

Background: The epigenetic effect of acrolein, an α,β-unsaturated aldehyde, remains unclear.
Results: Acrolein specifically inhibits acetylations of N-terminal tails of cytosolic but not nuclear histones and reduces their delivery into chromatin.
Conclusion: Acrolein differentially affects post-translational modifications of free histones and nucleosomal histones and compromises chromatin assembly.
Significance: Affecting chromatin assembly may represent a new model for the interaction between environmental factors and the genome function.
Keywords: Cigarette Smoke, Epigenetics, Histone Modification, Nucleosome, Transcription, Acrolein, Chromatin Assembly
Abstract
Chromatin structure and gene expression are both regulated by nucleosome assembly. How environmental factors influence histone nuclear import and the nucleosome assembly pathway, leading to changes in chromatin organization and transcription, remains unknown. Acrolein (Acr) is an α,β-unsaturated aldehyde, which is abundant in the environment, especially in cigarette smoke. It has recently been implicated as a potential major carcinogen of smoking-related lung cancer. Here we show that Acr forms adducts with histone proteins in vitro and in vivo and preferentially reacts with free histones rather than with nucleosomal histones. Cellular fractionation analyses reveal that Acr exposure specifically inhibits acetylations of N-terminal tails of cytosolic histones H3 and H4, modifications that are important for nuclear import and chromatin assembly. Notably, Acr exposure compromises the delivery of histone H3 into chromatin and increases chromatin accessibility. Moreover, changes in nucleosome occupancy at several genomic loci are correlated with transcriptional responses to Acr exposure. Our data provide new insights into mechanisms whereby environmental factors interact with the genome and influence genome function.
Introduction
It is becoming increasingly apparent that environmental factors alter epigenetic profiles, such as DNA methylation, chromatin modifications, and non-coding RNAs, thereby changing chromatin structure and gene expression (1). For example, exposure to metal toxicants, such as nickel, chromate, and arsenic, causes global changes in post-translational histone modifications (PTMs)3 (2). Chromatin structure can be regulated by nucleosome assembly, which ensures proper inheritance of chromatin structure, propagation of epigenetic marks, maintenance of genome integrity, and gene expression (3–5). Histone chaperones bind to histones and play critically important roles in histone dynamics, such as histone transfer, transport, or storage, thereby modulating chromatin assembly. In plants, different abiotic stresses down-regulate expression of histone chaperones, suggesting that aberrant nucleosome assembly is involved in the regulation of stress responses, although the mechanisms underlying this process have not been clarified yet (6). In addition to histone chaperones, appropriate covalent modifications of N-terminal tails of nascent histones H3 and H4 are required for histone nuclear import and assembly into chromatin. For example, H3 N-terminal tail acetylations and lysine 56 acetylation facilitate chromatin assembly, at least in part by enhancing the interaction between histone chaperone chromatin assembly factor-1 (CAF-1) and H3/H4 (4). Histone H4 Lys-5 and histone H4 Lys-12 (H4K12) acetylation, however, regulates the interaction between H3/H4 and importin 4, a nuclear transport receptor, and another chaperone anti-silencing function 1 (ASF1) (7–9). However, whether and how environmental factors influence PTMs of newly synthesized histones, leading to changes in nuclear import and chromatin assembly, remains unknown.
Acrolein (Acr) is an α,β-unsaturated aldehyde abundant in the environment. It is derived from automobile exhaust and industrial emissions and is enriched in cigarette smoke and fumes of heated cooking oil (10–13). It is also generated endogenously by oxidative stress. Acr has been implicated in the development of multiple sclerosis, Alzheimer disease, pulmonary disorders, and cardiovascular diseases (14–19). Specifically, Acr has surfaced as a potential major carcinogen associated with smoking-related lung cancer (11, 20). Polycyclic aromatic hydrocarbons have long been considered major carcinogens in cigarette smoke, evidenced by the p53 binding patterns of polycyclic aromatic hydrocarbons coinciding with the p53 mutational pattern in lung cancer (21). This idea has recently been challenged by the finding that Acr, which is 1000-fold more abundant than polycyclic aromatic hydrocarbons in cigarettes, also binds preferentially at p53 mutational hot spots (11). As a highly reactive aldehyde, Acr not only reacts with nucleophilic guanine bases in DNA (11, 20, 22–24); it also targets lysine, arginine, histidine, and cysteine residues within susceptible proteins (25, 26). By using antibodies against cyclic lysine adducts, several studies reported the formation of Acr-lysine adducts in vivo in the affected tissues of several degenerative diseases (27). The fact that histone proteins are rich in lysines raises the possibility that Acr also targets lysine residues in histone tails, affecting their PTMs. Because appropriate histone lysine modifications are crucial for genome function (28), the formation of Acr-histone adducts should have a significant impact on cellular processes.
In the current study, we investigated the formation of Acr-histone adducts in vitro and in cells as well as studied how these interactions affect histone PTMs. We also examined the effects of Acr on nucleosome assembly and chromatin accessibility. Finally, we analyzed the relationship between changes in chromatin organization and transcriptional response following Acr exposure. Because Acr can also be generated endogenously by oxidative stress (10, 12, 25), this work has the potential to shed new light on the investigation of environmental control of epigenetic mechanisms by a wide range of stress-inducing environmental factors.
EXPERIMENTAL PROCEDURES
Cell Culture
The immortalized human bronchial epithelial cell line BEAS-2B and the human lung adenocarcinoma cell line A549 were cultured in DMEM supplemented with 10% FBS. Cells (1 × 107) were grown overnight prior to exposure to Acr. To avoid side reactions with serum constituents, Acr was first diluted in DMEM without serum, followed by a second dilution in appropriate culture medium. Cells were washed with PBS and exposed to Acr in serum-free medium unless otherwise described. For “long-term” treatment, BEAS-2B cells were exposed to 5 μm Acr in DMEM with 5% FBS.
Antibodies
The antibodies were purchased as follows. Anti-Acr (ab48501), anti-histone H3 (ab1791), anti-acetyl-histone H4 Lys-12 (ab61238), and anti-β-actin (ab8226) were from Abcam; anti-dinitrophenyl (DNP; D9656), which reacts with DNP-BSA and DNP-rabbit serum albumin but not BSA or rabbit serum albumin, was from Sigma-Aldrich; anti-acetyl-histone H3 (06-599), anti-trimethyl-histone H3 Lys-4 (07-473), anti-dimethyl-histone H3 Lys-9 (07-441), anti-trimethyl-histone H3 Lys-27 (07-499), anti-acetyl-histone H3 Lys-18 (07-354), and anti-histone H4 (07-108) were from Millipore; anti-histone acetyltransferase 1 (HAT1) (sc376200), anti-CAF1 p150 (sc10772), and anti-GCN5 (sc20698) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); and anti-caspase-3 (CST9662) was from Cell Signaling Technology.
Cell Lysates, Acid Extraction, and Western Blot
After Acr exposure, cells were washed with ice-cold PBS and collected by centrifugation. Whole cell lysates were prepared by using Triton X-100 lysis buffer (50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mm NaCl, 10 mm MgCl2, 10 mm sodium butyrate, and protease inhibitors). For acid extraction, cells were resuspended in lysis buffer (10 mm HEPES, pH 7.5, 1.5 mm MgCl2, 10 mm KCl, 0.2 n HCl, 0.5 mm DTT, 10 mm sodium butyrate, and protease inhibitors), incubated on ice for 60 min, and centrifuged at 11,000 × g for 10 min. The supernatant was transferred to a new tube and neutralized with one-fifth volume of 1.5 m Tris-HCl buffer (pH 8.8). Western blot was performed as described elsewhere (29).
Nucleosome Reconstitution
Recombinant Xenopus histones H3, H2A, H2B, and H4 were expressed and purified separately from Escherichia coli as described by Luger and co-workers (30). The refolding of histone octamer and reconstitution of mononucleosome were performed essentially as described (30).
Nucleosome Preparation and ChIP
Mono- and dinucleosomes were isolated by micrococcal nuclease (MNase) digestion and sucrose gradient purification and subjected to ChIP analysis, as we described previously (31).
The following primers were used: EGR-2 (early growth response-2), 5′-CAGCGACGTCACGGGTATT-3′ (forward) and 5′-CGCCGAGCTATTAATCAATTGC-3′ (reverse); IAP (inhibitor of apoptosis), 5′-CCGCTGGAGTTCCCCTAAG-3′ (forward) and 5′-CGCACTCCTCCCAGTGGTT-3′ (reverse); S100A10, 5′-GCAGGGTCATCCAGCAAGTAA-3′ (forward) and 5′-GCGCAGAACCAGAGAAGCGAAGAA-3′ (reverse); MT1F (metallothionein-1F), 5′-AGCGGCCGGCTGTTG-3′ (forward) and 5′-ACTTTCCAAGAGAGAAGCGAAGAA-3′ (reverse); GAPDH body, 5′-AGGCTGTGGGCAAGGTCAT-3′ (forward) and 5′-CAGGTCCACCACTGACACGTT-3′ (reverse); Chr1, 5′-TCATTCCCACAAACTGCGTTG-3′ (forward) and 5′-TCCAACGAAGGCCACAAGA-3′ (reverse); H3.3B, 5′-ACGAAAGCCGCCAGGAA-3′ (forward) and 5′-CTGTAGCGATGAGGCTTCTTCA-3′ (reverse); OSTF1 (osteoclast-stimulating factor 1) transcriptional start site (TSS), 5′-GGCGGGCAGTAGGTCATC-3′ (forward) and 5′-TGTACTCATGGTGGCGTGGTG-3′ (reverse); SAT2 (spermidine/spermine N1-acetyltransferase family member 2), 5′-TGAATGGAATCGTCATCGAA-3′ (forward) and 5′-CCATTCGATAATTCCGCTTG-3′ (reverse); TM4SF1 (transmembrane 4 L six family member 1) TTS, 5′-AAGACAGGAAGCCGTTAGCA-3′ (forward) and 5′-GGCAGGAGGACCACGAGGAA-3′ (reverse); TRIM42 (tripartite motif-containing 42) TSS, 5′-AGTTTCCACCAACATACCAGC-3′ (forward) and 5′-TCCCAGGACTCTTGATGCCT-3′ (reverse).
MNase Digestion Assay
Nuclei were isolated from Acr-treated and control BEAS-2B cells as described (29). The nucleus suspension was adjusted to an A260 of 20. MNase was added (2, 6, or 18 × 10−3 units/μl, final concentration), and the suspension was incubated for 8 min at 37 °C. The reaction was stopped by adding EDTA (10 mm, final concentration). Proteinase K (1%, v/v) and SDS (1%, w/v) were added, and the mixture was incubated overnight at 37 °C. The DNA was purified by phenol/chloroform extraction and ethanol precipitation. Three μg of purified DNA was loaded on 2% agarose gel and visualized by ethidium bromide staining. The monomeric DNA band was excised, and the DNA was extracted for real-time quantitative PCR (qPCR) analysis.
Quantitative RT-PCR Assay and Flow Cytometry
The RT-qPCR assay and flow cytometry analysis were carried out as described (32, 33). The following primers were used: EGR-2, 5′-TGTGGAGGGCAAAAGGAGAT-3′ (forward) and 5′-CACAACCTGGAGACCCAACTC-3′ (reverse); IAP, 5′-TGGTTTCCAAGGTGTGAGTACTTG-3′ (forward) and 5′-GGGCTGTCTGATGTGGATAGC-3′ (reverse); S100A10, 5′-TTGTCCCAAAGGGTCGCTTA-3′ (forward) and 5′-TCCTGATCTGCTCATGAAATCCT-3′ (reverse); MT1F, 5′-CAGCGGCCGGCTGTT-3′ (forward) and 5′-AGAGACTGGACTTTCCAAGAGAGAAG-3′ (reverse). The other primers used are the same as listed for the ChIP assay.
Trypan Blue Staining and Cell Viability Assay
After exposure to Acr in medium containing various concentrations of serum, cells were trypsinized, centrifuged, and resuspended in culture medium. The total number of viable cells was determined by trypan blue staining, which stains dead cells and shows a blue color under the microscope. The cell viability was assessed by calculating at least 3 times the percentage of live cells within five randomly selected fields.
Cellular Fractionation
Cells (4 × 107) were suspended in 1.5 ml of hypotonic buffer (10 mm Tris-HCl, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm DTT, and protease inhibitors) for 10 min on ice and centrifuged at 2,500 × g for 10 min after passing four times through a 25-gauge needle. The supernatant was retained as the cytosolic fraction. The remaining pellet was resuspended in 0.5 pellet volume of low salt buffer (20 mm Tris-HCl, pH 7.4, 20 mm KCl, 1.5 mm MgCl2, 1 mm DTT, 0.2 mm EDTA, 25% glycerol, and protease inhibitors) and homogenized with a 25-gauge needle. The sample volume was carefully measured, and 0.5 volume of high salt buffer (20 mm Tris-HCl, pH 7.4, 1.2 m KCl, 1.5 mm MgCl2, 1 mm DTT, 0.2 mm EDTA, 25% glycerol, and protease inhibitors) was subsequently added to obtain a final KCl concentration of 0.42 m. After rotating for 30 min at 4 °C, the suspension was centrifuged at 14,000 × g for 15 min, and the supernatant was retained as nuclear extract. The insoluble pellet was resuspended in Tris buffer (10 mm Tris-HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2, and protease inhibitors) plus 1.5 mm CaCl2. After passing four times through a 20-gauge needle followed by four passes through a 25-gauge needle, the suspension was adjusted to A260 = 100 and digested with 12 × 10−2 units/μl MNase for 12 min at 37 °C. The reaction was stopped by adding EDTA (10 mm, final concentration). After sitting on ice for 30 min, the supernatant was collected as S1. The remaining pellet was resuspended in the Tris buffer plus 0.25 mm EDTA, incubated on ice for 15 min, and passed four times through a 25-gauge needle. After centrifugation at 14,000 × g for 10 min, the supernatant was collected as S2 and combined with S1 as the chromatin fraction.
Histone Carbonyl Assay
Histone carbonyl assays were performed as described by Thompson et al. (34) using 15 μg of acid-extracted total histones as substrates.
Statistical Analysis
Gel intensities were quantified using ImageJ image processing software (National Institutes of Health). Relevant results are represented as mean ± S.D. (error bars). Significance was assessed by Student's t test. p < 0.05 was considered statistically significant. The number of times that each experiment was repeated is indicated in the figure legends.
RESULTS
Acr-Histone Adduct Formation in Vitro and in Vivo
To examine if Acr reacts with histone proteins in vitro, we purified recombinant core histones H3, H4, H2A, and H2B (Fig. 1A) and incubated individual histones with 0, 50, and 100 μm Acr, concentrations that have frequently been used in studies of DNA damage, transcriptional response, and cytotoxicity (34–37). The formation of Acr-lysine adducts (i.e. formyl-dehydropiperidino-lysine) was examined via Western blot using anti-Acr antibodies. This antibody is specific for Acr-modified protein, especially the formyl-dehydropiperidino-lysine type derivative. As shown in Fig. 1B, Acr reacted with lysine residues on all four core histones, with the strongest interaction with H2B. Moreover, treatment with higher concentrations of Acr resulted in the formation of histone-histone cross-links as evidenced by the observation of double-sized shifted bands (Fig. 1B, lanes 3, 6, 9, and 12).
FIGURE 1.

Acr-histone adduct formation in vitro and in vivo. A, recombinant core histones expressed in E. coli were purified as described under “Experimental Procedures.” Purified histones H4, H2B, H2A, and H3 (2 μg each) were separated by 14% SDS-PAGE and detected by Coomassie Blue staining. B, representative immunoblot analysis of Acr-histone adduct formation in vitro. Recombinant core histones (2 μg each) were treated with 0, 50, and 100 μm Acr. Acr-histone adducts were detected by Western blot using anti-Acr antibodies (top). Coomassie Blue staining was used as a loading control (bottom). The band intensities were quantified using ImageJ software. Lane 2 (top) and lane 1 in (bottom) are used as references, which are set to 1, respectively. The bar graphs show relative quantification of the Acr reaction (100 μm) with each core histone normalized to input (lanes 1, 4, 7, and 10 in Coomassie Blue-stained gel, respectively; n = 2). The data are shown as mean ± S.D. (error bars). C, Coomassie Blue staining of total histones isolated by acid extraction from cells treated with or without 100 μm Acr. D, representative immunoblot analysis of Acr-histone adduct formation in vivo (n = 3). Acid-extracted histones were subjected to carbonyl assays (see “Experimental Procedures”). Carbonylated proteins were detected by Western blot with anti-DNP antibodies (left). The same membrane was reprobed with anti-H3 antibodies (right). The band intensities corresponding to H3 were evaluated. M, protein marker; ND, not defined.
Next, protein carbonyl assays were applied to test Acr-histone adduct formation in vivo (35). Carbonylated protein is a highly sensitive indicator of protein adduction because the reaction of Acr with protein nucleophiles predominantly proceeds via Michael addition to form carbonyl-retaining adducts, which can be detected by Western blot. Total histones were prepared by acid extraction from BEAS-2B cells treated with or without 100 μm Acr. Fig. 1C clearly shows the bands corresponding to H3, H2B, H2A, and H4, suggesting that acid-extracted histones are reasonably pure. We next incubated total histones with dinitrophenyl hydrazine solution (0.5% 2,4-dinitrophenyl hydrazine (w/v) in 10% trifluoroacetic acid (v/v)). After carbonyl derivatization, the carbonylated proteins were detected by Western blot using antibodies to DNP. Carbonyl assays showed that Acr reacts with two 17-kDa proteins as early as 30 min after the treatment (Fig. 1D, left). One of the 17-kDa protein bands overlapped with the H3 band when the same membrane was reprobed with anti-histone H3 antibodies (Fig. 1D, right). Similarly, the bottom band was overlapped with the H4 band (data not shown). We thus concluded that Acr can form adducts with H3 and H4 in vitro and in vivo.
Unmodified Free Histones Are Preferred Targets of Acr
In principle, Acr can adduct both non-nucleosomal free histones (predeposit form) and nucleosomal histones (deposited form). To determine whether Acr prefers free versus nucleosomal histones, we refolded histone octamers (Fig. 2A) and reconstituted nucleosomes on 147-bp monomeric DNA fragments (Fig. 2B). We then measured the levels of Acr-histone adducts in the context of nucleosomes following Acr treatment. In parallel, core histones were also exposed to Acr. As shown in Fig. 2C, more Acr-histone adducts and histone-histone cross-links were detected when free histones were used, indicating that Acr preferentially reacted with free histones rather than with nucleosomal histones (compare lanes 2 and 4).
FIGURE 2.

Non-nucleosomal free histones are preferred targets of Acr. A, recombinant histones H3, H2A, H2B, and H4 were mixed and refolded into octamer. The gel filtration-purified histone octamers were separated by 14% SDS-PAGE and detected by Coomassie Blue staining. M, protein marker. B, mononucleosome reconstitution was performed by mixing monomeric DNA and histone octamer at 2 m NaCl and slowly decreasing NaCl to 50 mm by gradient dialysis. The reconstituted mononucleosomes were separated by 5% native PAGE and detected by ethidium bromide staining. M, DNA marker; lane 1, free monomeric DNA, ∼150 bp; lane 2, reconstituted mononucleosomes, ∼450 bp. C, representative immunoblot analysis of Acr-histone adduct formation. Core histone mixture and nucleosomes reconstituted with 147-bp monomeric DNA fragments were treated with Acr (0 and 1 mm), followed by Western blot using anti-Acr antibodies (top). Silver staining was used as a loading control (bottom). The bar graphs show relative quantification of Acr-histone adducts in free histones and reconstituted nucleosomes normalized to input (bottom, lanes 1 and 3, respectively; n = 3). The data are shown as mean ± S.D. (error bars). **, p < 0.01. D, hyper- and hypoacetylated nucleosomes were isolated from HeLa cells treated with or without Acr (50 or 100 μm) and subjected to Western blot with antibodies against Acr (top), H3 (middle), and H3K9Ac and H3K14Ac (bottom), The left bar graphs show relative amount of Acr-histone adducts in hypoacetylated nucleosomes as compared with that in hyperacetylated nucleosomes. The right bar graphs show levels of H3K9Ac and H3K14Ac in hyperacetylated nucleosomes upon 100 μm Acr treatment. The average ± S.D. of three independent experiments is presented. *, p < 0.05.
To address the relationship between Acr-histone adduct formation and histone lysine acetylation status, we purified hypoacetylated and hyperacetylated monomers from HeLa cells (Fig. 2D, bottom) and treated them with a variety of Acr concentrations to examine the formation of Acr-histone adducts. Fig. 2D showed that the levels of H3K9 and H3K14 acetylations in hyperacetylated nucleosomes were not affected by Acr exposure (bottom; compare lanes 4–6), suggesting that Acr cannot react with already modified lysines. Accordingly, Acr reacted more strongly with histones in hypoacetylated nucleosomes than with those in hyperacetylated nucleosomes (Fig. 2D, left bar graphs). These results suggest that Acr is likely to target unmodified newly synthesized histones and that histone PTMs may prevent the amino acid residues from being targeted by Acr. Among a total of 56 lysines on human core histones, approximately 29 have not yet been observed as acetylated (38). Therefore, the observed Acr-histone adduct formation in hyperacetylated nucleosomes (Fig. 2D, lane 6) is very likely due to the reaction between Acr and these unmodified lysines. In addition, purified native nucleosomes contain a small amount of chromatin-associated nonhistone proteins. Thus, the observed Acr adducts in high molecular weight proteins (Fig. 2D, lanes 3 and 6) may represent histone-histone, histone-nonhistone protein cross-links or the reaction of Acr with high molecular weight chromatin-associated proteins.
Acr Drastically Down-regulates Levels of N-terminal Tail Acetylations of Cytosolic Histones
Formaldehyde-modified histones are resistant to PTMs in vitro (39). Because Acr reacts with histone lysine residues (Figs. 1 and 2), we hypothesized that the formation of Acr-histone lysine adducts inhibits appropriate PTMs on histone proteins. Because some FBS constituents can scavenge Acr, we cultured BEAS-2B cells in serum-free DMEM to maximize the effects of Acr. Acr-histone adducts were detected in BEAS-2B cells treated with 100 μm of Acr (Fig. 1D); we thus added Acr to BEAS-2B cells in serum-free medium to achieve a final concentration of 100 μm Acr. To determine the optimal length of exposure time, we first tested acute cytotoxicity induced by Acr using conventional trypan blue dye exclusion assays. Although about 50% of the cells died after a 4-h Acr exposure in serum-free medium, the vast majority of cells appeared alive after 2 h of Acr treatment (Fig. 3A). Analysis of apoptosis by a flow cytometry showed apoptotic fractions only after 4 h of Acr treatment (Fig. 3B). Moreover, no cleaved form of caspase-3, a marker of apoptosis, was observed when the cells were treated with Acr for 2 h (Fig. 3C). Thus, we chose a 2-h treatment to minimize the length of time that cells are under stress.
FIGURE 3.
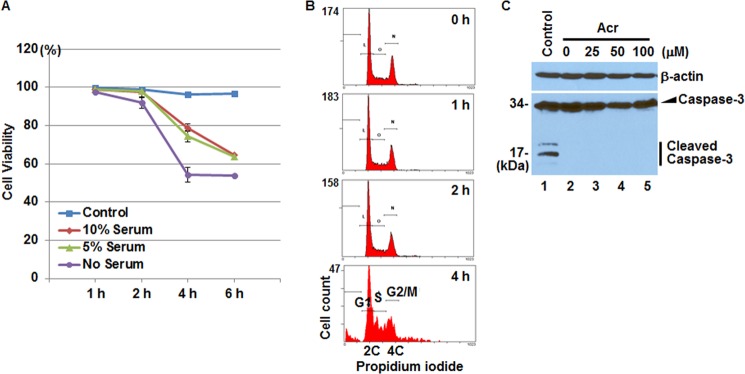
Cytotoxicity in BEAS-2B cells after 2-h treatment with Acr. A, cells were exposed to Acr (100 μm) for 1, 2, and 4 h in DMEM without or with various concentrations of serum. Cell viability was determined by trypan blue dye exclusion assays. Viability is represented as the percentage of live cells at each time point. Data represent the means ± S.D. (error bars) of triplicate tests. B, BEAS-2B cells were treated with or without Acr (100 μm) in serum-free DMEM for 1, 2, and 4 h. Treated cells were trypsinized and collected for flow cytometry analysis to monitor apoptosis. Apoptotic cells appeared only in 4-h Acr-treated cells. C, whole cell lysates were prepared from BEAS-2B cells treated with or without various concentrations of Acr for 2 h and subjected to Western blot with antibodies against β-actin and caspase-3. No cleaved caspase-3 was observed in two independent experiments. Whole cell lysates from BEAS-2B cells treated with 20 μm arsenite were used as the positive Caspase cleavage control (lane 1).
We isolated total histones from Acr-treated and untreated BEAS-2B cells and measured a number of different types of histone PTMs by Western blot. Unexpectedly, no apparent alterations were observed (Fig. 4A). Acr probably reacts with unmodified newly synthesized free histones (Fig. 2). Because such histones represent only a small percentage of total histones (40), the effects of Acr, if any, may not be detectable when total histones are examined. To confirm this, we isolated cell fractions (i.e. cytosolic, nuclear extract, and soluble chromatin fraction) (Fig. 4B) and subjected them to Western blot. As shown in Fig. 4C, the drastic decrease of H3K9, H3K14, H4K12 acetylations were observed only in the cytosolic fraction. Similar results were seen in the human lung adenocarcinoma cell line A549 (Fig. 4C), suggestive of a common effect among different cell types. To rule out the possibility that histone modification changes were attributable to the reduced expression of histone-modifying enzymes, we tested the expression of HAT1, which adds acetyl groups specifically to free histone H4 Lys-5 and Lys-12 (41). HAT1 expression was not decreased following 100 μm Acr exposure in either BEAS-2B or A549 cells (Fig. 4D), indicating that the observed reduction of histone acetylation by Acr exposure is probably due to a direct reaction of Acr with lysines on histones H3 and H4.
FIGURE 4.

Acr inhibits N-terminal tail acetylations of newly synthesized histones. A, BEAS-2B cells were exposed to various concentrations of Acr in serum-free medium for 2 h. Total histones were prepared by acid extraction and subjected to Western blot. No marked decrease of histone modification was observed in three independent experiments. B, experimental scheme for preparation of cell fractions. C, cytosolic cell fractions, nuclear extracts, and soluble chromatin fractions were isolated from BEAS-2B or A549 cells treated with or without Acr for 2 h and then subjected to Western blot. The bar graphs show relative quantification of histone modification levels normalized to H3. The data shown are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05; **, p < 0.01. Western blot analyses show drastic decrease of cytosolic H4K12Ac, H3K9Ac, and H3K14Ac by Acr exposure. D, whole cell lysates were prepared from Acr-treated and control cells and subjected to Western blot with antibodies against HAT1, a histone acetyltransferase specific for free histone H4 Lys-5 and -12. No decrease in HAT1 expression level was observed in two independent experiments. Coomassie Blue staining was used as the loading control.
Low Dose Exposure of Acr Reduces Cytosolic H4K12 Acetylation
To investigate how low dose exposure of Acr in the medium containing serum influences histone PTMs, we treated BEAS-2B cells continuously with 5 μm Acr in DMEM containing 5% FBS because this concentration is physiologically relevant in terms of the amounts of Acr generated by about one cigarette, depending on the type of cigarette (60 μg = 1.1 μmol/cigarette) (12, 13, 42). As shown in Fig. 5A, continuous treatment of cells with 5 μm Acr resulted in the gradual decrease of cytosolic H4K12 acetylation until day 7. Flow cytometry analysis showed no evidence of apoptosis under this condition (Fig. 5B). These results suggest that “long term,” low dose exposure to Acr at the levels that are relevant to cigarette smoking may alter modifications of nascent histones and affect their functions.
FIGURE 5.

Low dose exposure of Acr reduces H4K12 acetylation and facilitates chromatin accessibility. A, BEAS-2B cells were exposed to 5 μm Acr in 5% FBS-containing medium for 5–7 days. Cytosolic fractions were isolated and subjected to Western blot with antibodies against H4K12 acetylation, histone acetyltransferases GCN5 and HAT1, and histone H3. Each untreated control is used as a reference, which is set to 1. The bar graphs show relative levels of H4K12Ac (normalized to H3; n = 2). The data are mean ± S.D. (error bars). B, Acr-treated and control BEAS-2B cells were trypsinized and collected for flow cytometry analysis. No apoptosis was observed in 5 μm Acr-treated cells as late as 7 days. C, nuclei were prepared from Acr-treated and control cells and digested with MNase. Digested DNA was extracted and electrophoretically separated in a 2% agarose gel and visualized by ethidium bromide staining. Representative results from two independent experiments are shown. Increased chromatin accessibility is evident by an increased intensity of mononucleosome and disrupted dinucleosome bands (arrowheads) and a decreased intensity of polynucleosome bands (indicated by stars) in treated sample (lane 6) compared with the control (lane 5).
Nucleosome Assembly Is Compromised by Exposure to Acr
The normal acetylations of the N-terminal tails of H3 and H4 histones are critical for their nuclear import and assembly into chromatin (7–9). Thus, the reduction of cytosolic H3K9Ac, H3K14Ac, and H4K12Ac by Acr treatment may compromise chromatin assembly of newly synthesized histones. If so, we expect Acr exposure to affect deposition of H3 marked by H3K9Ac and H3K14Ac. To prove this possibility, we performed ChIP to measure the levels of H3K9 and H3K14 acetylations at several genomic loci. As shown in Fig. 6A, the acetylations of H3K9 and H3K14 were significantly decreased at the majority of loci tested. The reduction was most apparent at the promoters of S100A10, MT1F, and OSTF1 genes, the sites with the highest levels of H3K9 and H3K14 acetylations under untreated conditions (Fig. 6A). Interestingly, levels of another active transcription marker, H3K4me3, were about 2-fold up-regulated by exposure to 100 μm Acr in the S100A10 and MT1F promoter regions (Fig. 6B), suggesting that the reductions of H3K9 and H3K14 acetylations were due to the aberrant delivery of H3 marked with H3K9 and H3K14 acetylations but were not due to direct targeting of Acr to nucleosomal histones. Moreover, if histone delivery is blocked by Acr exposure, nucleosomal histone H3 (and H4) should be depleted after Acr exposure, although total H3 expression was not changed by Acr exposure (Fig. 4). To test this possibility, we isolated cell fractions from BEAS-2B cells treated with various concentrations of Acr for 2 h and measured the amount of nucleosomal H3. Fig. 6C shows that whereas levels of γ-tubulin were not changed, the amount of histone H3 in the chromatin fraction was decreased by about 20% at 100 μm Acr. These results further suggest that the delivery of histone H3 is compromised by Acr exposure.
FIGURE 6.

Acr exposure compromises chromatin assembly. A and B, BEAS-2B cells were treated with or without Acr (100 μm) for 2 h. Mono- and dinucleosomes were prepared by MNase digestion and subjected to ChIP assays with antibodies specific for H3K9Ac and H3K14Ac (A) and H3K4me3 (B), respectively. Representative data from two independent experiments are shown. The data shown are the mean ± S.D. (error bars) from qPCRs performed in triplicate. *, p < 0.05; **, p < 0.01. The loci tested are mostly promoters except for GAPDH (gene body), Chr1 (centromere), and TM4SF1 (gene end). Relative -fold change was calculated after normalization to input. C, reduced H3 levels in chromatin fraction after Acr exposure. The quantification of Western blot by ImageJ software is shown. The data are shown as mean ± S.D. of three independent experiments (*, p < 0.05 when compared with the untreated control). Although γ-tubulin was not changed, exposure to Acr (100 μm) resulted in reduction of nucleosomal H3. D, Acr exposure results in the increase of chromatin accessibility. BEAS-2B cells were treated with or without Acr (100 μm) in serum-free DMEM. Nuclei were isolated and digested with different concentrations of MNase. Digested DNA was extracted and electrophoretically separated in a 2% agarose gel and visualized by ethidium bromide staining. Representative results from two independent experiments are shown.
The reduced level of H3 in the chromatin fraction after Acr exposure resembles H3 depletion following the shutoff of its expression. Repression of histone expression facilitates chromatin accessibility (43–45). Therefore, we investigated the influence of Acr exposure on chromatin accessibility using MNase digestion assays. We isolated nuclei from BEAS-2B cells treated with 100 μm Acr for 0, 1, and 2 h and digested them with MNase. We then extracted DNAs and monitored the changes of protective nucleosome ladders by agarose gels. No alterations were observed when cells were treated for 1 h (Fig. 6D, lanes 1–6). However, Acr exposure for 2 h led to general increases in MNase sensitivity. This effect is most obvious with digestion by 2 × 10−3 units/μl MNase, as noted by the loss of bands indicative of polynucleosomes (larger than 5-mers) in Acr-treated cells (white arrowheads in Fig. 6D when comparing lane 10 with lane 7). Moreover, when digested with a higher concentration of MNase (12 × 10−3 units/μl), reduced intensity of dinucleosome bands was observed in Acr-treated cells (yellow arrowheads when comparing lane 12 with lane 9 in Fig. 6D). Furthermore, continuous treatment of cells with low dose (5 μm) Acr also increased chromatin accessibility by day 7 based on MNase digestion assays (Fig. 5C). Taken together, these results indicate that chromatin accessibility is facilitated by Acr exposure.
Changes in Nucleosome Occupancy Associate with Transcriptional Response to Acr Exposure
The general increase of chromatin accessibility by Acr suggests that nucleosome occupancy may change following Acr exposure. To test whether Acr exposure changes chromatin organization, we determined the nucleosome occupancy at several genomic loci, including several gene loci reportedly regulated by Acr exposure in A549 cells (37). We determined the changes in nucleosome occupancy by measuring the ratio of the abundance of target DNA sequences in MNase-digested mononucleosomes isolated from Acr-treated versus control cells (Fig. 7A). The abundance of specific target sequences was determined by qPCR (31). Nucleosomes within the GAPDH gene body, OSTF1 TSS, Chr1 α-satellite, and TRIM42 TSS, were lost to some extent by exposure to Acr (Fig. 7A). We also observed an increase of nucleosome occupancy at the promoter of the IAP gene. These data indicate that nucleosome occupancy could be altered by relatively brief (2-h) treatment of Acr.
FIGURE 7.

Correlation between changes in nucleosome occupancy and transcription by Acr exposure. A, MNase-digested monomeric DNA bands from untreated and 2-h treated BEAS-2B cells (Fig. 6D, lanes 9 and 12) were excised, and DNA was extracted. The abundance of a sequence in protected nucleosomes from Acr-treated cells relative to that from the control cells was determined by qPCR analysis. B, RT-qPCR measurements of transcripts of the indicated genes in Acr-treated and -untreated cells. Representative data from two independent experiments are shown. The data shown are the mean ± S.D. (error bars) from qPCRs performed in triplicate. *, p < 0.05; **, p < 0.01.
Nucleosomes compete with transcription factors for access to DNA and thus help to regulate transcription (43). To analyze the relationship between Acr-mediated changes in nucleosome occupancy and transcription, we determined the relative mRNA levels by RT-qPCR. As shown in Fig. 7B, the transcription levels of EGR-2, S100A10, GAPDH, and OSTF1 genes were increased to different extents upon Acr exposure. By contrast, the expressions of IAP and SAT2 were down-regulated by Acr treatment (Fig. 7B). Among them, the expressions of IAP, GAPDH, and OSTF1 genes were negatively correlated with changes in nucleosome occupancy. These data suggest that altered chromatin organization may contribute to changes in gene expression upon Acr exposure. Moreover, expression of some genes, such as the EGR-2 gene, displayed little or no correlation with changes in nucleosome occupancy (Fig. 7A), indicating that other mechanisms are also involved in Acr-mediated gene regulation.
DISCUSSION
In this study, we have addressed the formation of Acr-histone adducts and epigenetic effects of Acr on nucleosome assembly and chromatin organization. Acr formed adducts with lysine residues on recombinant core histones with the highest affinity for H2B (Fig. 1B). This is consistent with the fact that lysine residues are most abundant in H2B (∼15% of total) as compared with in other core histones (∼10%) (38). However, based on carbonylation assays, histones H3 and H4 seemed to be major targets of Acr in vivo. It would therefore be important to determine which of the core histones and lysines are preferred targets of Acr in vivo by methods such as mass spectrometry analysis. As an α,β-unsaturated aldehyde, Acr could react with any proteins containing nucleophilic lysine, arginine, cysteine, and histidine residues. However, in human lung cells, Acr exposure degraded some DNA repair proteins, such as XPA, XPC, hOGG1, PMS2, and MLH1 proteins, but had no effect on MSH2 and Ref1 (21), suggesting that there is protein specificity for Acr reactivity by as yet unknown mechanisms. Consistent with these reports, the lungs of mice after Acr exposure showed increased Acr-lysine adduction in several but not all proteins (46). We showed here that Acr preferentially reacts with unmodified free histones rather than with already modified nucleosomal histones (Fig. 2). Thus, the status of protein structure and PTMs may, at least in part, contribute to the specificity or affinity of Acr to proteins. Moreover, Acr exposure was not able to reduce the levels of H3K9 and H3K14 acetylations on hyperacetylated nucleosomes (Fig. 2D), suggesting that acetylated histones may prevent sites from being targeted by Acr. Consistent with this result, formaldehyde reacted only with unmodified and not acetylated histone H4, and the formation of formaldehyde-lysine adducts prevented those sites from being properly modified in vitro (39). Thus, protecting chromatin from being attacked by environmental factors, such as aldehydes, may represent a novel function of histone PTMs.
We have demonstrated that Acr reacts with histone proteins and specifically down-regulates the acetylations of N-terminal tails of cytosolic histones (Fig. 4). Appropriate acetylation of N-terminal tails of newly synthesized histones H3 and H4 is believed to be important for histone nuclear import and nucleosome assembly. In fact, Acr exposure led to compromise of the chromatin assembly. This conclusion is supported by the following results: (a) ChIP assays showed that the levels of histone H3 marked with H3K9 and H3K14 acetylations were drastically decreased at the majority of genomic loci tested, and that result seemed not to be due to direct reaction of Acr with nucleosomal histones because H3K4me3 was not down-regulated in parallel; (b) although the total H3 expression level was not changed, the H3 amount in chromatin fragments was depleted by Acr exposure; and (c) Acr treatment facilitated the accessibility of chromatin, as evidenced by partial MNase digestion assays (Figs. 5 and 6). It would be interesting in the future to test whether Acr exposure disrupts the associations of H3/H4 with nuclear import and histone chaperone proteins. Knockdown of histone chaperones ASF1 and CAF-1 results in changes in chromatin accessibility (47). This mechanism may not be involved in Acr-mediated abnormal nucleosome assembly because the expressions of CAF-1 p150 subunit, importin 4, and ASF1 were not affected by Acr exposure (37) (data not shown). However, it has not yet been determined if Acr could modify these proteins and affect their functions. Intriguingly, Acr exposure increased nucleosome occupancy at one promoter region tested, whereas all others were decreased or unchanged (Fig. 7A). This is not surprising, given that histone depletion does not necessarily mean that nucleosome occupancy will be decreased at all sites. Histone modification status, histone turnover rate, and binding of chromatin-associated proteins are believed to determine the loss/gain of nucleosome occupancy (43). The increase of nucleosome occupancy may represent repositioning of nucleosomes after the loss of neighboring nucleosomes.
The level of nucleosome occupancy is critical for transcriptional regulation. Among several genes tested, the expressions of IAP, GAPDH, and OSTF1 genes were negatively correlated with changes in their nucleosome occupancy (Fig. 7). Importantly, acetylations of active markers H3K9, H3K14, and H3K4me3 were down-regulated at OSTF1 TSS, whereas its transcription level is up-regulated by Acr exposure; conversely, although H3K4m3 is increased at the IAP promoter, the expression of IAP was down-regulated by Acr exposure (Figs. 6 and 7), suggesting that Acr-induced changes in nucleosome occupancy might be a major contributor of transcriptional response of these genes to Acr exposure. Fig. 6D shows that chromatin accessibility was not changed by the treatment of Acr for 1 h. Therefore, immediate early genes, such as EGR-2, are probably regulated by mechanisms other than changes in chromatin organization. Consistent with this idea, nucleosome occupancy at the EGR-2 promoter was not changed by Acr exposure, whereas EGR-2 expression was dramatically increased. The mechanism may involve damage to proteins that act at various signal transduction pathways. For instance, Acr formed adducts on IKKβ protein and inhibited the activation of NF-κB signaling (38). DNA damage within the promoter of genes caused down-regulation of susceptible genes during aging and oxidative stress (48). Thus, Acr-DNA adduct formation at regulatory regions may contribute to the Acr-induced gene regulation as well. In addition, it is important to explore how gene expression patterns are established and maintained after Acr is removed. Studies in yeast showed that depletion of H4 is only partially reversible when the H4 gene is reactivated, indicating that removing Acr may only partially reverse altered chromatin organization (49). Accordingly, the gene expressions that they control may be “permanently” dysregulated (50). Therefore, genes that are regulated by Acr exposure through altering chromatin organization may represent those stably heritable gene perturbations that are important for carcinogenesis and development of other Acr-related diseases.
In summary, we have shown that Acr, a major component of cigarette smoke, forms adducts with histone proteins and specifically inhibits appropriate covalent modifications of cytosolic histone H3 and H4. We further demonstrated that Acr exposure results in aberrant nucleosome assembly. The concept that a chemical toxicant reacts with newly synthesized free histones and regulates chromatin assembly is novel. Because of similar chemical properties, other aldehydes, such as formaldehyde and acetaldehyde, are likely to have a similar reaction with histone proteins. Other electrophiles, such as metabolically activated polycyclic aromatic hydrocarbons and alkylating agents, could also have similar effects, perhaps to a lesser extent. Moreover, aldehydes, including Acr, can be generated endogenously by oxidative stress, which often occurs in cells exposed to environmental agents (25). Thus, reaction with free histones, thereby compromising nucleosome assembly and altering chromatin organization and transcription, may represent an important mechanism whereby environmental factors interact with the genome and influence their biological functions.
Acknowledgments
We thank Cathy Klein for critical review, Hsiang-Tsui Wang for using acrolein and acrolein antibodies, and Mao-Wen Wang for assistance in FPLC.
This work was supported, in whole or in part, by National Institutes of Health, NIEHS, Center of Excellence Pilot Project Grant 5 P30 ES000260-47 (to C. J.).
- PTM
- post-translational modification
- Acr
- acrolein
- H3K9
- histone 3 Lys-9
- H4K9
- histone 4 Lys-9
- H3K14
- histone H3 Lys-14
- H3K4me3
- trimethylated histone H3 Lys-4
- DNP
- dinitrophenyl
- MNase
- micrococcal nuclease
- TSS
- transcriptional start site
- qPCR
- real-time quantitative PCR.
REFERENCES
- 1. Feil R., Fraga M. F. (2011) Epigenetics and the environment. Emerging patterns and implications. Nat. Rev. Genet. 13, 97–109 [DOI] [PubMed] [Google Scholar]
- 2. Zhou X., Li Q., Arita A., Sun H., Costa M. (2009) Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol. Appl. Pharmacol. 236, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Polo S. E., Almouzni G. (2006) Chromatin assembly. A basic recipe with various flavours. Curr. Opin. Genet. Dev. 16, 104–111 [DOI] [PubMed] [Google Scholar]
- 4. Li Q., Burgess R., Zhang Z. (2012) All roads lead to chromatin. Multiple pathways for histone deposition. Biochim. Biophys. Acta 1819, 238–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Groth A., Rocha W., Verreault A., Almouzni G. (2007) Chromatin challenges during DNA replication and repair. Cell 128, 721–733 [DOI] [PubMed] [Google Scholar]
- 6. Zhu Y., Dong A., Shen W. H. (2012) Histone variants and chromatin assembly in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 343–348 [DOI] [PubMed] [Google Scholar]
- 7. Ejlassi-Lassallette A., Mocquard E., Arnaud M. C., Thiriet C. (2011) H4 replication-dependent diacetylation and Hat1 promote S-phase chromatin assembly in vivo. Mol. Biol. Cell 22, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ejlassi-Lassallette A., Thiriet C. (2012) Replication-coupled chromatin assembly of newly synthesized histones. Distinct functions for the histone tail domains. Biochem. Cell Biol. 90, 14–21 [DOI] [PubMed] [Google Scholar]
- 9. Zhang H., Han J., Kang B., Burgess R., Zhang Z. (2012) Human histone acetyltransferase 1 protein preferentially acetylates H4 histone molecules in H3.1-H4 over H3.3-H4. J. Biol. Chem. 287, 6573–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang M. S., Wang H. T., Hu Y., Chen W. S., Akao M., Feng Z., Hu W. (2011) Acrolein induced DNA damage, mutagenicity and effect on DNA repair. Mol. Nutr. Food Res. 55, 1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feng Z., Hu W., Hu Y., Tang M. S. (2006) Acrolein is a major cigarette-related lung cancer agent. Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. U.S.A. 103, 15404–15409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faroon O., Roney N., Taylor J., Ashizawa A., Lumpkin M. H., Plewak D. J. (2008) Acrolein environmental levels and potential for human exposure. Toxicol. Ind. Health 24, 543–564 [DOI] [PubMed] [Google Scholar]
- 13. Stevens J. F., Maier C. S. (2011) Acrolein. Mol. Nutr. Food Res. 55, 1275–1276 [DOI] [PubMed] [Google Scholar]
- 14. Bein K., Leikauf G. D. (2011) Acrolein. A pulmonary hazard. Mol. Nutr. Food Res. 55, 1342–1360 [DOI] [PubMed] [Google Scholar]
- 15. Faroon O., Roney N., Taylor J., Ashizawa A., Lumpkin M. H., Plewak D. J. (2008) Acrolein health effects. Toxicol. Ind. Health 24, 447–490 [DOI] [PubMed] [Google Scholar]
- 16. Luo J., Hill B. G., Gu Y., Cai J., Srivastava S., Bhatnagar A., Prabhu S. D. (2007) Mechanisms of acrolein-induced myocardial dysfunction. Implications for environmental and endogenous aldehyde exposure. Am. J. Physiol. Heart Circ. Physiol. 293, H3673–H3684 [DOI] [PubMed] [Google Scholar]
- 17. Mercado C., Jaimes E. A. (2007) Cigarette smoking as a risk factor for atherosclerosis and renal disease. Novel pathogenic insights. Curr. Hypertens. Rep. 9, 66–72 [DOI] [PubMed] [Google Scholar]
- 18. Ning W., Li C. J., Kaminski N., Feghali-Bostwick C. A., Alber S. M., Di Y. P., Otterbein S. L., Song R., Hayashi S., Zhou Z., Pinsky D. J., Watkins S. C., Pilewski J. M., Sciurba F. C., Peters D. G., Hogg J. C., Choi A. M. (2004) Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. U.S.A. 101, 14895–14900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singh M., Dang T. N., Arseneault M., Ramassamy C. (2010) Role of by-products of lipid oxidation in Alzheimer's disease brain. A focus on acrolein. J. Alzheimers Dis. 21, 741–756 [DOI] [PubMed] [Google Scholar]
- 20. Hecht S. S. (2006) Smoking and lung cancer. A new role for an old toxicant? Proc. Natl. Acad. Sci. U.S.A. 103, 15725–15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Denissenko M. F., Pao A., Tang M., Pfeifer G. P. (1996) Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 274, 430–432 [DOI] [PubMed] [Google Scholar]
- 22. Wang H. T., Weng M. W., Chen W. C., Yobin M., Pan J., Chung F. L., Wu X. R., Rom W., Tang M. S. (2013) Effect of CpG methylation at different sequence context on acrolein- and BPDE-DNA binding and mutagenesis. Carcinogenesis 34, 220–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H. T., Hu Y., Tong D., Huang J., Gu L., Wu X. R., Chung F. L., Li G. M., Tang M. S. (2012) Effect of carcinogenic acrolein on DNA repair and mutagenic susceptibility. J. Biol. Chem. 287, 12379–12386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chung F. L., Young R., Hecht S. S. (1984) Formation of cyclic 1, N2-propanodeoxyguanosine adducts in DNA upon reaction with acrolein or crotonaldehyde. Cancer Res. 44, 990–995 [PubMed] [Google Scholar]
- 25. Stevens J. F., Maier C. S. (2008) Acrolein. Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 52, 7–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aldini G., Orioli M., Carini M. (2011) Protein modification by acrolein. Relevance to pathological conditions and inhibition by aldehyde sequestering agents. Mol. Nutr. Food Res. 55, 1301–1319 [DOI] [PubMed] [Google Scholar]
- 27. Burcham P. C., Kaminskas L. M., Tan D., Pyke S. M. (2008) Carbonyl-scavenging drugs and protection against carbonyl stress-associated cell injury. Mini Rev. Med. Chem. 8, 319–330 [DOI] [PubMed] [Google Scholar]
- 28. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 29. Jin C., Felsenfeld G. (2007) Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 21, 1519–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dyer P. N., Edayathumangalam R. S., White C. L., Bao Y., Chakravarthy S., Muthurajan U. M., Luger K. (2004) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 [DOI] [PubMed] [Google Scholar]
- 31. Jin C., Felsenfeld G. (2006) Distribution of histone H3.3 in hematopoietic cell lineages. Proc. Natl. Acad. Sci. U.S.A. 103, 574–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H., Smolen G. A., Beers L. F., Xia L., Gerald W., Wang J., Haber D. A., Lee S. B. (2008) Adenosine transporter ENT4 is a direct target of EWS/WT1 translocation product and is highly expressed in desmoplastic small round cell tumor. PLoS One 3, e2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H., Watford W., Li C., Parmelee A., Bryant M. A., Deng C., O'Shea J., Lee S. B. (2007) Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J. Clin. Invest. 117, 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson C. A., Burcham P. C. (2008) Protein alkylation, transcriptional responses and cytochrome c release during acrolein toxicity in A549 cells. Influence of nucleophilic culture media constituents. Toxicol. In Vitro 22, 844–853 [DOI] [PubMed] [Google Scholar]
- 35. Burcham P. C., Raso A., Thompson C. A. (2010) Toxicity of smoke extracts towards A549 lung cells. Role of acrolein and suppression by carbonyl scavengers. Chem. Biol. Interact. 183, 416–424 [DOI] [PubMed] [Google Scholar]
- 36. Burcham P. C., Raso A., Thompson C. A. (2010) Intermediate filament carbonylation during acute acrolein toxicity in A549 lung cells. Functional consequences, chaperone redistribution, and protection by bisulfite. Antioxid. Redox Signal. 12, 337–347 [DOI] [PubMed] [Google Scholar]
- 37. Thompson C. A., Burcham P. C. (2008) Genome-wide transcriptional responses to acrolein. Chem. Res. Toxicol. 21, 2245–2256 [DOI] [PubMed] [Google Scholar]
- 38. Basu A., Rose K. L., Zhang J., Beavis R. C., Ueberheide B., Garcia B. A., Chait B., Zhao Y., Hunt D. F., Segal E., Allis C. D., Hake S. B. (2009) Proteome-wide prediction of acetylation substrates. Proc. Natl. Acad. Sci. U.S.A. 106, 13785–13790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu K., Boysen G., Gao L., Collins L. B., Swenberg J. A. (2008) Formaldehyde-induced histone modifications in vitro. Chem. Res. Toxicol. 21, 1586–1593 [DOI] [PubMed] [Google Scholar]
- 40. Loyola A., Bonaldi T., Roche D., Imhof A., Almouzni G. (2006) PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell 24, 309–316 [DOI] [PubMed] [Google Scholar]
- 41. Verreault A., Kaufman P. D., Kobayashi R., Stillman B. (1998) Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr. Biol. 8, 96–108 [DOI] [PubMed] [Google Scholar]
- 42. Tirumalai R., Rajesh Kumar T., Mai K. H., Biswal S. (2002) Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol. Lett. 132, 27–36 [DOI] [PubMed] [Google Scholar]
- 43. Gossett A. J., Lieb J. D. (2012) In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet. 8, e1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han M., Chang M., Kim U. J., Grunstein M. (1987) Histone H2B repression causes cell-cycle-specific arrest in yeast. Effects on chromosomal segregation, replication, and transcription. Cell 48, 589–597 [DOI] [PubMed] [Google Scholar]
- 45. Wyrick J. J., Holstege F. C., Jennings E. G., Causton H. C., Shore D., Grunstein M., Lander E. S., Young R. A. (1999) Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 402, 418–421 [DOI] [PubMed] [Google Scholar]
- 46. Sithu S. D., Srivastava S., Siddiqui M. A., Vladykovskaya E., Riggs D. W., Conklin D. J., Haberzettl P., O'Toole T. E., Bhatnagar A., D'Souza S. E. (2010) Exposure to acrolein by inhalation causes platelet activation. Toxicol. Appl. Pharmacol. 248, 100–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adkins M. W., Tyler J. K. (2004) The histone chaperone Asf1p mediates global chromatin disassembly in vivo. J. Biol. Chem. 279, 52069–52074 [DOI] [PubMed] [Google Scholar]
- 48. Valacchi G., Pagnin E., Phung A., Nardini M., Schock B. C., Cross C. E., van der Vliet A. (2005) Inhibition of NFκB activation and IL-8 expression in human bronchial epithelial cells by acrolein. Antioxid. Redox Signal. 7, 25–31 [DOI] [PubMed] [Google Scholar]
- 49. Lu T., Pan Y., Kao S. Y., Li C., Kohane I., Chan J., Yankner B. A. (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891 [DOI] [PubMed] [Google Scholar]
- 50. Kim U. J., Han M., Kayne P., Grunstein M. (1988) Effects of histone H4 depletion on the cell cycle and transcription of Saccharomyces cerevisiae. EMBO J. 7, 2211–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]