M., Vischer S., Glennon M. C., Regazzi R., Deeney J. T., Corkey B. E. (1992) Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J. Biol. Chem. 267, 5802–5810 [PubMed] [Google Scholar]
- 6. Alquier T., Peyot M. L., Latour M. G., Kebede M., Sorensen C. M., Gesta S., Kahn C. R., Smith R. D., Jetton T. L., Metz T. O., Prentki M., Poitout V. (2009) Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 58, 2607–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., Matsumura F., Noguchi Y., Shinohara T., Hinuma S., Fujisawa Y., Fujino M. (2003) Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173–176 [DOI] [PubMed] [Google Scholar]
- 8. Yaney G. C., Corkey B. E. (2003) Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 46, 1297–1312 [DOI] [PubMed] [Google Scholar]
- 9. Ramanadham S., Bohrer A., Mueller M., Jett P., Gross R. W., Turk J. (1993) Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: prominence of plasmenylethanolamine molecular species. Biochemistry 32, 5339–5351 [DOI] [PubMed] [Google Scholar]
- 10. Turk J., Colca J. R., Kotagal N., McDaniel M. L. (1984) Arachidonic acid metabolism in isolated pancreatic islets. II. The effects of glucose and of inhibitors of arachidonate metabolism on insulin secretion and metabolite synthesis. Biochim. Biophys. Acta 794, 125–136 [DOI] [PubMed] [Google Scholar]
- 11. Wolf B. A., Turk J., Sherman W. R., McDaniel M. L. (1986) Intracellular Ca2+ mobilization by arachidonic acid. Comparison with myo-inositol 1,4,5-trisphosphate in isolated pancreatic islets. J. Biol. Chem. 261, 3501–3511 [PubMed] [Google Scholar]
- 12. Wolf B. A., Pasquale S. M., Turk J. (1991) Free fatty acid accumulation in secretagogue-stimulated pancreatic islets and effects of arachidonate on depolarization-induced insulin secretion. Biochemistry 30, 6372–6379 [DOI] [PubMed] [Google Scholar]
- 13. Bao S., Bohrer A., Ramanadham S., Jin W., Zhang S., Turk J. (2006) Effects of stable suppression of Group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. J. Biol. Chem. 281, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bao S., Song H., Wohltmann M., Ramanadham S., Jin W., Bohrer A., Turk J. (2006) Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J. Biol. Chem. 281, 20958–20973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kelly K. L., Laychock S. G. (1981) Prostaglandin synthesis and metabolism in isolated pancreatic islets of the rat. Prostaglandins 21, 759–769 [DOI] [PubMed] [Google Scholar]
- 16. Zeldin D. C., Foley J., Boyle J. E., Moomaw C. R., Tomer K. B., Parker C., Steenbergen C., Wu S. (1997) Predominant expression of an arachidonate epoxygenase in islets of Langerhans cells in human and rat pancreas. Endocrinology 138, 1338–1346 [DOI] [PubMed] [Google Scholar]
- 17. Falck J. R., Manna S., Moltz J., Chacos N., Capdevila J. (1983) Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem. Biophys. Res. Commun. 114, 743–749 [DOI] [PubMed] [Google Scholar]
- 18. Coleman R. A., Lewin T. M., Van Horn C. G., Gonzalez-Baro M. R. (2002) Do long chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J. Nutr. 132, 2123–2126 [DOI] [PubMed] [Google Scholar]
- 19. Ellis J. M., Mentock S. M., Depetrillo M. A., Koves T. R., Sen S., Watkins S. M., Muoio D. M., Cline G. W., Taegtmeyer H., Shulman G. I., Willis M. S., Coleman R. A. (2011) Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol. Cell. Biol. 31, 1252–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li L. O., Ellis J. M., Paich H. A., Wang S., Gong N., Altshuller G., Thresher R. J., Koves T. R., Watkins S. M., Muoio D. M., Cline G. W., Shulman G. I., Coleman R. A. (2009) Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J. Biol. Chem. 284, 27816–27826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 [DOI] [PubMed] [Google Scholar]
- 22. Cao Y., Dave K. B., Doan T. P., Prescott S. M. (2001) Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 61, 8429–8434 [PubMed] [Google Scholar]
- 23. Polokoff M. A., Bell R. M. (1978) Limited palmitoyl-CoA penetration into microsomal vesicles as evidenced by a highly latent ethanol acyltransferase activity. J. Biol. Chem. 253, 7173–7178 [PubMed] [Google Scholar]
- 24. Kim J. H., Lewin T. M., Coleman R. A. (2001) Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J. Biol. Chem. 276, 24667–24673 [DOI] [PubMed] [Google Scholar]
- 25. Hosaka K., Mishina M., Tanaka T., Kamiryo T., Numa S. (1979) Acyl-coenzyme-A synthetase I from Candida lipolytica. Purification, properties and immunochemical studies. Eur. J. Biochem. 93, 197–203 [DOI] [PubMed] [Google Scholar]
- 26. Igal R. A., Wang P., Coleman R. A. (1997) Triacsin C blocks de novo synthesis of glycerolipids and cholesterol esters but not recycling of fatty acid into phospholipid. Evidence for functionally separate pools of acyl-CoA. Biochem. J. 324, 529–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weintraub N. L., Fang X., Kaduce T. L., VanRollins M., Chatterjee P., Spector A. A. (1999) Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am. J. Physiol. 277, H2098–H2108 [DOI] [PubMed] [Google Scholar]
- 28. Edin M. L., Wang Z., Bradbury J. A., Graves J. P., Lih F. B., DeGraff L. M., Foley J. F., Torphy R., Ronnekleiv O. K., Tomer K. B., Lee C. R., Zeldin D. C. (2011) Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 25, 3436–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Magnes C., Sinner F. M., Regittnig W., Pieber T. R. (2005) LC/MS/MS method for quantitative determination of long chain fatty acyl-CoAs. Anal. Chem. 77, 2889–2894 [DOI] [PubMed] [Google Scholar]
- 30. Deutsch J., Grange E., Rapoport S. I., Purdon A. D. (1994) Isolation and quantitation of long chain acyl-coenzyme A esters in brain tissue by solid-phase extraction. Anal. Biochem. 220, 321–323 [DOI] [PubMed] [Google Scholar]
- 31. Minkler P. E., Kerner J., Ingalls S. T., Hoppel C. L. (2008) Novel isolation procedure for short-, medium-, and long chain acyl-coenzyme A esters from tissue. Anal. Biochem. 376, 275–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang M. J., Fujino T., Sasano H., Minekura H., Yabuki N., Nagura H., Iijima H., Yamamoto T. T. (1997) A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. U.S.A. 94, 2880–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mashek D. G., Li L. O., Coleman R. A. (2006) Rat long chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J. Lipid Res. 47, 2004–2010 [DOI] [PubMed] [Google Scholar]
- 34. Civelek V. N., Deeney J. T., Shalosky N. J., Tornheim K., Hansford R. G., Prentki M., Corkey B. E. (1996) Regulation of pancreatic beta-cell mitochondrial metabolism. Influence of Ca2+, substrate and ADP. Biochem. J. 318, 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karara A., Dishman E., Falck J. R., Capdevila J. H. (1991) Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J. Biol. Chem. 266, 7561–7569 [PubMed] [Google Scholar]
- 36. Weintraub N. L., Fang X., Kaduce T. L., VanRollins M., Chatterjee P., Spector A. A. (1997) Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circ. Res. 81, 258–267 [DOI] [PubMed] [Google Scholar]
- 37. Gross G. J., Gauthier K. M., Moore J., Falck J. R., Hammock B. D., Campbell W. B., Nithipatikom K. (2008) Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am. J. Physiol. Heart Circ. Physiol. 294, H2838–H2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brash A. R. (2001) Arachidonic acid as a bioactive molecule. J. Clin. Invest. 107, 1339–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Antinozzi P. A., Segall L., Prentki M., McGarry J. D., Newgard C. B. (1998) Molecular or pharmacologic perturbation of the link between glucose and lipid metabolism is without effect on glucose-stimulated insulin secretion. A re-evaluation of the long chain acyl-CoA hypothesis. J. Biol. Chem. 273, 16146–16154 [DOI] [PubMed] [Google Scholar]
- 40. Roduit R., Nolan C., Alarcon C., Moore P., Barbeau A., Delghingaro-Augusto V., Przybykowski E., Morin J., Massé F., Massie B., Ruderman N., Rhodes C., Poitout V., Prentki M. (2004) A role for the malonyl-CoA/long chain acyl-CoA pathway of lipid signaling in the regulation of insulin secretion in response to both fuel and nonfuel stimuli. Diabetes 53, 1007–1019 [DOI] [PubMed] [Google Scholar]
- 41. Carroll M. A., Balazy M., Margiotta P., Falck J. R., McGiff J. C. (1993) Renal vasodilator activity of 5,6-epoxyeicosatrienoic acid depends upon conversion by cyclooxygenase and release of prostaglandins. J. Biol. Chem. 268, 12260–12266 [PubMed] [Google Scholar]
- 42. Johnson D. G., Fujimoto W. Y., Williams R. H. (1973) Enhanced release of insulin by prostaglandins in isolated pancreatic islets. Diabetes 22, 658–663 [DOI] [PubMed] [Google Scholar]
- 43. Pek S., Tai T. Y., Elster A., Fajans (1975) Stimulation by prostaglandin E2 of glucagon and insulin release from isolated rat pancreas. Prostaglandins 10, 493–502 [DOI] [PubMed] [Google Scholar]
- 44. Pek S., Tai T. Y., Elster A. (1978) Stimulatory effects of prostaglandins E-1, E-2, and F-2-alpha on glucagon and insulin release in vitro. Diabetes 27, 801–809 [DOI] [PubMed] [Google Scholar]
- 45. Luo P., Chang H. H., Zhou Y., Zhang S., Hwang S. H., Morisseau C., Wang C. Y., Inscho E. W., Hammock B. D., Wang M. H. (2010) Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J. Pharmacol. Exp. Ther. 334, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Borradaile N. M., Han X., Harp J. D., Gale S. E., Ory D. S., Schaffer J. E. (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 47, 2726–2737 [DOI] [PubMed] [Google Scholar]
- 47. Watt M. J., Hoy A. J., Muoio D. M., Coleman R. A. (2012) Distinct roles of specific fatty acids in cellular processes. Implications for interpreting and reporting experiments. Am. J. Physiol. Endocrinol. Metab. 302, E1–E3 [DOI] [PMC free article] [PubMed] [Google Scholar]