

Background: Response regulator proteins within two-component signaling systems can autophosphorylate with small molecule phosphodonors.
Results: Autophosphorylation kinetics of the response regulator PhoB exhibit multiple features indicative of positive cooperativity between active sites.
Conclusion: PhoB autophosphorylation kinetics reflect formation of heterodimers (between phosphorylated and unphosphorylated monomers) with enhanced kinetics compared with monomers.
Significance: Response regulator dimers with a single phosphoryl group may contribute to transcription regulation.
Keywords: Bacterial Signal Transduction, Cooperativity, Enzyme Kinetics, Protein Phosphorylation, Protein-Protein Interactions, Dimerization, PhoB, Phosphoramidate, Response Regulator, Two-component System
Abstract
Response regulator proteins within two-component signal transduction systems are activated by phosphorylation and can catalyze their own covalent phosphorylation using small molecule phosphodonors. To date, comprehensive kinetic characterization of response regulator autophosphorylation is limited to CheY, which follows a simple model of phosphodonor binding followed by phosphorylation. We characterized autophosphorylation of the response regulator PhoB, known to dimerize upon phosphorylation. In contrast to CheY, PhoB time traces exhibited an initial lag phase and gave apparent pseudo-first order rate constants that increased with protein concentration. Furthermore, plots of the apparent autophosphorylation rate constant versus phosphodonor concentration were sigmoidal, as were PhoB binding isotherms for the phosphoryl group analog BeF3−. Successful mathematical modeling of the kinetic data necessitated inclusion of the formation of a PhoB heterodimer (one phosphorylated and one unphosphorylated monomer) with an enhanced rate of phosphorylation. Specifically, dimerization constants for the PhoB heterodimer and homodimer (two phosphorylated monomers) were similar, but the rate constant for heterodimer phosphorylation was ∼10-fold higher than for the monomer. In a test of the model, disruption of the known PhoBN dimerization interface by mutation led to markedly slower and noncooperative autophosphorylation kinetics. Furthermore, phosphotransfer from the sensor kinase PhoR was enhanced by dimer formation. Phosphorylation-mediated dimerization allows many response regulators to bind to tandem DNA-binding sites and regulate transcription. Our data challenge the notion that response regulator dimers primarily form between two phosphorylated monomers and raise the possibility that response regulator heterodimers containing one phosphoryl group may participate in gene regulation.
Introduction
Two-component signal transduction systems, present in both eukaryotic and prokaryotic microorganisms as well as plants, are nearly ubiquitous in bacteria with tens of thousands of two-component system proteins currently annotated in sequence databanks (1, 2). The essential components of any two-component system are a sensor kinase and a partner response regulator. Detection of an environmental stimulus regulates the sensor kinase, often leading to autophosphorylation of a conserved histidine. The phosphoryl group is subsequently transferred to a conserved aspartate residue on the partner response regulator. Phosphorylation switches the response regulator to an active conformation (3), allowing the response regulator to exert its downstream effects. Many response regulators control transcription (4), typically by dimerization and binding to a symmetric DNA-binding site (5). To terminate the signal, response regulators dephosphorylate through autodephosphorylation (6), phosphatase-enhanced dephosphorylation (7, 8), or reverse phosphotransfer (9).
All response regulators contain a conserved receiver domain, and most also contain one or more variable output domains. The receiver domain (Fig. 1) has a conserved (βα)5 tertiary structure and contains the dimerization interface for many response regulators as well as the conserved active site that catalyzes both phosphorylation and dephosphorylation (5, 10). The five invariable residues that compose the active site include a conserved aspartate that is the site of phosphorylation, two acidic residues that coordinate a divalent cation required for catalysis, a threonine/serine, and a lysine. In addition to catalyzing phosphotransfer from the sensor kinase, response regulators can also catalyze their own autophosphorylation with small molecule phosphodonors, including phosphoramidate (H3N+–PO32−, PAM),2 acetyl phosphate, and monophosphoimidazole (11–15).
FIGURE 1.
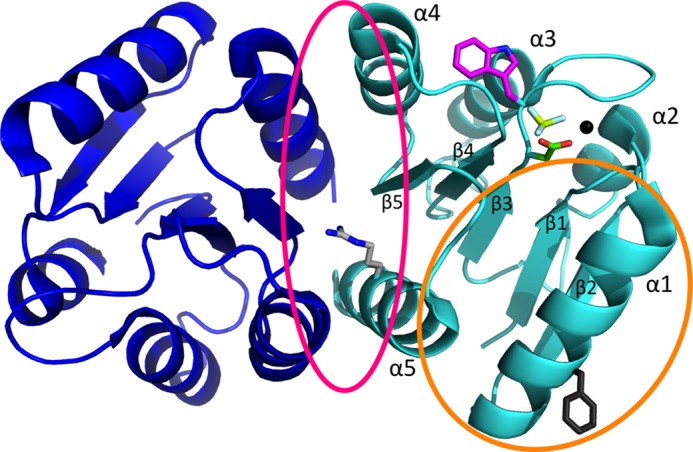
Ribbon representation of the physiologically relevant PhoBN dimer (dimer 1). The crystal structure shown is of PhoBN bound to the phosphoryl group analog BeF3− (Protein Data Bank code 1ZES). The two (βα)5 monomers that compose the dimer are colored blue (1ZES chain B) and cyan (1ZES chain C). Phosphoryl group analog BeF3− (yellow and white) is bound to Asp-53 (green). Also shown are the active site magnesium ion (black sphere) and the side chain for the Trp-54 fluorophore (magenta). The dimer 1 interface (pink circle) is composed of residues on helix α4, strand β5, and helix α5 and includes Arg-115 (light gray). The nonphysiological dimer 2 interface (orange circle) is composed of residues on helix α1, β5α5 loop, and helix α5 and includes Phe-20 (dark gray).
Although autophosphorylation has been qualitatively demonstrated for many response regulators, CheY is the only response regulator for which extensively characterized autophosphorylation kinetics have been published (11, 13, 15, 16). The model of CheY autophosphorylation kinetics (in Scheme 1 with the phosphodonor PAM (12)) involves initial binding of phosphodonor to CheY to form a noncovalent complex (dissociation constant KS), followed by phosphorylation (rate constant kphos) and subsequent autodephosphorylation (rate constant kdephos).
SCHEME 1.
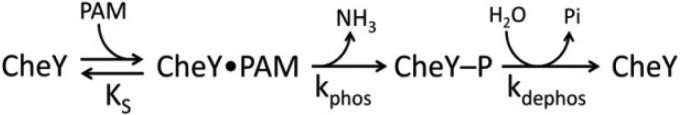
Kinetic model of autophosphorylation of the response regulator CheY.
For CheY, the pre-steady state pseudo-first order rate constant for the accumulation of phosphorylated CheY (CheY-P) upon mixing CheY with phosphodonor remains linear with respect to PAM or acetyl phosphate concentration up to the highest concentrations of phosphodonor tested (100 mm), indicating very weak binding (KS >500 mm) (11, 15, 16). Thus for CheY, it is only possible to measure the apparent bimolecular rate constant, kphos/KS, for accumulation of CheY-P, which is ∼10 m−1 s−1 for autophosphorylation with either acetyl phosphate or PAM (11, 13, 15, 16). However, CheY is not a typical response regulator in several respects. CheY consists only of a receiver domain and thus does not contain a variable output domain. Also, CheY does not act as a transcriptional regulator and does not dimerize upon phosphorylation.
To further explore response regulator autophosphorylation kinetics, we sought to characterize a more typical response regulator. PhoB is a member of the largest class of response regulators, termed the OmpR/PhoB family, which contains ∼30% of annotated response regulators (4). Like many response regulators, PhoB dimerizes through its receiver domain, allowing two output domains to come together and bind to a DNA-binding site on the promoter to regulate transcription (17). PhoB can autophosphorylate with acetyl phosphate or PAM (18–21) and contains an active site tryptophan (Fig. 1) that can be used as a fluorescent probe for both metal binding and autophosphorylation (20).
Analysis of structures of numerous receiver domains shows that phosphorylation or binding of a phosphoryl group analog to the active site Asp leads to conformational rearrangements on the α4-β5-α5 surface (3, 5, 22). These conformational shifts occur via a conserved mechanism of allosteric communication between the site of phosphorylation and the functionally relevant α4-β5-α5 surface. In dimerizing response regulators like PhoB (20, 23, 24), this conformational change allows two monomers to dimerize along the α4-β5-α5 surface (Fig. 1) (5). The coupling of phosphorylation and conformational change within the receiver domain occurs in both directions. Just as phosphorylation of response regulators promotes the active conformation, increasing the population of response regulator in the active conformation (e.g. binding target ligand) increases the rate of autophosphorylation (16, 25).
The PhoB receiver domain (PhoBN) has been demonstrated to form two different types of dimers. The two PhoBN dimers have been extensively characterized both structurally and biochemically (26–29). As expected, a dimer along the α4-β5-α5 surface (here termed dimer 1) forms in the presence of a phosphoryl group analog (29) and represents the functional, phosphorylation-mediated dimer. Unexpectedly, metal binding alone supports a modified version of dimer 1 (27). Phosphorylated full-length PhoB (PhoBF) dimer (dimer 1) has a dimerization constant of 5.1 μm (28) measured by analytical ultracentrifugation. A second PhoBN dimer, here termed dimer 2, forms along the α1-β5α5 loop-α5 surface (Fig. 1) and has been observed under crystallographic conditions in either the absence of metal and phosphorylation (26) or in the presence of just metal (single crystallographic conditions resulted in PhoBN monomers in both dimer 1 and dimer 2 within the asymmetric unit (27)). Dimer 2 may not be physiologically relevant due to a dimerization constant (>350 μm (28)) that is substantially higher than the in vivo concentration of PhoB (30).
Here, we present kinetic characterization of autophosphorylation of the Escherichia coli response regulator PhoB with the phosphodonor PAM. PhoB autophosphorylation exhibited several kinetic features that were fundamentally different from those of CheY. In contrast to the linear relationship between the pseudo-first order rate constant for accumulation of CheY-P and PAM concentration, this relationship for PhoB appeared sigmoidal and to approach saturation. Furthermore, unlike CheY, apparent pseudo-first order rate constants for PhoB autophosphorylation showed a protein concentration dependence. Additional biochemical evidence, supported by mathematical modeling, indicated a link between formation of PhoB dimer 1 and autophosphorylation kinetics. We propose that a heterodimer formed between a phosphorylated monomer and an unphosphorylated monomer of PhoB plays a role in PhoB autophosphorylation kinetics.
EXPERIMENTAL PROCEDURES
Cloning and Site-directed Mutagenesis
Genes encoding E. coli full-length PhoB (PhoBF, residues 1–256) and the N-terminal receiver domain of PhoB (PhoBN, residues 1–127) were amplified using standard PCR methods from the pENTR-b0399 (phoB) plasmid (31), provided by Dr. Michael Laub (Massachusetts Institute of Technology). The gene encoding E. coli CheY was amplified using standard PCR methods from pRS3 (32). PCR products were inserted into the pET28a vector (Novagen) to generate constructs (PhoBF, pET28a·phoBF; PhoBN, pET28a·phoBN; and CheY, pKC1) that encode proteins with a thrombin-cleavable His6 tag at the N terminus. After thrombin cleavage, the resultant proteins contain an additional Gly-Ser-His at the N terminus. Addition of the three small residues is unlikely to affect the activity of the PhoB constructs because the N terminus is on the opposite side of the protein from the site of phosphorylation and is in a portion of PhoB that does not undergo a conformational change between the inactive and the active conformation. The presence of the same three residues in CheY did not affect the rates of autophosphorylation by PAM (data not shown) or autodephosphorylation (33). Site-directed mutagenesis was performed using QuikChange technology (Agilent Technologies). Wild type and mutagenized plasmids were transformed into E. coli strain BL21(DE3) for protein expression and purification.
The Gateway Entry vector for phoR (pENTR-b0400 (31)) encoding the soluble portion of E. coli PhoR (residues 193–431, throughout referred to as PhoR)) and the Gateway Destination vector (pML375) were provided by Dr. Michael Laub (Massachusetts Institute of Technology). The gene encoding PhoR was transferred from the entry vector into the destination vector using standard Gateway protocols (Invitrogen) to generate a construct (pDEST·phoR) that encodes a protein with a thrombin-cleavable His6 tag at the N terminus.
Synthesis of Phosphoramidate
The potassium salt of PAM was synthesized as described (34).
Protein Overexpression and Purification
For the purification of CheY, PhoBF, PhoBN, PhoBN F20D, and PhoR, 1 liter of LB (+30 μg/ml kanamycin) was inoculated with 2.5 ml of an overnight culture of the appropriate expression strain and grown at 37 °C to an absorbance at 600 nm of 0.4. The culture was then transferred to room temperature, induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and grown for 8–10 h. Cells were collected by centrifugation and resuspended in lysis buffer (50 mm Hepes, pH 7.0, 300 mm NaCl, 10 mm imidazole). The cells were lysed by sonication, and cellular debris was removed by ultracentrifugation (45 min, 4 °C, 37,000 × g). The supernatant was passed over a nickel-NTA-agarose (Qiagen) column, and the column was subsequently washed with wash buffer (50 mm Hepes, pH 7.0, 300 mm NaCl, 20 mm imidazole). Protein was eluted with elution buffer (50 mm Hepes, pH 7.0, 300 mm NaCl, 150 mm imidazole). All eluted proteins except PhoR were incubated with human α-thrombin (1 unit/ml sample, Hematologic Technologies, Inc.) overnight at 4 °C, followed by gel filtration over a Superdex 75 16/60 column in 50 mm Hepes, pH 7.0, 100 mm KCl, 2 mm DTT, 10% (v/v) glycerol to remove the thrombin and cleaved His6 tag (for CheY, DTT was omitted from the gel filtration buffer). PhoR was further purified using a Superose 12 gel filtration column in 50 mm Tris, pH 7.5, 500 mm NaCl, and 10% (v/v) glucose. Aliquots of purified proteins were flash-frozen in dry ice/ethanol and stored at −80 °C.
PhoBN R115D was expressed and purified similarly to the above procedure with slight modifications based on a published purification protocol (28). One liter of LB (+30 μg/ml kanamycin) with 1 m sorbitol and 10 mm betaine was inoculated with 2.5 ml of an overnight culture in LB and grown at 37 °C to an absorbance at 600 nm of 0.4. The culture was transferred to room temperature, induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside, and grown for 17–20 h. The cell pellet was resuspended in lysis buffer containing 140 μg of Halt protease inhibitor mixture (Thermo Scientific) followed by sonication, nickel-NTA chromatography, and thrombin cleavage as above. The Superdex 75 gel filtration buffer contained 50 mm Hepes, pH 7.0, 2 mm DTT, and 10% (v/v) glycerol.
Fluorescence Measurements
All fluorescence measurements were made using a PerkinElmer Life Sciences LS-50B fluorimeter equipped with a magnetic stirring device and FLWinLab software. Tryptophan fluorescence was measured at an excitation wavelength of 295 nm and an emission wavelength of 346 nm. Sample temperature was held at 25 ± 0.5 °C by a circulating water bath.
Equilibrium Fluorescence Measurements
Tryptophan fluorescence was used to monitor binding of divalent cation (Mn2+ or Mg2+) or beryllium trifluoride (BeF3−) to various PhoBN constructs. All ligand binding assays were performed in triplicate in a 1 × 1-cm quartz cuvette at an initial volume of 1.5 ml.
Divalent cation binding was assayed similarly to a published protocol (35). PhoBN (5 μm) or PhoBN F20D (5 μm) in 100 mm Hepes, pH 7.0, was allowed to come to temperature equilibrium with constant magnetic stirring, and the initial fluorescence emission was measured. Then small volumes of a concentrated stock of MnCl2 or MgCl2 were incrementally added to the solution. After each addition, the solution was incubated for 2 min to ensure a homogeneous mixture. Ten fluorescence measurements were taken (1 measurement/s), and the values were averaged and corrected for dilution. Altogether, fluorescence was measured for 15 concentrations of divalent cation ranging between 5 and 1000 μm MnCl2 and between 0.25 and 5 mm MgCl2.
For BeF3− binding, PhoBN (5 μm) or PhoBN F20D (5 μm) was incubated in 100 mm Hepes, pH 7.0, 3 mm MnCl2, and 10 mm NaF with constant stirring to reach temperature equilibrium. Small volumes of a concentrated stock of BeCl2 were incrementally added to give concentrations between 1 and 100 μm. After each addition of BeCl2, the solution was incubated for 2 min. BeCl2 reacts with NaF to form a series of beryllium fluoride adducts, which should be dominated by BeF3− at 10 mm NaF (36). Thus, the concentration of BeF3− is assumed to be equal to the concentration of BeCl2. Ten independent fluorescence measurements were taken (1 measurement/s) for each concentration of BeCl2, and the values were averaged and corrected for dilution. To correct for nonspecific interactions between PhoBN and BeCl2, titrations were also performed in the absence of NaF. There was a small increase in fluorescence upon the first addition of BeCl2, which was subtracted from the fluorescence intensity for the titrations that contained 10 mm NaF.
The above protocols were modified slightly for measurements of ligand binding to PhoBN R115D. PhoBN R115D exhibited a modest time-dependent quench in fluorescence, which stabilized within 15 min, in response to changes in solution ionic strength. The initial solutions containing 5 μm PhoBN R115D in buffer were thus allowed to incubate for 30 min at 25 °C. An additional control titration was carried out with KCl (0–15 mm) in lieu of Mn2+, Mg2+, or BeF3− to quantify the ionic strength response. For all titrations, solutions were incubated for 15 min after addition of ligand and before fluorescence measurements were taken. The changes in fluorescence due to changes in ionic strength as determined by KCl titration were subtracted from the fluorescence intensity at each concentration of ligand. For divalent metal and BeF3− binding, the changes in fluorescence due to changes in ionic strength were less than 5% of the total change in fluorescence.
For both divalent cation and BeF3− binding, plots of (I0 − I) versus ligand concentration were constructed, where I0 is the fluorescence intensity with no added ligand, and I is the fluorescence intensity at a given concentration of ligand. Plots of BeF3− binding to PhoBN and PhoBN F20D were sigmoidal, and data were fit to Equation 1 (GraphPad Prism) to determine n, the Hill coefficient, and K½, the concentration of BeCl2 required to acquire half (I0 − Imax).
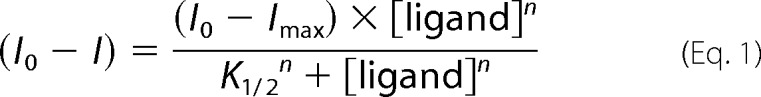 |
For determining all other ligand binding constants, a standard one-site binding equation was used.
Fluorescence Measurements of Reaction Kinetics
Tryptophan fluorescence was used to continuously monitor autophosphorylation of both CheY and various PhoB constructs with PAM. Observed rate constants for autophosphorylation of CheY at a given concentration of PAM decrease with increasing ionic strength of the solution (13). Thus, all reactions were performed at a constant ionic strength of 400 mm, which includes the concentration of buffer, divalent cation, PAM, and KCl.
Fluorescence measurement of CheY autophosphorylation was similar to previous reports (12, 15, 16). Briefly, a rapid mixing device (Applied Photophysics RX2000) was used to mix equal volumes of 10 μm CheY (in 6 mm MnCl2 and 100 mm Hepes, pH 7.0) and PAM (20–300 mm in 100 mm Hepes, pH 7.0). Both CheY and PAM solutions were supplemented with KCl to give a final ionic strength of 400 mm. Because CheY and PAM solutions were mixed at a one-to-one (v/v) ratio, the concentrations of CheY, MnCl2, and PAM were decreased by half in the final reaction. Fluorescence was continuously monitored, and time points were recorded every 20 ms. Reactions were performed in duplicate for each experimental condition.
Autophosphorylation reactions for PhoBF, PhoBN, and PhoBN F20D were carried out in a 1 × 0.4-cm quartz cuvette with manual mixing to initiate the reaction. The beginnings of the time traces were identical when measured using a stopped-flow apparatus or the standard cuvette. However, the ends of the time traces showed variability when measured using the stopped-flow apparatus, likely due to diffusion over the longer period of time required to achieve steady state for PhoB (up to 35 min). PhoBF (5 μm), PhoBN (2.5–20 μm), or PhoBN F20D (5 μm) was incubated in 100 mm Hepes, pH 7.0, 3 mm MnCl2, and varying concentrations of KCl to reach a constant ionic strength of 400 mm (after PAM addition) with magnetic stirring to reach temperature equilibrium. The autophosphorylation reaction was initiated by addition of PAM (10–150 mm final concentration) to bring the final volume to 750 μl, followed by repetitive pipetting to mix (<5 s), and fluorescence was continuously monitored. Time points were recorded every 20 or 100 ms. Reactions were performed in triplicate for each experimental condition.
Autophosphorylation time traces of PhoBN R115D were measured in a 1 × 1-cm quartz cuvette at a final volume of 1.5 ml. PhoBN R115D (10 μm) was incubated in 6 mm MnCl2, 100 mm Hepes, pH 7.0, and 282 mm KCl at a volume of 750 μl for 30 min with magnetic stirring. Because of the effect of ionic strength on PhoBN R115D fluorescence (see above), it was necessary to keep the ionic strength of the whole system at 400 mm throughout the experiment. For that reason, the autophosphorylation reaction was initiated by addition of an equal volume of PAM (20–300 mm) in 100 mm Hepes, pH 7.0, and varying concentrations of KCl to maintain a constant ionic strength of 400 mm, followed by mixing by repetitive pipeting (<5 s), and fluorescence was continuously monitored. Because PhoBN R115D and PAM solutions were mixed at a one-to-one (v/v) ratio, the concentrations of PhoBN R115D, MnCl2, and PAM were decreased by half. Time points were recorded every 0.5 s. Reactions were performed in triplicate for each experimental condition.
Analysis of Kinetic Data
Autophosphorylation reactions were performed under pseudo-first order conditions, with a large molar excess of PAM over protein. Fluorescence time traces reflected a continually changing reaction rate, as the amount of unreacted CheY or PhoB decreased, although PAM concentration was essentially unchanged. All time traces were normalized by dividing the fluorescence intensities by the initial fluorescence intensity (I0) to generate relative fluorescence intensity (RFI). Replicate time courses for the same protein or PAM concentration were averaged. The averaged, normalized fluorescence intensities are referred to as “time traces” for the rest of this study. Initial curve fits were made to a single exponential decay (GraphPad Prism). For CheY and PhoBN R115D, the time traces gave excellent fits to a single exponential, and the resulting pseudo-first order rate constants were termed observed rate constants (kobs). For PhoBF, PhoBN, and PhoBN F20D, there were modest but systematic deviations from a single exponential decay. Therefore, we also tried fitting these data to a two-phase exponential decay, but the fits were indistinguishable from a single exponential. As described under “Results,” there was utility in using the single exponential fits for initial data analysis, so the rate constants derived from imperfect single exponential fits were termed apparent rate constants (kapp). The simple kinetic model shown in Scheme 1 implicates the relationship between the pseudo-first order rate constant kobs and individual kinetic parameters, shown in Equation 2 (12). For CheY·Mn2+, plots of kobs versus PAM concentration were linear, consistent with results obtained with CheY·Mg2+ (11, 15, 16). This result indicates that the dissociation constant between CheY·Mn2+ and PAM is much larger than the highest concentration of PAM used in the experiment (KS ≫ [PAM]), reducing Equation 2 to Equation 3. Thus the slope of the line is equal to the apparent bimolecular rate constant (kphos/KS). For PhoBN R115D, plots of kobs versus the concentration of PAM were also linear, and the slope was similarly used to determine kphos/KS. Plots of the pseudo-first order rate constant kapp versus the concentration of PAM for PhoBF, PhoBN, and PhoBN F20D appeared sigmoidal and were thus fit using a modified Hill equation (Graph Pad Prism), Equation 4, where kmax is the maximum change in apparent rate constant; K½ is the PAM concentration required to reach the mid-point of the dose-response curve; n is the Hill coefficient; and kmin is the minimum apparent rate constant.
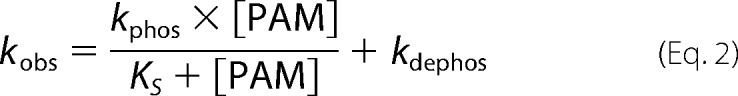 |
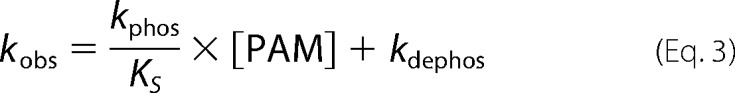 |
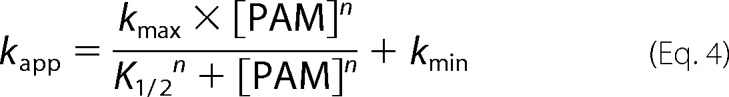 |
Mathematical Modeling of PhoBN Autophosphorylation Time Traces
Mathematical modeling of PhoBN autophosphorylation time traces was performed using DynaFit software (37) to potentially explain several features of the kinetic data, including an apparent lag at the beginning of the PhoBN autophosphorylation time traces and the observation that protein concentration affected the apparent pseudo-first order rate constant for autophosphorylation. As described under “Results,” numerous models of increasing complexity were tried before choosing the model described below. In particular, the protein concentration dependence of normalized PhoBN time traces constrained the model significantly because it necessitated the inclusion of a reaction step whereby multiple molecules of PhoBN interact to give reaction species with altered phosphorylation kinetics. Thus, based on a large body of evidence documenting the dimerization of PhoBF and PhoBN (20, 23, 24, 26–29), a comprehensive model of autophosphorylation kinetics of PhoB that included multiple versions of PhoB dimer 1 with the possibility of differing autophosphorylation kinetics was considered (Scheme 2). In the model, a monomer (m) of PhoB can bind to a molecule of PAM with dissociation constant KS(m) and then phosphorylate with rate constant kphos(m), analogous to the first two steps in Scheme 1 for CheY. A phosphorylated PhoB monomer can then interact with either another phosphorylated PhoB monomer to form a homodimer (PhoB-P/PhoB-P) or with an unphosphorylated PhoB monomer to form a heterodimer (PhoB/PhoB-P), with dimerization constants Kdimer(hom) and Kdimer(het), respectively. The heterodimer can then bind to a molecule of PAM with dissociation constant KS(het) and phosphorylate with rate constant kphos(het). All three phosphorylated species of PhoB can also dephosphorylate, with rate constants kdephos(hom), kdephos(het), and kdephos(m). Dimerization of two unphosphorylated PhoB monomers (dimer 2) was excluded from the model due to a measured dimerization constant (378 μm (28)) that was much greater than the highest concentration of PhoB used in our experiments.
SCHEME 2.

Reactions included in mathematical model of autophosphorylation of PhoB.
To fit the kinetic data to the model, the normalized fluorescence intensities for PhoBN autophosphorylation time traces were first multiplied by the protein concentration used in the experiment. DynaFit requires a conversion factor that translates the concentration of different PhoB species to the measured fluorescence responses. We established conversion factors based on the assumption that tryptophan fluorescence was quenched solely as a result of the conformational change that occurs upon PhoB phosphorylation or formation of the heterodimer but not by PAM binding. Thus, the species that are not primarily in the active conformation (PhoB and PhoB·PAM) were assigned a conversion factor equal to the initial fluorescence intensity divided by the protein concentration. The species that are primarily in the active conformation (PhoB-P, PhoB/PhoB-P, PhoB·PAM/PhoB-P, and PhoB-P/PhoB-P) were assigned a conversion factor equal to the minimum fluorescence intensity for autophosphorylation of 5 μm PhoBN with 150 mm PAM (i.e. the time trace with the maximum quench in fluorescence intensity) divided by the protein concentration. The described assumptions for the conversion factors gave good fits. Alternative assumptions were also tested (e.g. PAM binding to PhoBN resulted in quenched tryptophan fluorescence). However, alternative conversion factors could not by themselves account for fundamental features of the kinetics, such as the lag phase observed in the time traces or the protein concentration dependence of the apparent pseudo-first order rate constant.
An initial model composed of the nine reactions described above gave good fits for 10 of the 12 autophosphorylation time traces representing different concentrations of protein or PAM. However, there were systematic deviations for the two experimental conditions with the slowest time scales for autophosphorylation (10 and 20 mm PAM). Specifically, the rate and equilibrium constants predicted by DynaFit gave modeled time traces that had the expected initial lag phases; however, the modeled plateaus at long reaction times were up to 30% below the experimental data. Slow formation of a PhoB aggregate (PhoBa) was added to the model with a rate constant of kagg(m). To compensate for the higher plateau observed in the experimental time traces, the conversion factor for PhoBa was set to three times the initial fluorescence intensity divided by the protein concentration. This larger conversion factor is consistent with light scattering due to formation of a soluble aggregate.
Next, we set the following four parameters to experimentally measured values. Kdimer(hom) was set to the published value of 5.1 μm (28). (For all equilibrium constants, we assumed rapid equilibrium and set the kon rate constant equal to 1 × 106 m−1 s−1 (38).) The three autodephosphorylation rate constants were determined by monitoring the disappearance of [32P]PhoBN-P or [32P]PhoBN R115D-P over time (see “Experimental Procedures” below, data not shown). The rate constant kdephos(hom) was set equal to the rate constant determined for [32P]PhoBN-P (0.0003 s−1). Because PhoBN R115D cannot form dimer 1, kdephos(m) was set equal to the rate constant determined for [32P]PhoBN R115D-P (0.0002 s−1). Because the model did not include formation of the unphosphorylated PhoB dimer, autodephosphorylation of the heterodimer was equivalent to dimer dissociation followed by monomer dephosphorylation. For that reason, kdephos(het) was also set equal to the rate constant determined for [32P]PhoBN R115D-P. Note that the measured kdephos values for PhoBN are in close agreement with previously reported values for kdephos for PhoBF (30, 35, 39).
The remaining six rate and equilibrium constants (KS(m), kphos(m), Kdimer(het), KS(het), kphos(het), and kagg(m)) were then manually varied until the modeled time traces were similar to the raw time traces for autophosphorylation of 5 μm PhoBN with 60 mm PAM. These modeled values were then used as initial constants for global analysis of all of the time traces for PhoBN autophosphorylation (i.e. 2.5–20 μm PhoBN with 60 mm PAM and 5 μm PhoBN with 10–150 mm PAM). Nonlinear least square regressions were performed to simultaneously fit the six adjustable rate and equilibrium constants: KS(m), kphos(m), Kdimer(het), KS(het), kphos(het), and kagg(m). Perturbing the initial values for the rate and equilibrium constants did not affect the resultant output. The residuals that were output by DynaFit were divided by protein concentration to allow for comparison between protein concentrations.
Radiolabeling of PhoR Using [γ-32P]ATP
His6-tagged PhoR (230 μg) was reacted with 1 mm [γ-32P]ATP (18 Ci/mmol) in 35 mm Tris, pH 8.0, 35 mm KCl, and 3.5 mm MgCl2 in a total volume of 200 μl for 30 min at room temperature. The sample was passed over 100 μl of prewashed nickel-NTA resin in a small centrifugal 0.22-μm filtration device and washed 10 times with 400 μl of wash buffer (50 mm Hepes, pH 7.0, 300 mm NaCl, 20 mm imidazole) to remove excess [γ-32P]ATP. The [32P]PhoR-P was eluted 3× with 400 μl of elution buffer (50 mm Hepes, pH 7.0, 300 mm NaCl, 150 mm imidazole) and concentrated. The concentration of [32P]PhoR-P was determined using a Bradford assay with BSA standards. Purified aliquots of [32P]PhoR-P were stored at −20 °C.
Phosphotransfer from [32P]PhoR-P to PhoBN
[32P]PhoR-P was thawed and diluted into 35 mm Tris, pH 8.0, 35 mm KCl, and 3 mm MnCl2 to give a final protein concentration of 1 μm and a final volume of 120 μl. A 7.5-μl aliquot was removed and added to 7.5 μl of 2× SDS-PAGE buffer for a zero time point. Either PhoBN or PhoBN R115D was added at a 10-fold molar excess (10 μm) and mixed, and a series of 7.5-μl time points up to 10 min were removed and immediately combined with 2× SDS-PAGE buffer. Samples were electrophoresed on 18% polyacrylamide gels, and the gels were subsequently dried. The amount of [32P]PhoR-P at each time point was determined by phosphorimaging. The percentage of [32P]PhoR-P remaining was plotted against time and then fit to a one-phase exponential decay to determine the rate constant for phosphotransfer.
Rate Constants for [32P]PhoB-P Autodephosphorylation
His6-tagged PhoR (6 μm) was reacted with 1 mm [γ-32P]ATP (18 Ci/mmol) in 35 mm Tris, pH 8.0, 35 mm KCl, and 3 mm MnCl2 in a total volume of 50 μl for 15 min at room temperature. The sample was passed over 100 μl of prewashed nickel-NTA resin in a small centrifugal 0.22-μm filtration device to bind His6-tagged PhoR and washed 10 times with 400 μl of buffer containing 35 mm Tris, pH 8.0, 35 mm KCl, and 3 mm MnCl2 to remove excess [γ-32P]ATP. A 100-μl sample of either PhoBN (30 μm) or PhoBN R115D (30 μm) in 35 mm Tris, pH 8.0, 35 mm KCl, and 3 mm MnCl2 was added to the column and incubated for 5 min at room temperature. The sample was allowed to filter over the nickel-NTA resin, and the nonbound material was collected. 7.5-μl samples were removed and added to an equal volume of 2× SDS-PAGE buffer at the following time points: 5, 10, 15, 30, 45, 60, 90, and 120 min. Samples were electrophoresed on 18% polyacrylamide gels. The gels were dried, and the amount of [32P]PhoBN-P at each time point was determined by phosphorimaging. The percentage of [32P]PhoBN-P remaining was plotted against time and then fit to a one-phase exponential decay to determine the rate constant for autodephosphorylation.
RESULTS
Optimization of PhoB Autophosphorylation Reaction Conditions
A divalent cation bound in the receiver domain active site is essential for catalysis of both response regulator phosphorylation and dephosphorylation (12, 40). Therefore, as a prerequisite for measuring PhoB autophosphorylation kinetics, it was necessary to characterize the metal binding properties of the various PhoB proteins (full-length PhoB (PhoBF), the N-terminal receiver domain (PhoBN), PhoBN F20D, and PhoBN R115D) so that the occupancy of the metal-binding site was known for subsequent autophosphorylation experiments. Tryptophan fluorescence measurements indicated that binding between Mg2+ and PhoBN occurred with a dissociation constant (KD) of 1.6 ± 0.2 mm, in agreement with the reported KD for PhoBF Mg2+ binding of 2.0 ± 0.5 mm (35). However, the concentration of Mg2+ required to reach high levels of metal-bound PhoB (> 95%) led to visible precipitation of both PhoBN and PhoBF within the time scale of the autophosphorylation reactions.
To avoid complications associated with precipitation, we searched for another metal ion that bound to PhoB and could support autophosphorylation. Although Mg2+ is most likely the divalent cation utilized by PhoB in vivo, Mg2+ and Mn2+ have similar size and preferred coordination number, geometry, and ligand type (41). Furthermore, Mn2+ supports autophosphorylation of the response regulator Spo0F with PAM (42), autophosphorylation of several sensor kinases, and phosphotransfer from a sensor kinase to a response regulator in vitro (43–45). Binding of Mn2+ to both PhoBF and PhoBN was much tighter than Mg2+, with a KD of 82 ± 6 and 70 ± 4 μm, respectively. Mn2+ bound to PhoBN F20D and PhoBN R115D with similar affinities (KD of 51 ± 5 and 20 ± 8 μm, respectively). Furthermore, Mn2+ was highly capable of mediating PhoBN autophosphorylation, and the concentration of Mn2+ required for >95% occupancy of the metal-binding site (3 mm MnCl2) did not lead to visible precipitation. Thus, the standard conditions for all autophosphorylation reactions in this study included 3 mm MnCl2 to ensure high occupancy of the metal-binding site. Autophosphorylation of CheY was also readily catalyzed by 3 mm Mn2+ (see below); thus, all autophosphorylation experiments were done in parallel for PhoB and CheY to rule out the possibility that any novel characteristics of PhoB autophosphorylation were due to the use of Mn2+ as opposed to Mg2+.
The visible precipitation that occurred between Mg2+ and PhoB was more severe for PhoBF than for PhoBN. Although no visible precipitation occurred in solutions containing Mn2+ and either PhoBF or PhoBN, we wanted to take every measure to minimize formation of aggregates during autophosphorylation experiments. For that reason, the majority of the autophosphorylation reactions was performed using the highly soluble PhoBN. As expected from previous work (46), the removal of the DNA-binding domain did not affect autophosphorylation of PhoB (see below).
Normalized Autophosphorylation Time Traces for PhoBN Displayed a Visible Lag Phase and a Protein Concentration Dependence
Autophosphorylation of both PhoBN and CheY at various PAM and protein concentrations was monitored by continuous measurement of tryptophan fluorescence. In all cases, PAM concentration exceeded protein concentration by at least 3 orders of magnitude, so the reactions occurred under pseudo-first order conditions. For both CheY and PhoBN, autophosphorylation was initiated by the addition of PAM, which resulted in a time-dependent quench in fluorescence that eventually leveled out (Fig. 2, A–D). The fluorescence intensity is a relative measure of the progress of the reaction from all unphosphorylated CheY or PhoBN (intensity at zero time) to a steady state level of phosphorylated CheY or PhoBN (intensity after steady state is achieved). Achievement of steady state indicates equal rates of autophosphorylation and autodephosphorylation. Because the absolute magnitude of the initial fluorescence intensity varied for individual reactions (especially for different protein concentrations), time traces (Fig. 2) were normalized by dividing by the initial fluorescence intensities and are reported as RFI.
FIGURE 2.

Normalized CheY and PhoBN autophosphorylation time traces at various substrate or protein concentrations. Fluorescence measurement of autophosphorylation of 5 μm CheY (A) and 5 μm PhoBN (B) at the following PAM concentrations: 10 mm (purple), 20 mm (gray), 40 mm (dark blue), 60 mm (light blue), 80 mm (orange), 100 mm (green), 125 mm (black), and 150 mm (red). Insets to A and B show a magnified view of the first portions of the autophosphorylation time traces. C, autophosphorylation of 2.5 μm (green), 5 μm (orange), and 10 μm (purple) CheY with 60 mm PAM. D, autophosphorylation of 2.5 μm (green), 5 μm (orange), 10 μm (purple), 15 μm (red), and 20 μm (blue) PhoBN with 60 mm PAM. RFI is the relative fluorescence intensity and represents the normalized tryptophan fluorescence intensity obtained by dividing all fluorescence intensities by the initial fluorescence intensity. Reactions were carried out in duplicate for A and C and in triplicate for B and D. Time traces represent the mean of multiple reactions, with standard deviations (not shown for clarity) that were less than 3% of the mean. Note that the time scales of all panels are different.
As expected, the time traces for CheY·Mn2+ autophosphorylation (Fig. 2A) displayed similar properties as those reported for CheY·Mg2+ (11, 13). The shapes of the time traces for CheY·Mn2+ autophosphorylation were the same as for CheY·Mg2+ and occurred on a similar time scale (20–100 s). Also, the maximum quench in fluorescence due to autophosphorylation was ∼50% for both CheY·Mn2+ and CheY·Mg2+. Because of the pseudo-first order reaction conditions employed ([PAM] ≫ [CheY]) and the fact that fluorescence is a measure of the relative progress of the reaction, increasing the concentration of PAM resulted in a shift of the time traces to shorter reaction times (Fig. 2A), but varying the concentration of CheY did not affect the time frame of the normalized time traces (Fig. 2C).
However, comparison of autophosphorylation time traces for CheY (Fig. 2A) and PhoBN (Fig. 2B) revealed three evident differences. First, autophosphorylation of PhoBN was markedly slower than autophosphorylation of CheY, although the PhoBN time traces were still dependent on PAM concentration. For example, at 60 mm PAM, it took ∼500 s for autophosphorylation of PhoBN to reach steady state, while it only took ∼35 s for CheY. Second, the PhoBN autophosphorylation time traces displayed a visible lag phase (Fig. 2B, inset) that was not present in time traces for CheY autophosphorylation (Fig. 2A, inset). This lag was most evident during the first 100 s of the autophosphorylation time traces for 40 and 60 mm PAM. Finally, the half-life of the normalized autophosphorylation time traces of PhoBN clearly decreased with increasing protein concentrations (2.5–20 μm) (Fig. 2D); however, CheY autophosphorylation time traces did not show a protein concentration dependence (Fig. 2C).
Both the lag and the protein concentration dependence of normalized time traces suggested that PhoBN autophosphorylation occurred with a more complex kinetic mechanism than CheY. Protein concentration dependence was inconsistent with a reaction in which a single molecule of PhoBN participates but was consistent with a reaction that involves interactions between two or more PhoBN molecules. The PhoBN protein concentration dependence, in particular, suggested that interactions between PhoBN monomers play a role in the kinetic mechanism of PhoBN autophosphorylation.
Initial Analysis of PhoBN Autophosphorylation Suggested Positive Cooperativity with Respect to PAM
To analyze the kinetics of autophosphorylation more quantitatively, we fit the autophosphorylation time traces to a one-phase exponential decay and plotted the resultant rate constants against PAM concentration. For CheY, the time traces fit extremely well to a one-phase exponential decay (Fig. 3A), with low residuals (difference between data and curve fitting) (< ±0.25%) that displayed no obvious pattern. The CheY rate constants from these fits were termed kobs. For CheY·Mn2+, the plot of kobs versus PAM concentration was linear, consistent with results obtained with CheY·Mg2+ (11, 15, 16), and the slope of the plot was equal to kphos/KS. The kphos/KS for reaction of CheY·Mn2+ with PAM was 2.2 ± 0.04 m−1 s−1, which was ½ to ⅓ the kphos/KS of 6.1 m−1 s−1 for reaction of CheY·Mg2+ (after corrections for ionic strength (15)).
FIGURE 3.

Analysis of CheY and PhoBN time traces by fitting to a single exponential decay. Autophosphorylation time traces for reaction of 5 μm CheY (A) or 5 μm PhoBN (B) with 60 mm PAM are shown in gray. The best fit to a one-phase exponential decay is shown in dashed black lines. The rate constant from the fit is kobs for CheY and kapp for PhoBN. Residuals (the difference between the raw data and the one-exponential decay) are shown at the bottom. C, plot of kobs versus concentration of PAM for 5 μm CheY fit with linear regression (see Equation 3). The slope of the line gave kphos/KS, and the y intercept gave kdephos. Error bars represent standard deviation from duplicate experiments. D, plot of kapp versus concentration of PAM for 5 μm PhoBN fit with sigmoidal dose-response curve (see Equation 4). Error bars represent standard deviation from triplicate experiments. Reactions were performed in duplicate for A and C and in triplicate for B and D. Time traces represent the mean of multiple reactions, with standard deviations (not shown for clarity) that were less than 3% of the mean. Most error bars in C and D were smaller than data points.
In contrast to CheY and consistent with the visible lag, the PhoBN autophosphorylation time traces exhibited systematic deviations from a one-phase exponential decay (Fig. 3B). The residuals for PhoBN followed a pattern and were 4–52 times larger than for CheY throughout the entire time course (Fig. 3, A and B, lower panel). Despite the systematic deviations from a single exponential decay, R2 values for fitting PhoBN autophosphorylation time traces with a one-phase exponential decay were greater than 0.98. Rate constants obtained from the single exponential fits were used to estimate the overall rate of acquisition of steady state; however, due to the systematic deviations, we labeled these apparent rate constants kapp and used these values as an initial tool for analyzing the data.
A plot of kapp versus PAM concentration for PhoBN (Fig. 3D) differed from the plot of kobs versus PAM concentration for CheY (Fig. 3C) in two ways. First, the plot of kapp versus PAM concentration for PhoBN fit to a sigmoidal dose-response curve with a Hill coefficient of 2.1 ± 0.2, which suggested that autophosphorylation of PhoBN exhibited some sort of positive cooperativity. The apparent positive cooperativity indicated communication between PhoBN active sites, such that autophosphorylation of one active site led to enhanced autophosphorylation kinetics at other sites, another suggestion that multimerization affects the rate of autophosphorylation of PhoBN. Second, in contrast to kobs for CheY, which showed no signs of saturating at the PAM concentrations used, kapp for PhoBN appeared to saturate at high PAM concentrations.
Previous work on PhoB autophosphorylation showed that removal of the DNA-binding domain did not affect the rate of PhoB autophosphorylation with 20 mm PAM (46). As expected, PhoBF autophosphorylation time traces with multiple PAM concentrations also showed a visible lag (Fig. 4A) and protein concentration dependence (data not shown) similar to PhoBN. Also, plots of kapp versus PAM concentration for PhoBF (Fig. 4B) were nearly superimposable on PhoBN (Fig. 3D, Hill coefficient of 2.1 ± 0.3) and also appeared saturable. Thus, the kinetic properties exhibited by PhoBN were not due to removal of the DNA-binding domain.
FIGURE 4.
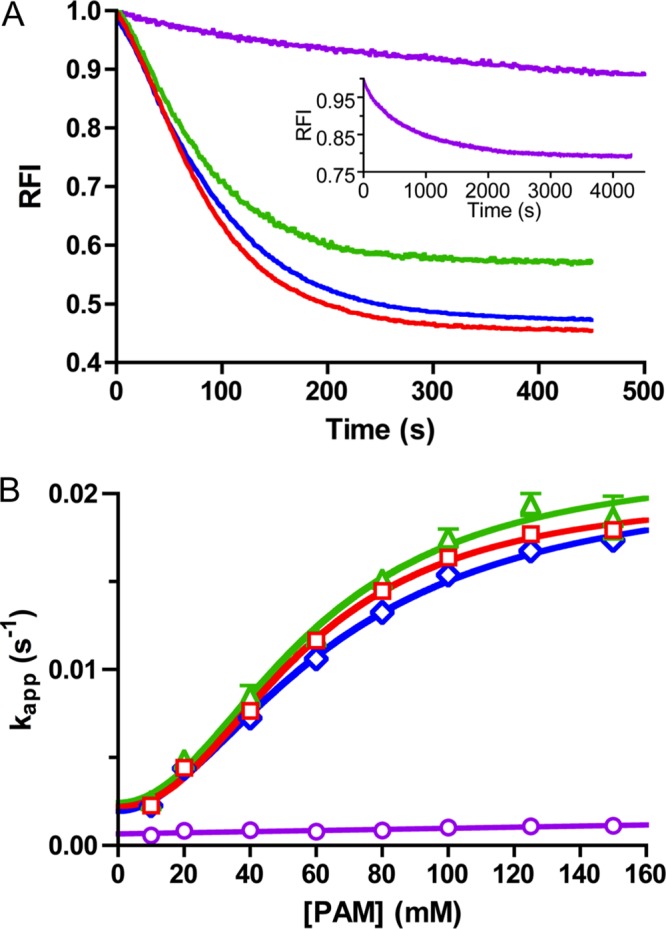
Autophosphorylation kinetics of wild type PhoB (PhoBF and PhoBN) and PhoBN dimer interface mutants. A, autophosphorylation time traces for reaction of 5 μm PhoBF (green), 5 μm PhoBN F20D (blue), or 5 μm PhoBN R115D (purple) with 60 mm PAM. For comparison, the autophosphorylation time trace for reaction of 5 μm PhoBN with 60 mm PAM from Fig. 2B is shown in red. The inset is an expanded time scale to show the full autophosphorylation time trace for PhoBN R115D. PhoBF contains one extra tryptophan in the output domain that is not present in PhoBN, leading to a smaller maximal quench in fluorescence for PhoBF. Time traces represent the mean of three reactions, with standard deviations (not shown for clarity) that were less than 3% of the mean. B, plot of kapp versus concentration of PAM for 5 μm PhoBF (green triangles), 5 μm PhoBN F20D (blue diamonds), 5 μm PhoBN R115D (purple circles), or 5 μm PhoBN as shown in Fig. 3D for comparison (red squares) fit with sigmoidal dose-response curve (see Equation 4) for PhoBF and PhoBN F20D or linear regression (see Equation 3) for PhoBN R115D. Error bars represent standard deviation from triplicate experiments.
Beryllium Trifluoride Binding to PhoBN Displayed Positive Cooperativity
The plot of kapp versus PAM concentration for PhoBN (Fig. 3) used rate constants derived from curve fits that systematically deviated from a single exponential decay. Thus, we sought an alternative method of assessing communication between PhoBN active sites by examining binding of a phosphoryl group analog. BeF3− binds to the active site of response regulators and mimics the Asp-phosphate linkage of phosphorylated response regulators (22, 29, 47). A plot of the quench in PhoBN tryptophan fluorescence versus BeF3− concentration (Fig. 5) was also sigmoidal with a Hill coefficient of 1.8 ± 0.2. The positive cooperativity exhibited in BeF3− binding lends credibility to the apparent cooperativity of PhoBN autophosphorylation kinetics (Fig. 3D). Also, with a K½ for BeF3− binding to PhoBN in the low micromolar range (44 ± 4 μm), the concentrations of BeF3− required to reach saturation were approximately 3 orders of magnitude lower than PAM concentrations needed, further reducing the possibility that a small amount of invisible aggregation could be affecting the PhoBN fluorescence signal.
FIGURE 5.
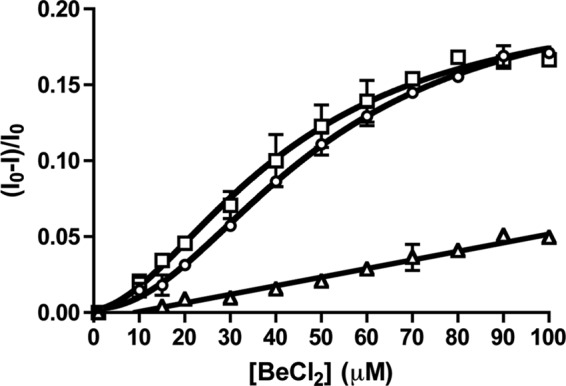
Beryllium trifluoride binding to PhoB constructs. Beryllium chloride titration with 5 μm PhoBN (square), 5 μm PhoBN F20D (circle), or 5 μm PhoBN R115D (triangle) was measured by fluorescence. The buffer contains 10 mm NaF. Error bars represent standard deviation from triplicate experiments.
Mathematical Modeling Supported a Link between Dimerization and Autophosphorylation of PhoBN
In an attempt to account for the lag in the time traces and the protein concentration dependence of PhoBN autophosphorylation, we utilized mathematical modeling software to construct a model of autophosphorylation. We began with a model that was similar to the kinetic model of CheY autophosphorylation (Scheme 1) and included PAM binding, phosphorylation, and dephosphorylation of PhoBN. This model was unable to account for either the lag of the autophosphorylation time traces or the protein concentration dependence, even when slow PAM binding or a variety of fluorescence intensity conversion factors were considered, suggesting a more complex kinetic mechanism of PhoBN autophosphorylation. The protein concentration dependence of PhoBN autophosphorylation, the visible lag in the PhoBN autophosphorylation time traces, the cooperative relationship between kapp and PAM concentration, and the sigmoidicity of BeF3− binding to PhoBN all suggested that there was a relationship between multimerization and autophosphorylation of PhoBN. Specifically, the protein concentration dependence of autophosphorylation rate constants demanded that multiple PhoBN molecules participated in the autophosphorylation reaction, and the lag suggested the existence of a step early in the reactions that, once completed, resulted in faster autophosphorylation. PhoB is known to dimerize when phosphorylated, so we next included formation of a homodimer (PhoBN-P/PhoBN-P) in the model. This, too, was unable to account for the protein concentration dependence of autophosphorylation and the lag in the PhoBN autophosphorylation time traces, suggesting an even more complex mechanism.
To account for all aspects of the autophosphorylation data, formation of a PhoBN heterodimer (PhoBN-P/PhoBN) was fundamental to the model. The resultant 10-step kinetic model (Scheme 2, Fig. 6, and see “Experimental Procedures” for extensive details) included the formation of both the PhoBN homodimer (PhoBN-P/PhoBN-P) and the PhoBN heterodimer (PhoBN-P/PhoBN) and the possibility of differing autophosphorylation kinetics (KS and kphos) for the PhoBN monomer and the PhoBN heterodimer. Four out of the 10 kinetic parameters were set to constants obtained experimentally to limit the number of variables that were subject to change by DynaFit software.
FIGURE 6.
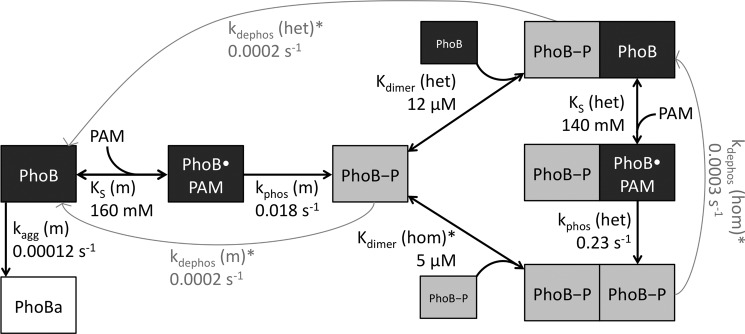
Model linking PhoBN heterodimer formation and autophosphorylation. A monomer of PhoBN binds a small molecule phosphodonor (PhoB·PAM) and phosphorylates (PhoB-P). The phosphorylated monomer can bind another phosphorylated monomer to form a homodimer. The phosphorylated monomer can also bind an unphosphorylated monomer to form a heterodimer, which can then bind PAM and phosphorylate. All three phosphorylated species of PhoBN can autodephosphorylate. Rate and equilibrium constants predicted by mathematical modeling are included by corresponding arrows. Kinetic constants marked with an asterisk were measured experimentally.
All 12 time traces for autophosphorylation (i.e. 5 μm PhoBN with 10–150 mm PAM (Fig. 2B) and 2.5–20 μm PhoBN with 60 mm PAM (Fig. 2D)) were simultaneously fit to the model by global analysis using DynaFit software (Fig. 7). The global analysis of the PhoBN autophosphorylation time traces succeeded in fitting all data, and a single set of predicted kinetic constants (Fig. 6) gave simulated time traces that overlaid well with all 12 of the PhoBN autophosphorylation time traces. Formation of the PhoB aggregate was an extremely slow process with a rate constant (kagg(m)) at least 2 orders of magnitude slower than either the rate constant for phosphorylation of the monomer (kphos(m)) or the heterodimer (kphos(het)). Furthermore, excluding aggregation from the model did not greatly affect 10 of the 12 autophosphorylation time traces. This was consistent with slow formation of a very small amount of soluble aggregate.
FIGURE 7.

Mathematical modeling by global kinetic analysis of autophosphorylation of PhoBN. Autophosphorylation time traces for reaction of 2.5–20 μm PhoBN with 60 mm PAM (A) and for reaction of 5 μm PhoBN with 10–150 mm PAM truncated to 500 s (B) with overlays of modeled fits from global kinetic analysis by DynaFit (black dashed lines) and residuals. Colors in A are described in Fig. 2D. Colors in B are described in Fig. 2B. RFIP represents the normalized tryptophan fluorescence intensity obtained by dividing all fluorescence intensities by the initial fluorescence intensity then multiplying by the protein concentration.
The rate and equilibrium constants generated by computer modeling were consistent with a link between PhoBN dimerization and autophosphorylation. Specifically, formation of a PhoBN heterodimer (PhoBN-P/PhoBN) and different autophosphorylation properties of the heterodimer compared with the monomer were fundamental to the model. Not only was inclusion of the heterodimer essential to fit the data, it was essential that the dimerization constant for the heterodimer (Kdimer(het)) had to be in the low micromolar range, similar to the dimerization constant for formation of the homodimer (Kdimer(hom), 5.1 μm (28)). In addition, kphos(het) had to be at least 10-fold faster than kphos(m). Dimerization constants in the same range as the protein concentration used in the experiments account for the protein concentration dependence of kapp. The difference between kphos(m) and kphos(het) accounts for the lag in the autophosphorylation time traces. Even modest modifications (±50%) to the rate and equilibrium constants predicted by DynaFit led to poor fits to the autophosphorylation data.
Disruption of the Physiologically Relevant Dimer 1 Interface of PhoBN Impeded Autophosphorylation
Crystallographic evidence shows that PhoBN is able to dimerize at two different dimerization interfaces (26, 27, 29), the α4-β5-α5 surface (dimer 1) and the α1-β5α5 loop-α5 surface (dimer 2) (Fig. 1). To more directly test the potential link between dimer formation and autophosphorylation of PhoBN, we characterized autophosphorylation kinetics of PhoBN variants containing previously characterized amino acid substitutions known to disrupt dimer formation (28). Replacement of Arg-115 with Asp (PhoBN R115D) disrupted the electrostatic interaction between residues Arg-115 and Asp-101 in the dimer 1 interface. Replacement of Phe-20 with Asp (PhoBN F20D) disrupted the hydrophobic dimerization interface of dimer 2.
Autophosphorylation of PhoBN F20D (dimer 2 mutant) with varying PAM concentrations displayed kinetics similar to wild type PhoBN. The autophosphorylation time traces exhibited a visible lag at the beginning of the time traces similar to wild type PhoBN (Fig. 4A). A plot of kapp versus PAM concentration for PhoBN F20D also fit to a sigmoidal curve with a Hill coefficient of 1.8 ± 0.2 (Fig. 4B), implying positive cooperativity of autophosphorylation similar to wild type PhoBN. Therefore, the cooperativity of autophosphorylation of wild type PhoBN was not likely due to formation of a dimer along the nonphysiological dimer 2 interface.
In contrast, autophosphorylation of PhoBN R115D (dimer 1 mutant) with PAM occurred over a much longer time scale than wild type PhoBN and did not exhibit a visible lag at the beginning of the time traces (Fig. 4A, inset). The plot of kobs versus PAM concentration for PhoBN R115D (Fig. 4B) showed no signs of sigmoidicity and fit to a linear regression. Thus, disruption of the physiologically relevant PhoBN dimer 1 interface resulted in slow autophosphorylation kinetics that were not cooperative. This result supported the link between dimerization and autophosphorylation of PhoBN and, furthermore, identified the dimer 1 interface as the functionally relevant dimerization surface in autophosphorylation. Putting the mutant results together with previous observations and modeling, we propose that autophosphorylation kinetics of wild type PhoBN reflect the formation of a heterodimer along the dimer 1 interface, which leads to an enhanced rate constant for phosphorylation of the PhoBN heterodimer compared with the PhoBN monomer.
Beryllium Trifluoride Binding to the Physiologically Relevant Dimer 1 Mutant PhoBN R115D Was Not Cooperative
To further investigate the role of dimer formation on the cooperativity of wild type PhoBN kinetics, we characterized the BeF3− binding properties of PhoBN R115D and PhoBN F20D. Binding of BeF3− to PhoBN F20D was cooperative with a Hill coefficient (2.0 ± 0.1) and binding constant (51 ± 3 μm) similar to wild type PhoBN (Fig. 5). In contrast, binding of BeF3− to PhoBN R115D was not cooperative and displayed a much weaker binding constant than wild type PhoBN (Fig. 5). Because disruption of dimer 1 eliminated the cooperativity of BeF3− binding to PhoBN, the cooperativity of BeF3− binding to wild type PhoBN was likely due to the formation of a dimer along the dimer 1 interface.
Phosphotransfer from [32P]PhoR-P to Wild Type PhoBN Was Faster Than to PhoBN R115D
To determine whether there was a link between the formation of the PhoBN dimer and phosphotransfer from the histidine kinase PhoR, the rate constants for phosphotransfer from [32P]PhoR-P to wild type PhoBN and PhoBN R115D were determined by monitoring the time-dependent disappearance of [32P]PhoR-P in the presence of a 10-fold molar excess PhoBN (Fig. 8). Phosphotransfer from [32P]PhoR-P to wild type PhoBN was approximately five times faster than phosphotransfer from [32P]PhoR-P to PhoBN R115D, with bimolecular rate constants of 77,000 ± 5000 and 16,000 ± 1,000 m−1 s−1, respectively. Therefore, both PhoBN autophosphorylation with PAM and phosphotransfer from [32P]PhoR-P to PhoBN were enhanced by the formation of dimer 1. Furthermore, accumulation of the radiolabeled phosphoryl group on PhoBN R115D occurred in a time-dependent manner to a similar extent as wild type PhoBN (Fig. 8) indicating functional PhoBN R115D protein.
FIGURE 8.
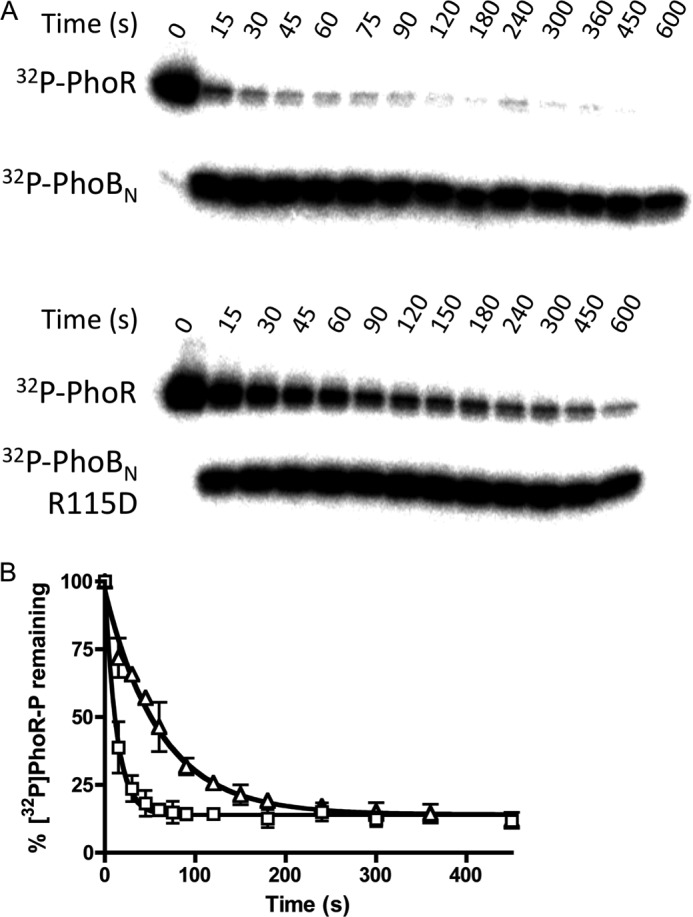
Phosphotransfer from [32P]PhoR-P to either wild type PhoBN or PhoBN R115D. A, phosphorimage of SDS-polyacrylamide gel analysis of phosphotransfer reactions. Each lane represents a different time point between 0 and 600 s as shown. B, quantitative analysis of the phosphorimage as shown in A for wild type PhoBN (squares) and PhoBN R115D (triangles), fit to a one-phase exponential decay. Each point represents the average and standard deviation of replicate experiments.
DISCUSSION
A central observation here was that in contrast to CheY, PhoBN and PhoBF autophosphorylation kinetics exhibited properties indicative of communication between active sites from different monomers. Numerous lines of evidence supported the notion that association of PhoB monomers to form heterodimers (one phosphorylated and one unphosphorylated monomer) affected autophosphorylation kinetics, including the protein concentration dependence of the normalized autophosphorylation time traces (Fig. 2D), the systematic deviation of the autophosphorylation time traces from a one-phase exponential (Fig. 2B), and the sigmoidicity of the plot of kapp versus PAM concentration (Fig. 3D). Binding of the phosphoryl group analog BeF3− to PhoBN was sigmoidally dependent on BeF3− concentration (Fig. 5) also indicating communication between PhoBN active sites. As predicted by a reaction scheme in which heterodimers play a role in autophosphorylation kinetics, disruption of the dimer 1 dimerization interface by the previously characterized R115D amino acid substitution (28) greatly reduced the rate of PhoBN autophosphorylation (Fig. 4B) and eliminated cooperativity in both autophosphorylation and BeF3− binding (Fig. 5).
Successful Mathematical Modeling of Autophosphorylation Data Provided Insights into the PhoB Reaction Mechanism
After testing multiple models of increasing complexity, we developed a scheme that linked autophosphorylation and dimerization of PhoBN (Fig. 6). A mathematical model based on this scheme and utilizing a single set of kinetic and equilibrium constants (some experimentally determined, some computationally inferred) was able to faithfully reproduce all the experimentally observed effects of changing either PhoBN or PAM concentration (Fig. 7).
First, the difference between kphos(m) and kphos(het) accounted for the lag in the autophosphorylation time traces and deviation from a single phase exponential. Specifically, PhoB autophosphorylation kinetics were initially dominated by monomers (kphos(m)) and hence were slow. After creation of phosphorylated monomers facilitated heterodimer formation with an excess of unphosphorylated PhoB monomers, faster autophosphorylation kinetics (kphos(het)) became evident. The model predicted that under the experimental conditions employed, there was a flux from monomers through heterodimers to homodimers but not necessarily a large accumulation of heterodimers. Second, a predicted dimerization constant (Kdimer(het)) for heterodimer formation in the same range as the protein concentration used in the experiments accounted for the protein concentration dependence of the normalized PhoBN time traces observed in Fig. 2D. Changing PhoBN concentration around Kdimer(het) altered heterodimer formation and hence the experimentally measured rate constants for the reaction. Additional insights gleaned from the mathematical model into the detailed mechanism of PhoB positive cooperativity are described below.
Dimerization Constant for the PhoBN-P/PhoBN Heterodimer Was in the Low Micromolar Range
A surprising prediction from the kinetic modeling was that the dimerization constant for formation of the PhoBN-P/PhoBN heterodimer (Kdimer(het), 12 μm) was similar to that measured for formation of the PhoBN-P/PhoBN-P homodimer (Kdimer(hom), 5.1 μm (28)). Receiver domain phosphorylation results in a conserved allosteric activating conformational change that includes changes in the α4-β5-α5 surface (3, 5). For multiple response regulators in the OmpR/PhoB family, the changes in the α4-β5-α5 surface result in formation of a dimer (3, 5, 20, 23, 24). Thus, it might be expected that acquisition of the active conformation is a prerequisite for dimerization and that formation of a PhoB heterodimer would involve association of a phosphorylated PhoB monomer with an unphosphorylated monomer that is in the active conformation. In such a circumstance, Kdimer(het) would be a composite of both the equilibrium constants for the conformational change and for dimerization (linked equilibria (48)), described by Kdimer, link in Equation 5, where Kdimer, link is the apparent dimerization constant for heterodimer formation; Kconf is the conformational equilibrium constant for PhoB in the active conformation, and Kdimer, act is the associative dimerization constant of PhoB in the active conformation,
Although it is difficult to measure the fraction of the response regulator population in the active conformation, it is estimated that no more than 10% of unphosphorylated response regulator molecules are in the active conformation (16, 49). Therefore, if Kconf is less than 0.1, we would predict Kdimer(het) to be at least 10-fold larger than Kdimer(hom), which does not contain a significant conformational component because the phosphorylated monomers are predominantly in the active conformation. The similarity between the predicted Kdimer(het) and the measured Kdimer(hom) suggests that acquisition of the active conformation is not a prerequisite for PhoB heterodimer formation. Although there is no direct evidence for the formation of a PhoBN heterodimer, a modified version of dimer 1 has been crystallized in the absence of BeF3− (27). Both monomers in the structure were in the inactive conformation, implying that PhoBN dimer 1 formation does not require acquisition of the active conformation before association.
Accelerated Autophosphorylation of the PhoBN-P/PhoBN Heterodimer
The second important prediction from the mathematical modeling of PhoBN autophosphorylation was that the rate constant for phosphorylation of the heterodimer (kphos(het)) was approximately 10 times higher than the rate constant for phosphorylation of the monomer (kphos(m)), implying that formation of the heterodimer of PhoBN enhanced the rate of autophosphorylation. This result is consistent with other observations that have linked an activated conformation to faster autophosphorylation kinetics. Addition of FliM (a ligand that pushes CheY toward an active-like conformation (50)) to CheY greatly enhances the rate of autophosphorylation compared with no ligand present (16). Similarly, addition of DNA that binds to OmpR increases the rate of autophosphorylation compared with no ligand present (25). We propose that formation of the heterodimer of PhoBN pushes the conformational equilibrium of the unphosphorylated monomer in the heterodimer toward the active conformation, which results in a faster rate of autophosphorylation of the PhoBN heterodimer compared with the PhoBN monomer.
Physiological Implications of Cooperative Kinetics
We have presented extensive yet indirect evidence for formation of a PhoB heterodimer. It is not presently known whether other response regulators form heterodimers and, if so, whether heterodimer formation leads to enhanced phosphorylation kinetics. However, there is at least one published observation consistent with heterodimer formation by another response regulator. An apparent heterodimer of ComE was trapped by cross-linking and separated from the cross-linked homodimer by SDS-PAGE (51).
How the network properties of different two-component systems can result in different output behaviors, including all-or-none versus graded responses, is the focus of both theoretical and experimental studies (52, 53). Response regulators that form heterodimers with enhanced phosphorylation kinetics may contribute to an all-or-none output response. Our data demonstrating that phosphotransfer from the sensor kinase PhoR to PhoB was enhanced by dimer 1 formation (Fig. 8) would predict a sigmoidal relationship between phosphorylated PhoR (PhoR-P) and phosphorylated PhoB (PhoB-P) (i.e. a sudden nonlinear output response over a small change in input). PhoB is an autoregulated response regulator, where PhoB-P regulates transcription of both phoR and phoB, leading to a positive feedback loop for phoB regulation (54). In combination, the positive cooperativity and auto-regulation could lead to a physiological response that acts as an on/off switch. At low concentrations of PhoR-P, there would be little PhoB-P and thus a low level of transcriptional activation within the cell. Eventually, a threshold concentration of PhoR-P will be reached, and PhoB-P will abruptly accumulate, leading to an abrupt cellular response. There is precedent for such behavior in the all-or-none response of PhoB to phosphorylation by noncognate histidine kinases (55). Because the PhoRB system regulates transcription of genes encoding proteins involved in phosphorus assimilation, the potential on/off switch may act as a way to dampen responses to minor fluctuations in the environmental concentration of phosphate.
Because formation of dimer 1 of PhoB orients the DNA-binding domains to interact with the symmetric DNA-binding sites (29), it is possible that a PhoB heterodimer would have similar affinity for DNA and thus could regulate transcription in vivo. A recent study that characterizes the relationship between unphosphorylated and phosphorylated PhoB concentrations in vivo and transcriptional activity showed that, even at full transcriptional activity, the concentration of phosphorylated PhoB was less than the total concentration of PhoB (30). Although there are multiple explanations for this phenomenon, it could be due to a physiologically active heterodimer. If the heterodimer is active in vivo, there are many potential consequences. First, because the heterodimer only requires phosphorylation of a single monomer of PhoB, it will accumulate at lower concentrations of PhoB-P than the homodimer, leading to an increased sensitivity of response for the heterodimer. Second, some response regulators (e.g. OmpR (56), BvgA (57), and NtrC (58)) bind to different promoters with different affinities, which allows stepwise responses to changing environmental circumstances. High affinity promoters are populated at low response regulator concentrations, and promoters with low binding affinities are not populated until the concentration of response regulator is sufficiently high. The PhoB heterodimer and homodimer might exhibit different promoter binding affinities, which would facilitate a more complex regulatory scheme. Third, when the cellular signal is turned off, dephosphorylation of the PhoB homodimer may result in a functional heterodimer that can induce transcription for a longer timeframe than if no active heterodimer is formed.
Autophosphorylation of PhoBN Was Different Than CheY
Positive cooperativity was not the only parameter that differed between CheY and PhoB. Although CheY autophosphorylation was 15–30 times faster than PhoBN autophosphorylation, PhoBN appeared to bind PAM slightly tighter than CheY. Based on our mathematical modeling, PAM binds to both the heterodimer and the monomer with binding constants (KS(het), KS(m)) of ∼150 mm, whereas the PAM binding affinity for CheY is estimated to be greater than 500 mm (11, 15, 16). These differences in rate and binding constants were noteworthy due to the similarities in tertiary structure, active site geometry, and divalent metal coordination of PhoBN and CheY. There must be other characteristics that differ between PhoBN and CheY that can explain the differences in rate and binding constants. One hypothesis based on work on CheY where positions near to the site of phosphorylation were substituted is that a more positive charge at the active site leads to faster autophosphorylation kinetics (15). We looked at the electrostatic surface potential of the Mg2+-bound forms of PhoBN (Protein Data Bank 2IYN (27)) and CheY (Protein Data Bank 2CHE (59)) using Adaptive Poisson-Boltzman Solver software (60). The surface of CheY is more positively charged than PhoBN due to residues with varying charges in nonconserved positions structurally adjacent to the active site. Because the small molecule phosphodonor is intrinsically negatively charged, the overall positive charge of the surface of CheY may lead to the faster rate of autophosphorylation compared with PhoBN.
Acknowledgments
We thank Robert Immormino for assistance with mathematical modeling and for cloning and purification of PhoR; Kimberly Coggan for cloning of cheY; Aaron Moore for cloning of phoBF and phoBN; Brian Crane, Robert Immormino, Stephani Page, and Stephanie Thomas for helpful discussions; and Marti Head and Peggy Cotter for comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM050860 from NIGMS.
- PAM
- phosphoramidate
- CheY-P
- phosphorylated CheY
- PhoBF
- full-length PhoB
- PhoBN
- PhoB N-terminal receiver domain
- PhoB-P
- phosphorylated PhoB
- nickel-NTA
- nickel-nitrilotriacetic acid
- RFI
- relative fluorescence intensity.
REFERENCES
- 1. Capra E. J., Laub M. T. (2012) Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 66, 325–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ulrich L. E., Zhulin I. B. (2010) The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38, D401–D407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao R., Stock A. M. (2009) Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63, 133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galperin M. Y. (2010) Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 13, 150–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao R., Stock A. M. (2010) Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hess J. F., Bourret R. B., Oosawa K., Matsumura P., Simon M. I. (1988) Protein phosphorylation and bacterial chemotaxis. Cold Spring Harbor Symp. Quant. Biol. 53, 41–48 [DOI] [PubMed] [Google Scholar]
- 7. Silversmith R. E. (2010) Auxiliary phosphatases in two-component signal transduction. Curr. Opin. Microbiol. 13, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huynh T. N., Stewart V. (2011) Negative control in two-component signal transduction by transmitter phosphatase activity. Mol. Microbiol. 82, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Georgellis D., Kwon O., De Wulf P., Lin E. C. (1998) Signal decay through a reverse phosphorelay in the Arc two-component signal transduction system. J. Biol. Chem. 273, 32864–32869 [DOI] [PubMed] [Google Scholar]
- 10. Bourret R. B. (2010) Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Da Re S. S., Deville-Bonne D., Tolstykh T., V ron M., Stock J. B. (1999) Kinetics of CheY phosphorylation by small molecule phosphodonors. FEBS Lett. 457, 323–326 [DOI] [PubMed] [Google Scholar]
- 12. Lukat G. S., McCleary W. R., Stock A. M., Stock J. B. (1992) Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl. Acad. Sci. U.S.A. 89, 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mayover T. L., Halkides C. J., Stewart R. C. (1999) Kinetic characterization of CheY phosphorylation reactions: comparison of P-CheA and small molecule phosphodonors. Biochemistry 38, 2259–2271 [DOI] [PubMed] [Google Scholar]
- 14. Silversmith R. E., Appleby J. L., Bourret R. B. (1997) Catalytic mechanism of phosphorylation and dephosphorylation of CheY: kinetic characterization of imidazole phosphates as phosphodonors and the role of acid catalysis. Biochemistry 36, 14965–14974 [DOI] [PubMed] [Google Scholar]
- 15. Thomas S. A., Immormino R. M., Bourret R. B., Silversmith R. E. (2013) Nonconserved active site residues modulate CheY autophosphorylation kinetics and phosphodonor preference. Biochemistry 52, 2262–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schuster M., Silversmith R. E., Bourret R. B. (2001) Conformational coupling in the chemotaxis response regulator CheY. Proc. Natl. Acad. Sci. U.S.A. 98, 6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Makino K., Shinagawa H., Amemura M., Kimura S., Nakata A., Ishihama A. (1988) Regulation of the phosphate regulon of Escherichia coli. Activation of pstS transcription by PhoB protein in vitro. J. Mol. Biol. 203, 85–95 [DOI] [PubMed] [Google Scholar]
- 18. Barbieri C. M., Stock A. M. (2008) Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal. Biochem. 376, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiratsu K., Nakata A., Shinagawa H., Makino K. (1995) Autophosphorylation and activation of transcriptional activator PhoB of Escherichia coli by acetyl phosphate in vitro. Gene 161, 7–10 [DOI] [PubMed] [Google Scholar]
- 20. McCleary W. R. (1996) The activation of PhoB by acetyl phosphate. Mol. Microbiol. 20, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 21. McCleary W. R., Stock J. B. (1994) Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 269, 31567–31572 [PubMed] [Google Scholar]
- 22. Lee S.-Y., Cho H. S., Pelton J. G., Yan D., Berry E. A., Wemmer D. E. (2001) Crystal structure of activated CheY: comparison with other activated receiver domains. J. Biol. Chem. 276, 16425–16431 [DOI] [PubMed] [Google Scholar]
- 23. Fiedler U., Weiss V. (1995) A common switch in activation of the response regulators NtrC and PhoB: phosphorylation induces dimerization of the receiver modules. EMBO J. 14, 3696–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao R., Tao Y., Stock A. M. (2008) System-level mapping of Escherichia coli response regulator dimerization with FRET hybrids. Mol. Microbiol. 69, 1358–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ames S. K., Frankema N., Kenney L. J. (1999) C-terminal DNA binding stimulates N-terminal phosphorylation of the outer membrane protein regulator OmpR from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 96, 11792–11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Solá M., Gomis-Rüth F. X., Serrano L., González A., Coll M. (1999) Three-dimensional crystal structure of the transcription factor PhoB receiver domain. J. Mol. Biol. 285, 675–687 [DOI] [PubMed] [Google Scholar]
- 27. Solà M., Drew D. L., Blanco A. G., Gomis-Rüth F. X., Coll M. (2006) The cofactor-induced pre-active conformation in PhoB. Acta Crystallogr. D Biol. Crystallogr. 62, 1046–1057 [DOI] [PubMed] [Google Scholar]
- 28. Mack T. R., Gao R., Stock A. M. (2009) Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J. Mol. Biol. 389, 349–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bachhawat P., Swapna G. V., Montelione G. T., Stock A. M. (2005) Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure 13, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao R., Stock A. M. (2013) Probing kinase and phosphatase activities of two-component systems in vivo with concentration-dependent phosphorylation profiling. Proc. Natl. Acad. Sci. U.S.A. 110, 672–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skerker J. M., Prasol M. S., Perchuk B. S., Biondi E. G., Laub M. T. (2005) Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 3, e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silversmith R. E. (2005) High mobility of carboxyl-terminal region of bacterial chemotaxis phosphatase CheZ is diminished upon binding divalent cation or CheY-P substrate. Biochemistry 44, 7768–7776 [DOI] [PubMed] [Google Scholar]
- 33. Bourret R. B., Thomas S. A., Page S. C., Creager-Allen R. L., Moore A. M., Silversmith R. E. (2010) Measurement of response regulator autodephosphorylation rates spanning six orders of magnitude. Methods Enzymol. 471, 89–114 [DOI] [PubMed] [Google Scholar]
- 34. Sheridan R. C., McCullough J. F., Wakefield Z. T. (1972) in Inorganic Syntheses (Cotton F. A., ed) Vol. 13, pp. 23–26, McGraw-Hill, New York [Google Scholar]
- 35. Zundel C. J., Capener D. C., McCleary W. R. (1998) Analysis of the conserved acidic residues in the regulatory domain of PhoB. FEBS Lett. 441, 242–246 [DOI] [PubMed] [Google Scholar]
- 36. Martin R. B. (1988) Ternary hydroxide complexes in neutral solutions of Al3+ and F. Biochem. Biophys. Res. Commun. 155, 1194–1200 [DOI] [PubMed] [Google Scholar]
- 37. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 38. Janin J., Chothia C. (1990) The structure of protein-protein recognition sites. J. Biol. Chem. 265, 16027–16030 [PubMed] [Google Scholar]
- 39. Fisher S. L., Kim S. K., Wanner B. L., Walsh C. T. (1996) Kinetic comparison of the specificity of the vancomycin resistance VanS for two response regulators, VanR and PhoB. Biochemistry 35, 4732–4740 [DOI] [PubMed] [Google Scholar]
- 40. Lukat G. S., Stock A. M., Stock J. B. (1990) Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry 29, 5436–5442 [DOI] [PubMed] [Google Scholar]
- 41. da Silva J. J., Williams R. J. (2001) The Biological Chemistry of the Elements: The Inorganic Chemistry of Life, 2nd ed., Oxford University Press, New York [Google Scholar]
- 42. Zapf J. W., Hoch J. A., Whiteley J. M. (1996) A phosphotransferase activity of the Bacillus subtilis sporulation protein Spo0F that employs phosphoramidate substrates. Biochemistry 35, 2926–2933 [DOI] [PubMed] [Google Scholar]
- 43. Psakis G., Mailliet J., Lang C., Teufel L., Essen L. O., Hughes J. (2011) Signaling kinetics of cyanobacterial phytochrome Cph1, a light regulated histidine kinase. Biochemistry 50, 6178–6188 [DOI] [PubMed] [Google Scholar]
- 44. Gilles-Gonzalez M. A., Gonzalez G. (1993) Regulation of the kinase activity of heme protein FixL from the two-component system FixL/FixJ of Rhizobium meliloti. J. Biol. Chem. 268, 16293–16297 [PubMed] [Google Scholar]
- 45. McCleary W. R., Zusman D. R. (1990) Purification and characterization of the Myxococcus xanthus FrzE protein shows that it has autophosphorylation activity. J. Bacteriol. 172, 6661–6668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barbieri C. M., Mack T. R., Robinson V. L., Miller M. T., Stock A. M. (2010) Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 285, 32325–32335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan D., Cho H. S., Hastings C. A., Igo M. M., Lee S.-Y., Pelton J. G., Stewart V., Wemmer D. E., Kustu S. (1999) Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. U.S.A. 96, 14789–14794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weber G. (1972) Ligand binding and internal equilibria in proteins. Biochemistry 11, 864–878 [DOI] [PubMed] [Google Scholar]
- 49. Volkman B. F., Lipson D., Wemmer D. E., Kern D. (2001) Two-state allosteric behavior in a single-domain signaling protein. Science 291, 2429–2433 [DOI] [PubMed] [Google Scholar]
- 50. Dyer C. M., Dahlquist F. W. (2006) Switched or not? the structure of unphosphorylated CheY bound to the N terminus of FliM. J. Bacteriol. 188, 7354–7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hung D. C., Downey J. S., Kreth J., Qi F., Shi W., Cvitkovitch D. G., Goodman S. D. (2012) Oligomerization of the response regulator ComE from Streptococcus mutans is affected by phosphorylation. J. Bacteriol. 194, 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kierzek A. M., Zhou L., Wanner B. L. (2010) Stochastic kinetic model of two component system signalling reveals all-or-none, graded and mixed mode stochastic switching responses. Mol. Biosyst. 6, 531–542 [DOI] [PubMed] [Google Scholar]
- 53. Goulian M. (2010) Two-component signaling circuit structure and properties. Curr. Opin. Microbiol. 13, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shinagawa H., Makino K., Nakata A. (1983) Regulation of the pho regulon in Escherichia coli K-12. Genetic and physiological regulation of the positive regulatory gene phoB. J. Mol. Biol. 168, 477–488 [DOI] [PubMed] [Google Scholar]
- 55. Zhou L., Grégori G., Blackman J. M., Robinson J. P., Wanner B. L. (2005) Stochastic activation of the response regulator PhoB by noncognate histidine kinases. J. Integr. Bioinform. 2:11 [Google Scholar]
- 56. Head C. G., Tardy A., Kenney L. J. (1998) Relative binding affinities of OmpR and OmpR-phosphate at the ompF and ompC regulatory sites. J. Mol. Biol. 281, 857–870 [DOI] [PubMed] [Google Scholar]
- 57. Decker K. B., James T. D., Stibitz S., Hinton D. M. (2012) The Bordetella pertussis model of exquisite gene control by the global transcription factor BvgA. Microbiology 158, 1665–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Magasanik B. (1996) in Escherichia coli and Salmonella: Cellular and Molecular Biology (Neidhardt F. C., ed) 2nd Ed., pp 1344–1356, American Society for Microbiology, Washington, D. C [Google Scholar]
- 59. Stock A. M., Martinez-Hackert E., Rasmussen B. F., West A. H., Stock J. B., Ringe D., Petsko G. A. (1993) Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry 32, 13375–13380 [DOI] [PubMed] [Google Scholar]
- 60. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]