

Background: α1-Protease inhibitor Portland (α1PDX) and serpin B8 are proprotein convertase (PC) inhibitors whose specificity and selectivity for PCs are not understood.
Results: α1PDX-serpin B8 and furin-PC chimeras revealed new serpin and protease (re)active-site and exosite determinants of reactivity.
Conclusion: α1PDX reactive-site and exosite determinants may be exploited for engineering specificity and selectivity for inhibiting PCs.
Significance: Specific PC inhibitors will advance understanding of PC function and therapeutics.
Keywords: Protease, Protease Inhibitor, Protein Engineering, Serine Protease, Serpin, α1PDX, Furin, Proprotein Convertase, Serpin B8
Abstract
α1-Protease inhibitor Portland (α1PDX) is an engineered serpin family inhibitor of the proprotein convertase (PC), furin, that exhibits high specificity but limited selectivity for inhibiting furin over other PC family proteases. Here, we characterize serpin B8, a natural inhibitor of furin, together with α1PDX-serpin B8 and furin-PC chimeras to identify determinants of serpin specificity and selectivity for furin inhibition. Replacing reactive center loop (RCL) sequences of α1PDX with those of serpin B8 demonstrated that both the P4–P1 RXXR recognition sequence as well as the P1′–P5′ sequence are critical determinants of serpin specificity for furin. Alignments of PC catalytic domains revealed four variable active-site loops whose role in furin reactivity with serpin B8 was tested by engineering furin-PC loop chimeras. The furin(298–300) loop but not the other loops differentially affected furin reactivity with serpin B8 and α1PDX in a manner that depended on the serpin RCL-primed sequence. Modeling of the serpin B8-furin Michaelis complex identified serpin exosites in strand 3C close to the 298–300 loop whose substitution in α1PDX differentially affected furin reactivity depending on the furin loop and serpin RCL-primed sequences. These studies demonstrate that RCL-primed residues, strand 3C exosites, and the furin(298–300) loop are critical determinants of serpin reactivity with furin, which may be exploited in the design of specific and selective α1PDX inhibitors of PCs.
Introduction
Furin is a member of the proprotein convertase (PC)2 family of calcium-dependent serine proteases that have structural homology to subtilisin/kexin-type proteases and are characterized by their recognition of a distinctive P4 Arg-X-Arg/Lys-P1 Arg consensus cleavage site (1, 2). PCs perform the intracellular and pericellular processing of a large number of peptide and protein precursors transiting the constitutive and regulated protein secretion pathways, including prohormones, growth factors, and their receptors, matrix metalloproteases and integrins. PCs are thus pivotal in the control of cell signaling, proliferation, motility, and adhesion (3, 4). PC dysfunction is associated with a broad spectrum of diseases, including cancer, autoimmunity, and Alzheimer disease. In addition, a number of significant human infectious organisms take advantage of host PC activity to promote their growth (5). PCs have been considered promising therapeutic drug targets, and the development of specific inhibitors is being intensely pursued (6, 7). However, a limitation has been the lack of detailed understanding of PC substrate specificity compounded by the similarity of PC catalytic domains (8, 9). Moreover, most PC specificity studies have focused on the unprimed substrate binding pockets of the catalytic site and neglected the primed binding pockets (10).
Protein active site-directed inhibitors of PCs have been engineered by substituting the P4 Arg-X-X-P1 Arg sequence into the reactive sites of different families of protein protease inhibitors, including leech eglin-C (11), turkey ovomucoid (12), α2-macroglobulin (13), and the serpin α1-protease inhibitor or α1PDX (14). However, the engineered PC target sequence is recognized by all PC family members and therefore such inhibitors are of limited use as therapeutics or research tools. Of particular note, protein protease inhibitors of the serpin family have been reported to be natural inhibitors of PCs, although their physiologic targets have not been defined (15–18). Serpins inhibit their target proteases by a distinctive suicide substrate mechanism that leads to the formation of a stable serpin-proteinase covalent inhibitory complex (19, 20). Unlike most other protein protease inhibitor families, the specificity and selectivity of serpins for inhibiting their target proteases is achieved through recognition determinants in an exposed reactive center loop as well as in exosites surrounding this loop (20). Serpin B8 (also known as proteinase inhibitor 8, or PI8) is an intracellular serpin that was first shown to inhibit furin in vitro because of the presence in its RCL of two Arg-X-X-Arg sequences (15). It is the only human serpin that carries a PC recognition sequence in its RCL. Serpin B8 has been detected in a wide variety of human tissues (21) and been shown to inhibit furin in platelets (22). Similar natural serpin inhibitors of furin exist in insects and cephalochordates (16–18). An understanding of the determinants of specificity and selectivity of these natural serpins for their target PCs could therefore provide insights into the design of specific serpin inhibitors of PCs.
In the present study, we have sought to gain insights into designing specificity and selectivity in the engineered serpin inhibitor of furin, α1PDX, by characterizing α1PDX-serpin B8 chimeras as well as chimeras of furin in which variable active-site loops of the protease were replaced with those of other PCs. We demonstrate that in addition to the P4 Arg-X-X-P1 Arg recognition sequence in the serpin RCL, the primed RCL sequence plays a critical role in α1PDX reactivity with furin and its ability to discriminate among different PC family members. Serpin exosites in strand 3 of β-sheet C are also shown to influence α1PDX reactivity and selectivity for furin. The contribution of reactive-site and exosite determinants of α1PDX to reactivity with furin is further shown to be dependent on the furin(298–300) loop sequence. Our findings demonstrate the potential for engineering specific and selective α1PDX inhibitors of furin and other PCs by targeting RCL and exosite determinants of this serpin.
EXPERIMENTAL PROCEDURES
Proteins
cDNA encoding serpin B8 was kindly provided by Dr. Walter Kisiel (University of New Mexico, Albuquerque, NM). Two stable monomeric forms of recombinant serpin B8 were engineered either by mutating the five surface cysteines at positions 5, 59, 340, 348, and 368 (as judged by molecular modeling) to serines (serpin B8-5S) or by mutating the five surface cysteines to serine and the five buried cysteines, residues 98, 108, 128, 317, and 368, to alanine (serpin B8-5S5A). Alanine rather than serine substitutions were found to be essential for expression of the latter variant. Wild-type and mutant forms of serpin B8 were expressed with a C-terminal His tag in Sf9 insect cells using the baculovirus expression system (Invitrogen). Cells were lysed by sonication after suspension in 20 mm Hepes buffer, pH 7.5, containing 50 mm NaCl, 20 mm imidazole, 6 mm MgCl2, 1 mm CaCl2, DNase and protease inhibitor mixture (Sigma). Serpin B8 was purified by first applying the cell lysate to a nickel affinity column (ProBond, Invitrogen) equilibrated in 20 mm Hepes, 100 mm NaCl, pH 7.5, washing with 5 volumes of 20 mm Hepes, 500 mm NaCl, pH 7.5, and then step eluting the protein with equilibrating buffer containing 500 mm imidazole. Pooled and concentrated protein fractions were then applied to a Superdex 75 column (GE Healthcare) equilibrated and eluted in 20 mm Hepes, 100 mm NaCl, pH 7.5, buffer. Protein concentration was determined from the 280 nm absorbance using an extinction coefficient of 32,900 m−1 cm−1 calculated from the amino acid sequence (23). Both serpin B8 forms migrated on SDS-PAGE as monomeric forms with the expected molecular weight under both nonreducing and reducing conditions, and proved to be very stable.
The full-length furin cDNA was kindly provided by Dr. Gary Thomas (University of Pittsburgh, Pittsburgh, PA). Furin was expressed as a 53-kDa truncated form that included the first 579 residues up to the end of the P-domain, preceded by a FLAG sequence and followed by a C-terminal His tag. Recombinant furin was produced as a secreted protein in Hi5 insect cells using the baculovirus expression system (Invitrogen). The culture media containing the expressed protein was first concentrated and buffer exchanged into 20 mm Hepes, 50 mm NaCl, 20 mm imidazole, 5 mm CaCl2, 5% glycerol, pH 7.5, by repeated ultrafiltration. Furin was purified by nickel affinity chromatography as with serpin B8 except that the exchange buffer was used for equilibration of the column. This was followed by gel filtration chromatography on Superdex 75 in the same equilibrating buffer without imidazole. Furin samples were supplemented with glycerol to 50% prior to storage at −80 °C. Protein concentration was determined from the 280 nm absorbance using a calculated extinction coefficient of 87,820 m−1 cm−1 (23).
α1PDX was engineered from an α1PI C232S variant by changing both P1 Met and P4 Ala residues to Arg as in previous studies (14, 24). Wild-type and chimeric forms of α1PDX were produced in Escherichia coli BL21 cells using the T7 expression system (Invitrogen) and refolded from inclusion bodies as described (25). After refolding, α1PDX was purified by ion exchange chromatography on DEAE-Sepharose at pH 6.5 followed by MonoQ (GE Healthcare) at pH 7.0, using linear sodium chloride gradients to elute the protein (24). The protein concentration was obtained from the 280 nm absorbance using an extinction coefficient of 27,000 m−1 cm−1 (26).
Mutagenesis of serpin B8, furin, or α1PDX cDNAs was done by PCR using specifically designed oligonucleotides from Integrated DNA Technologies and Pfu Turbo DNA polymerase from Stratagene according to the manufacturer's instructions. All mutations were confirmed by DNA sequencing. SDS-PAGE analysis indicated that all purified wild-type and mutant recombinant proteins were routinely >95% pure.
Furin Activity Assay
Furin activity was assayed by monitoring the linear rate of cleavage of the fluorogenic substrates, Boc-Arg-Val-Arg-Arg-7-amido-methylcoumarin or Pyr-Arg-Thr-Lys-Arg-amido-methylcoumarin (Bachem) at 100 μm, in 100 mm Hepes buffer, pH 7.5, containing 1 mm CaCl2, 1 mm β-mercaptoethanol, 0.5% Triton X-100, 0.1% polyethylene glycol 8000 at 25 °C. Excitation and emission wavelengths were 380 and 460 nm, respectively. Michaelis-Menten parameters were determined from the dependence of initial rates of substrate cleavage on substrate concentration in the range 0.15–10 × Km.
Furin Active-site Titration
Fixed concentrations of wild-type or variant furins (100 nm) were incubated overnight at 25 °C with increasing concentrations of Dec-Arg-Val-Lys-Arg-chloromethylketone (Bachem) in furin assay buffer adjusted to pH 5.5 to minimize hydrolysis of the chloromethylketone inhibitor but allow complete inhibition of furin based on the measured ka for furin inhibition at this pH (∼104 m−1 s−1). Residual furin activity was then assayed and plotted as a function of the inhibitor concentration. Plots showed linear decreases in furin activity with end points of zero activity in all cases. The abscissa intercept of the linear regression fit of the data yielded the functional furin concentration. Furin and furin variants were found to be fully active.
Stoichiometry of Serpin-Furin Reactions
The stoichiometry of inhibition for reactions of furin and furin variants with serpins was determined in furin assay buffer at 25 °C by incubating a fixed concentration of furin with increasing concentrations of serpin for a time allowing >95% inhibition based on measured values of ka. Residual furin activity was assayed and plotted as a function of the molar ratio of serpin to protease. Linear regression fits yielded the stoichiometry of inhibition from the abscissa intercept.
Kinetics of Furin Inhibition by Serpins
The kinetics of serpin B8 or α1PDX inhibition of furin were studied in furin assay buffer at 25 °C by continuous or discontinuous assays of the loss of furin activity under pseudo-first order reaction conditions as in previous studies, with the serpin concentration at least 10-fold higher than the protease concentration (24). In the continuous assay, serpin inhibition of furin was monitored in the presence of 100 μm furin fluorogenic substrate from the exponential decrease in the rate of cleavage of substrate as described for the furin activity assay. The rate of fluorescence increase due to furin cleavage of the substrate in the absence of inhibitor remained linear over the range of fluorescence amplitudes observed for furin inhibition reactions, indicating no significant substrate depletion. For reactions with t½ > 1 min, furin inhibition was followed by a discontinuous assay in which reactions were quenched at different reaction times by 10-fold dilution with substrate and the initial rate of substrate cleavage was measured as in the furin activity assay. Observed pseudo-first order rate constants (kobs) were obtained by fitting progress curves of substrate hydrolysis in the continuous assay or the fractional loss in furin activity with time for the discontinuous assay by a single exponential equation. Apparent second order association rate constants were obtained from slopes of linear regression fits of the dependence of kobs on the effective inhibitor concentration; i.e. after correcting for the competitive effect of substrate in the case of continuous assays by dividing by the factor, 1 + [S]o/Km, where [S]o is the substrate concentration and Km is the Michaelis constant for substrate hydrolysis. At least five different inhibitor concentrations ranging from 5 to 50 nm for the fastest reactions (second order rate constants of ∼106 m−1 s−1) or 200–800 nm for the slowest reactions (second order rate constants 103-104 m−1 s−1) were employed. To correct for the fraction of serpin that is cleaved by furin along a competing substrate pathway, the apparent association rate constant was multiplied by the stoichiometry of inhibition to yield ka, the association rate constant for reaction along the inhibitory pathway.
SDS-PAGE
The purity of recombinant proteins and the ability of serpins to form SDS-stable complexes with proteases were analyzed by SDS-PAGE under non-reducing conditions using the Laemmli buffer system (27).
Protein Structure Modeling
Models of the serpin B8 structure were obtained using the SWISS-MODEL tool for protein homology modeling. The serpin B8 amino acid sequence was threaded using as templates the x-ray crystal structures of the serpins, maspin (Protein Data Bank (PDB) entry 1XQJ), antithrombin (PDB entry 1EO3), and α1-antitrypsin (PDB entry 1ATU). Modeling of the serpin B8-furin Michaelis complex was done using the program DeepView (Swiss PDB-Viewer). The serpin B8 model and the x-ray structure of the furin catalytic domain (PDB entry 1P8J) were superimposed on the Michaelis complex structures of antithrombin-factor Xa (PDB entry 2GD4) antithrombin-thrombin (PDB entry 1SR5), and α1PI-trypsin (PDB entry 1OPH). To superimpose the subtilisin-type fold of furin on the chymotrypsin family protease components of the Michaelis complexes, the structurally conserved catalytic triads were aligned.
RESULTS
Characterization of the Serpin B8-Furin Reaction
To provide insights into engineering a specific and selective serpin inhibitor of PCs, we first characterized serpin B8, a natural intracellular inhibitor of furin. The propensity of wild-type serpin B8 to form disulfide-linked multimers through its 10 cysteine residues prompted us to engineer stable monomeric forms of the serpin by mutating the cysteines to serine or alanine (see “Experimental Procedures”). The ability of the two stable serpin B8 variants to inhibit furin was assessed by continuous monitoring of the kinetics of furin cleavage of a reporter fluorogenic substrate under pseudo-first order conditions. Reactions performed at increasing serpin B8 concentrations produced a family of inhibition progress curves that were characterized by increasing rates of furin inhibition and decreasing fluorescence amplitudes (Fig. 1). Observed pseudo-first order rate constants (kobs) derived from the progress curves showed a proportional dependence on serpin B8 concentration for both serpin B8 variants (Fig. 1), yielding similar apparent second order rate constants of ∼105 m−1 s−1 for inhibition of furin. Stoichiometries of inhibition (SI), assessed from titrations of the loss in furin activity as a function of increasing molar ratios of serpin B8 to furin, yielded values of 1.5 and 2.2 mol of inhibitor/mol of protease for serpin B8–5S and serpin B8–5S5A, respectively (Fig. 1). The observation of stoichiometries greater than 1 suggested that a fraction of the serpin was being cleaved by furin as a substrate, in accordance with the branched pathway suicide substrate mechanism by which serpins inhibit proteases (19). This was verified by SDS-PAGE analysis of the products of the reaction, which showed both serpin-protease complex and cleaved serpin products (Fig. 2). Correction of apparent second rate constants for the different extents of substrate reaction by multiplying by the SI yielded similar association rate constants for reaction along the inhibitory pathway (ka) of ∼2 × 105 m−1 s−1 for the two serpin B8 variants that were in reasonable agreement with reported values for wild-type serpin B8 (15).
FIGURE 1.
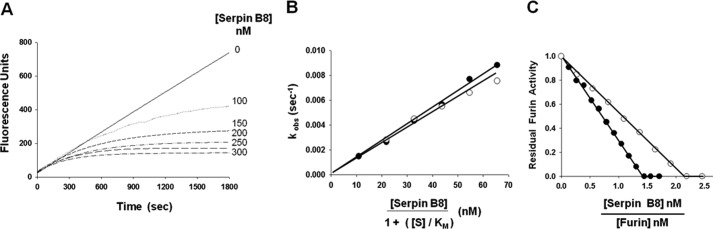
Kinetics and stoichiometry of serpin B8 inhibition of furin. A, progress curves for inhibition of 2.6 nm furin by increasing concentrations of serpin B8-5S monitored from the exponential decrease in rate of furin cleavage of fluorogenic substrate as a function of time. B, dependence of observed pseudo-first order rate constants (kobs) derived from exponential fits of progress curves in panel A on the serpin B8 concentration corrected for fluorogenic substrate competition as described under “Experimental Procedures.” Data are shown for reactions of serpin B8-5S (●) and serpin B8-5S5A (○). The slope of linear regression fits of the data (solid lines) provided the apparent second order association rate constants (ka,app) for the inhibition reaction. C, stoichiometric titrations of furin with serpin B8 analyzed from the furin activity remaining after complete reaction with increasing molar ratios of serpin B8 to furin. Shown are reactions with serpin B8-5S (●) and serpin B8-5S5A (○). The SI was obtained from the abscissa intercept of linear regression fits of titrations (solid lines).
FIGURE 2.
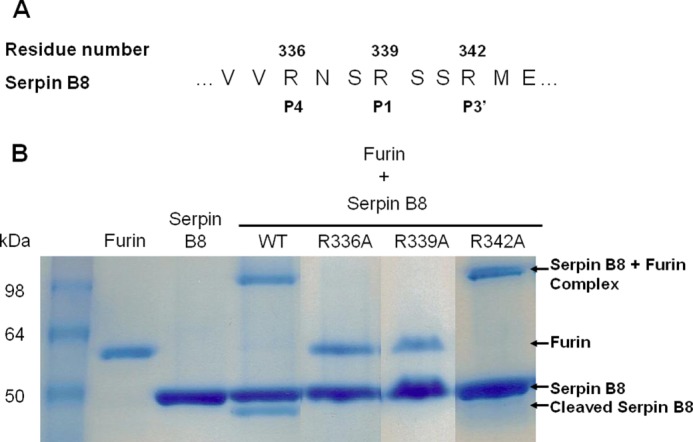
SDS-PAGE analysis of reactions of parent and RCL variant forms of serpin B8-5S with furin. A, sequence of the reactive center loop P6–P5′ region of serpin B8-5S showing the two RXXR sequences at P4–P1 and P1–P3′. B, SDS-PAGE analysis of the reactions of parent and three single Arg → Ala variant forms of serpin B8-5S (4 μg) with furin (1 μg) as indicated. Protein bands were stained with Coomassie Blue. Labels on the right indicate the position of serpin-protease complex and cleaved serpin bands. Molecular weight standards are in the leftmost lane.
Two Arg-X-X-Arg substrate recognition sequences for PCs exist in the RCL of serpin B8, one that aligns with the P4–P1 sequence and the other with the P1–P3′ sequence of other serpins (Fig. 2). To confirm that the former is the functional site for inhibition of furin and determine whether the latter might be an alternate site of cleavage, single alanine mutations of arginine residues 336, 339, and 342 were introduced in serpin B8-5S to eliminate one or both target sequences. Of the three variants tested, only the R342A variant that retained the P4 Arg336-P1 Arg339 sequence was a functional inhibitor of furin (Fig. 2), confirming the importance of the furin recognition sequence in the P4-P1 positions for inhibitor function. Surprisingly, the R336A variant containing the alternative P1 Arg339-P3′ Arg342 furin recognition sequence was completely unreactive as an inhibitor or substrate of furin. This suggested that the cleaved serpin B8 produced in the reaction of the parent serpin B8-5S with furin does not derive from an alternative cleavage after Arg342, but rather arises from cleavage after P1 Arg339. Interestingly, the R342A variant differed from the parent serpin B8-5S in producing only inhibitory complexes with no cleaved serpin (Fig. 2) and inhibiting furin with a 3-fold lower ka (Table 1). This indicated that the P3′ Arg342 is an important determinant of serpin B8 reactivity with furin both as an inhibitor and a substrate.
TABLE 1.
Kinetic constants and stoichiometries of inhibition for reactions of furin with wild-type and variant forms of serpin B8 and α1PDX
Association rate constants (ka) and stoichiometries of inhibition (SI) for serpin-furin reactions were measured at pH 7.5, 25 °C, as described in the legend to Fig. 1 and under “Experimental Procedures.” Errors represent S.E. from linear regression fits of data. The SI for the serpin B8 R342A mutant was assumed to be 1 based on the absence of detectable cleaved serpin product in the reaction by SDS-PAGE analysis (Fig. 2).
| Inhibitor variant | ka | SI |
|---|---|---|
| m−1 s−1 | ||
| Serpin B8-5S | 2.4 ± 0.3 × 105 | 1.5 ± 0.1 |
| Serpin B8-5S5A | 2.3 ± 0.2 × 105 | 2.2 ± 0.1 |
| Serpin B8-5S-R342A | 7.6 ± 0.5 × 104 | 1 |
| α1PDX | 1.1 ± 0.1 × 106 | 1.1 ± 0.1 |
| α1PDX-serpin B8 P6–P1 | 2.5 ± 0.3 × 106 | 1.3 ± 0.1 |
| α1PDX-serpin B8 P1′–P5′ | 5.5 ± 0.8 × 104 | 1.2 ± 0.1 |
| α1PDX-serpin B8 P6–P5′ | 1.1 ± 0.1 × 105 | 1.4 ± 0.1 |
| α1PDX-YE | 2.1 ± 0.3 × 105 | 1.9 ± 0.1 |
| α1PDX-serpin B8 P6–P5′–YE | 1.5 ± 0.2 × 103 | 1.3 ± 0.2 |
RCL Determinants of Serpin Specificity for Furin
We next investigated whether we could transfer the determinants of serpin B8 specificity for furin to α1PDX, an engineered inhibitor of furin derived from the extracellular serpin, α1PI, by replacing the P4 and P1 RCL residues with Arg to generate the RXXR recognition sequence of PCs (14). α1PDX inhibited furin with a ka of 1 × 106 m−1 s−1 and a stoichiometry of inhibition of 1, in good agreement with previous studies (28) (Table 1). α1PDX is thus a ∼5-fold faster inhibitor of furin than serpin B8. To determine whether this difference in specificity for furin was encoded in the RCL sequence, we characterized α1PDX-serpin B8 chimeras in which the unprimed (P6–P1), the primed (P1′–P5′), or the full (P6–P5′) RCL sequences of α1PDX were replaced with those of serpin B8 (Fig. 3). Replacing just the P6–P1 RCL sequence of α1PDX increased ka for furin inhibition 2-fold over that of α1PDX. By contrast, replacing the primed P1′–P5′ or full P6–P5′ RCL sequences of α1PDX with that of serpin B8 decreased ka for furin inhibition 20- and 10-fold, respectively, from that of α1PDX (Fig. 3 and Table 1). The inhibition stoichiometries were not significantly affected by these RCL changes. These findings suggested that the unprimed RCL sequence of serpin B8 confers a modestly greater reactivity and the primed RCL sequence a substantially lower reactivity for furin than the corresponding sequences of α1PDX.
FIGURE 3.
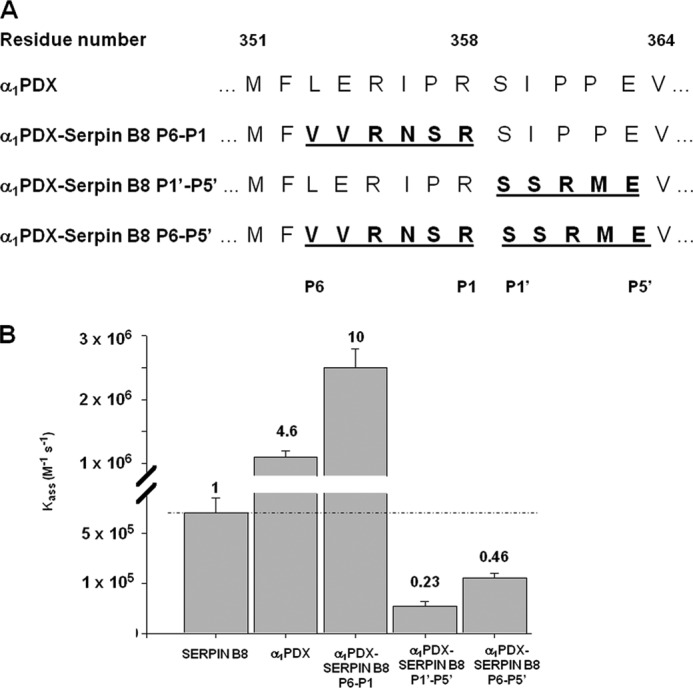
Comparison of the reactivities of serpin B8, α1PDX, and α1PDX-serpin B8 RCL chimeras with furin. A, RCL sequences of α1PDX and α1PDX-serpin B8 chimeras in which α1PDX P6–P1, P1′–P5′, and P6–P5′ residues were replaced with those of serpin B8. B, bar graph comparing ka for the reactions of serpin B8-5S, α1PDX, and the three α1PDX-serpin B8 chimeras with furin from Table 1. Values at the top of bars are ka expressed relative to that for the serpin B8 reaction.
Surface Loops in the Furin Catalytic Domain Affect Serpin Reactivity
To assess whether furin determinants exist to regulate the specificity and selectivity of serpin B8 for furin and other PCs, we examined sequence alignments of the catalytic domains of seven human PCs. The alignments revealed a high degree of sequence conservation as previously noted (10), except for four surface loops surrounding the active-site, which showed high sequence variability. These loops comprise residues 187–189, 298–300, 345–350, and 357–359 (Figs. 4 and 5). To determine whether these loops contributed to the high reactivity of furin with serpin B8, we expressed four furin variants in which the residues of each variable loop were all mutated to Ala. The effect of the loop mutations on furin catalytic activity was first tested by studying the kinetics of cleavage of two synthetic RXXR fluorogenic substrates. The four loop mutants each showed decreases in the overall specificity constants, i.e. kcat/Km, for hydrolysis of the two substrates ranging from 1.1- to 1.2-fold for the 345–350 loop mutant to 6–8-fold for the 187–189 loop mutant (Fig. 6). These decreases resulted mostly from changes in kcat (Table 2).
FIGURE 4.

Sequence alignments of PC catalytic domains. Sequence alignments of the catalytic domains of seven human PC family members with black, gray, and white coloring reflecting high conservation, moderate conservation, and poor conservation of individual residues, respectively. The inverted triangles indicate the catalytic triad residues. The indicated four surface loops surrounding the enzyme active site, comprising residues 187–189, 298–300, 345–350, and 357–359, show high sequence variability.
FIGURE 5.

Location of furin variable surface loops in the furin catalytic domain. The murine furin catalytic domain structure (PDB 1P8J) is depicted in ribbons showing the four variable loops surrounding the catalytic cleft in stick representation: loop 187–189 (yellow), loop 298–300 (green), loop 345–350 (cyan), and loop 357–359 (purple). The catalytic triad residues are shown in stick (red). The 298–300 loop lies adjacent to the S2′ binding pocket (29). The figure was prepared using PyMol software.
FIGURE 6.

Reactivity profiles of furin variable loop mutants with synthetic substrate and serpin inhibitors. Bar graphs depicting the effects of mutating the four variable surface loops of furin to alanines on kcat/Km for furin cleavage of a tetrapeptide fluorogenic substrate and on ka for furin reactions with serpin B8-5S, α1PDX, and α1PDX-serpin B8 P6–P1, P1′–P5′, and P6–P5′ chimeras. kcat/Km values are taken from Table 2 and ka values are from Table 3. Numbers at the top of bars represent kinetic constants expressed relative to wild-type furin values.
TABLE 2.
Kinetic constants for the catalytic reaction of furin and furin alanine loop mutants with two RXXR fluorogenic substrates
Kinetic constants were measured at pH 7.5, 25 °C from the dependence of initial rates of substrate hydrolysis on substrate concentration as described under “Experimental Procedures.” Errors represent S.E. from nonlinear regression fits of data by the Michaelis-Menten equation.
| Furin surface loop variant | Furin residues mutated into alanines | Boc-RVRR-amc |
Pyr-RTKR-amc |
||||||
|---|---|---|---|---|---|---|---|---|---|
| KM | kcat | kcat/KM | kcat/KM variant/wt ratio | KM | kcat | kcat/KM | kcat/KM variant/wt ratio | ||
| μm | s−1 | m−1 s−1 | μm | s−1 | m−1 s−1 | ||||
| Wild-type | 28.2 ± 1.3 | 0.27 ± 0.01 | 9.2 ± 1.0 × 103 | 1 | 4.7 ± 0.5 | 0.16 ± 0.00 | 3.4 ± 0.7 × 104 | 1 | |
| 187–189 | TQM | 25.1 ± 2.7 | 0.03 ± 0.00 | 1.1 ± 0.2 × 103 | 0.12 ± 0.03 | 9.0 ± 1.0 | 0.05 ± 0.00 | 6.1 ± 1.1 × 103 | 0.18 ± 0.07 |
| 298–300 | REH | 20.2 ± 1.2 | 0.09 ± 0.01 | 4.5 ± 0.7 × 103 | 0.49 ± 0.13 | 5.0 ± 0.4 | 0.10 ± 0.00 | 2.1 ± 0.3 × 104 | 0.62 ± 0.21 |
| 345–350 | NQNEKQ | 23.3 ± 1.9 | 0.19 ± 0.03 | 8.1 ± 1.6 × 103 | 0.88 ± 0.27 | 5.4 ± 0.3 | 0.15 ± 0.03 | 2.7 ± 0.6 × 104 | 0.73 ± 0.31 |
| 357–359 | RQK | 8.7 ± 0.7 | 0.03 ± 0.00 | 2.9 ± 0.3 × 103 | 0.32 ± 0.07 | 5.8 ± 0.6 | 0.08 ± 0.01 | 1.3 ± 0.2 × 104 | 0.38 ± 0.14 |
The effect of the loop mutations on the kinetics and stoichiometry of furin inhibition by serpin B8 and α1PDX was next determined. Mutations of three of the furin loops, 187–189, 345–350, and 357–359, resulted in decreases in ka for reaction of furin with the two serpins that differed by at most ∼2-fold from the decreases observed in kcat/Km for substrate hydrolysis, i.e. 1.6–4-fold (Fig. 6 and Table 3). By contrast, the 298–300 loop mutant affected furin reactivity with the two serpins quite differently. ka for reaction with serpin B8 was thus decreased 4-fold, whereas ka for reaction with α1PDX was increased ∼2-fold relative to wild-type furin, both in contrast to the ∼2-fold reduction in kcat/Km for hydrolysis of the RXXR substrate. This suggested that the furin(298–300) loop differentially affects reactivity with the two serpins through determinants that do not involve the RCL RXXR recognition sequence.
TABLE 3.
Kinetic constants and stoichiometries of inhibition for the reactions of serpin B8, α1PDX, and α1PDX-serpin B8 chimeras with furin and furin alanine loop mutants
Values of ka and SI ±S.E. for serpin-furin reactions were measured at pH 7.5, 25 °C as in Table 1. SIs for the reactions of α1PDX-serpin B8 P6–P5′-YE chimera with the furin loop mutants could not be determined because of enzyme instability over the long times required for complete reaction.
| Furin surface loop variant | Serpin B8 |
α1PDX |
α1PDX-serpin B8 P6–P1 |
α1PDX-serpin B8 P1′–P5′ |
α1PDX-serpin B8 P6–P5′ |
α1PDX-YE |
α1PDX-serpin B8 P6–P5′-YE |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ka | SI | ka | SI | ka | SI | ka | SI | ka | SI | ka | SI | ka | SI | |
| m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | ||||||||
| Wild-type | 2.4 ± 0.3 × 105 | 1.5 ± 0.1 | 1.1 ± 0.1 × 106 | 1.1 ± 0.1 | 2.5 ± 0.3 × 106 | 1.3 ± 0.1 | 5.5 ± 0.8 × 104 | 1.2 ± 0.1 | 1.1 ± 0.1 × 105 | 1.4 ± 0.1 | 2.1 ± 0.3 × 105 | 1.9 ± 0.1 | 1.5 ± 0.2 × 103 | 1.3 ± 0.2 |
| 187–189 | 5.6 ± 0.9 × 104 | 1.2 ± 0.2 | 3.8 ± 0.8 × 105 | 0.9 ± 0.1 | 3.5 ± 0.3 × 105 | 1.3 ± 0.1 | 2.0 ± 0.3 × 104 | 1.0 ± 0.0 | 2.5 ± 1.0 × 104 | 1.5 ± 0.1 | 4.2 ± 0.7 × 104 | 1.0 ± 0.1 | 5.5 ± 0.4 × 102 | NDa |
| 298–300 | 6.1 ± 1.3 × 104 | 1.5 ± 0.3 | 1.8 ± 0.1 × 106 | 1.2 ± 0.1 | 2.7 ± 0.3 × 106 | 1.4 ± 0.0 | 2.3 ± 0.2 × 104 | 1.0 ± 0.0 | 1.9 ± 0.9 × 104 | 1.0 ± 0.1 | 1.3 ± 0.1 × 105 | 0.8 ± 0.2 | 1.5 ± 0.1 × 103 | ND |
| 345–350 | 1.5 ± 0.2 × 105 | 1.1 ± 0.1 | 5.2 ± 0.5 × 105 | 1 | 6.7 ± 0.9 × 105 | 1.0 ± 0.1 | 4.7 ± 1.1 × 104 | 1.1 ± 0.2 | 8.9 ± 1.7 × 104 | 1.8 ± 0.1 | 1.7 ± 0.3 × 105 | 1.3 ± 0.1 | 1.4 ± 0.0 × 103 | ND |
| 357–359 | 5.6 ± 1.1 × 104 | 1.3 ± 0.2 | 7.1 ± 1.0 × 105 | 0.8 ± 0.1 | 8.5 ± 0.9 × 105 | 1.0 ± 0.0 | 5.2 ± 1.1 × 104 | 0.9 ± 0.1 | 7.4 ± 1.9 × 104 | 1.3 ± 0.1 | 6.9 ± 1.0 × 104 | 0.8 ± 0.2 | 1.0 ± 0.2 × 103 | ND |
a ND, not determined.
To assess whether the serpin determinants that mediate this differential reactivity with furin reside in the RCL, we compared the reactivity profiles of the furin loop mutants with α1PDX and the α1PDX-serpin B8 RCL chimeras (Fig. 6 and Table 3). Swapping the full serpin B8 RCL into α1PDX dramatically altered α1PDX reactivity with the furin(298–300) loop mutant, but minimally affected reactivity with the other furin loop mutants. ka for the α1PDX RCL chimera-furin reaction was thus decreased 4-fold by the furin loop mutation, in marked contrast to the increase in ka observed for the α1PDX-furin reaction but similar to the decrease in ka observed for the serpin B8-furin reaction. Significantly, replacing just the P1′-P5′ residues of α1PDX with those of serpin B8 resulted in a change in ka for inhibition the furin(298–300) loop mutant relative to furin like that of the full RCL swap, whereas replacing just the P1–P6 α1PDX residues with those of serpin B8 resulted in no change in ka for mutant and wild-type furin reactions. These findings suggested that the furin(298–300) loop affects α1PDX reactivity largely through interactions with the primed side of the RCL.
PC Variability of the 298–300 Loop Influences Serpin Reactivity
The observation that replacing the furin(298–300) loop sequence but not the other furin variable loops with alanines produced a large differential reactivity of furin with serpin B8 and α1PDX, suggested that the 298–300 loop might encode specificity determinants that govern PC reactivity with serpins. To test this possibility, we constructed a panel of furin-PC chimeras in which the amino acid sequence of the furin(298–300) loop was substituted with the homologous sequences from other PCs (Fig. 7 and Table 4). The kinetic parameters for the reaction of the chimeras with one fluorogenic substrate showed alterations in kcat/Km that varied from 3-fold lower to 1.3-fold higher than wild-type furin. These variations mostly reflected differences in kcat.
FIGURE 7.

Reactivity profiles of furin(298–300) loop PC chimeras with synthetic substrate and serpin inhibitors. Bar graphs showing the effects of substituting the 298–300 loop of furin with the corresponding loop sequences of other PC family members on kcat/Km for furin reaction with a tetrapeptide fluorogenic substrate, and on ka for furin reactions with serpin B8-5S5A, with α1PDX, and with the α1PDX-serpin B8 P6–P5′ chimera. Values of kcat/Km are taken from Table 4 and values of ka are from Table 5. Values at the top of bars are kinetic constants expressed relative to the value for wild-type furin.
TABLE 4.
Kinetic constants for the catalytic reaction of furin-PC(298–300) loop chimeras with RXXR fluorogenic substrate
Kinetic parameters were determined at pH 7.5, 25 °C as in Table 2.
| Furin-PC chimeras | Amino acid sequence at loop 298–300 | Boc-RVRR-amc |
|||
|---|---|---|---|---|---|
| KM | kcat | kcat/KM | kcat/KM variant/wt ratio | ||
| μm | s−1 | m−1 s−1 | |||
| Wild-type | REH | 28.2 ± 1.3 | 0.27 ± 0.01 | 9.2 ± 1.0 × 103 | 1 |
| F-PC1 | RQG | 27.6 ± 2.3 | 0.17 ± 0.01 | 6.2 ± 1.0 × 103 | 0.67 ± 0.18 |
| F-PC2 | SY- | 20.0 ± 1.5 | 0.09 ± 0.01 | 4.6 ± 0.8 × 103 | 0.50 ± 0.14 |
| F-PC4 | LHY | 34.3 ± 2.4 | 0.24 ± 0.03 | 7.0 ± 1.3 × 103 | 0.76 ± 0.22 |
| F-PC5/6 | RSK | 45.7 ± 2.7 | 0.13 ± 0.02 | 2.8 ± 0.5 × 103 | 0.30 ± 0.09 |
| F-PACE4 | REG | 27.0 ± 1.5 | 0.27 ± 0.01 | 9.7 ± 1.0 × 103 | 1.1 ± 0.23 |
| F-PC7 | QHN | 23.9 ± 1.8 | 0.29 ± 0.06 | 1.2 ± 0.3 × 104 | 1.3 ± 0.47 |
| 298–300 | AAA | 20.5 ± 2.9 | 0.09 ± 0.01 | 4.4 ± 0.8 × 103 | 0.48 ± 0.14 |
Notably, the furin(298–300) loop chimeras showed significantly greater reductions in reactivity with serpin B8 than with the synthetic substrate (Fig. 7 and Table 5) but showed either minimal change or up to 2.4-fold enhancements in reactivity with α1PDX relative to wild-type furin. Again, substituting the serpin B8 P6–P5′ residues into α1PDX switched the reactivity profile with the furin chimeras to resemble that of serpin B8. As a result, the reactivities of each furin(298–300) loop chimera exhibited marked differences of 10–95-fold between α1PDX and the α1PDX-serpin B8 RCL chimera. Such findings supported the idea that the 298–300 loop sequence was an important determinant of the differential reactivities of furin with α1PDX and serpin B8.
TABLE 5.
Kinetic constants and SI for reactions of serpin B8, α1PDX, and α1PDX-serpin B8 chimeras with furin and furin-PC(298–300) loop chimeras
Values of ka and SI ±S.E. for serpin-furin reactions were measured at pH 7.5, 25 °C as in Table 1. SIs for the reactions of α1PDX-serpin B8 P6–P5′-YE chimera with the furin-PC loop chimeras could not be determined because of enzyme instability over the long times required for complete reaction.
| Furin-PC chimeras | Serpin B8 |
α1PDX |
α1PDX-serpin B8 P6–P5′ |
α1PDX-YE |
α1PDX-serpin B8 P6–P5′-YE |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| ka | SI | ka | SI | ka | SI | ka | SI | ka | SI | |
| m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | ||||||
| Wild-type | 2.3 ± 0.2 × 105 | 2.2 ± 0.1 | 1.1 ± 0.1 × 106 | 1.1 ± 0.1 | 1.1 ± 0.1 × 105 | 1.4 ± 0.1 | 2.1 ± 0.7 × 105 | 1.9 ± 0.1 | 1.5 ± 0.2 × 103 | 1.3 ± 0.2 |
| F-PC1 | 9.8 ± 1.2 × 104 | 1.4 ± 0.1 | 2.6 ± 0.5 × 106 | 1.7 ± 0.2 | 7.1 ± 1.4 × 104 | 1.3 ± 0.1 | 9.3 ± 1.7 × 105 | 1.3 ± 0.1 | 5.6 ± 0.1 × 103 | NDa |
| F-PC2 | 6.4 ± 1.4 × 104 | 1.6 ± 0.2 | 1.1 ± 0.2 × 106 | 1.5 ± 0.1 | 5.3 ± 0.5 × 104 | 0.9 ± 0.1 | 1.7 ± 0.2 × 105 | 1.2 ± 0.1 | 9.2 ± 0.7 × 102 | ND |
| F-PC4 | 1.3 ± 0.1 × 105 | 1.6 ± 0.1 | 1.0 ± 0.2 × 106 | 1.4 ± 0.2 | 6.4 ± 0.9 × 104 | 1.6 ± 0.1 | 2.4 ± 0.5 × 105 | 1.3 ± 0.2 | 3.4 ± 0.2 × 103 | ND |
| F-PC5/6 | 4.9 ± 0.6 × 104 | 1.1 ± 0.1 | 2.2 ± 0.4 × 106 | 1.5 ± 0.2 | 6.4 ± 1.5 × 104 | 0.9 ± 0.1 | 9.4 ± 1.6 × 105 | 1.2 ± 0.2 | 2.6 ± 0.3 × 104 | ND |
| F-PACE4 | 9.4 ± 0.9 × 104 | 1.8 ± 0.1 | 1.6 ± 0.3 × 106 | 1.9 ± 0.3 | 1.3 ± 0.1 × 105 | 0.8 ± 0.2 | 2.4 ± 0.5 × 105 | 1.4 ± 0.3 | 1.0 ± 0.1 × 103 | ND |
| F-PC7 | 2.5 ± 0.4 × 105 | 2.1 ± 0.1 | 1.0 ± 0.2 × 106 | 1.4 ± 0.1 | 6.0 ± 0.7 × 104 | 1.3 ± 0.1 | 2.4 ± 0.2 × 105 | 1.1 ± 0.0 | 3.0 ± 0.1 × 103 | ND |
| 298–300 AAA | 6.1 ± 1.3 × 104 | 1.5 ± 0.3 | 1.8 ± 0.1 × 106 | 1.2 ± 0.1 | 1.9 ± 0.8 × 104 | 1.0 ± 0.1 | 1.3 ± 0.1 × 105 | 0.8 ± 0.2 | 1.5 ± 0.1 × 103 | ND |
a ND, not determined.
Strand 3C Residues Influence α1PDX Reactivity with Furin
Because our data suggested that the 298–300 loop of furin affects serpin reactivity through interactions with the RCL primed sequence, we investigated whether this loop might also affect serpin reactivity through interactions with exosites outside the RCL. To localize such exosites, we constructed a molecular model of the serpin B8-furin Michaelis complex using the x-ray crystal structure of the furin catalytic domain together with the structures of several serpin-protease Michaelis complexes as a template (Fig. 8). Placement of the subtilisin family protease, furin, in the Michaelis complexes with chymotrypsin family proteases was based on the common catalytic triad structures of subtilisin and chymotrypsin family proteases. The furin(298–300) loop was observed to closely approach residues Phe198 and Glu200 in strand 3 of β-sheet C of serpin B8 in all models.
FIGURE 8.
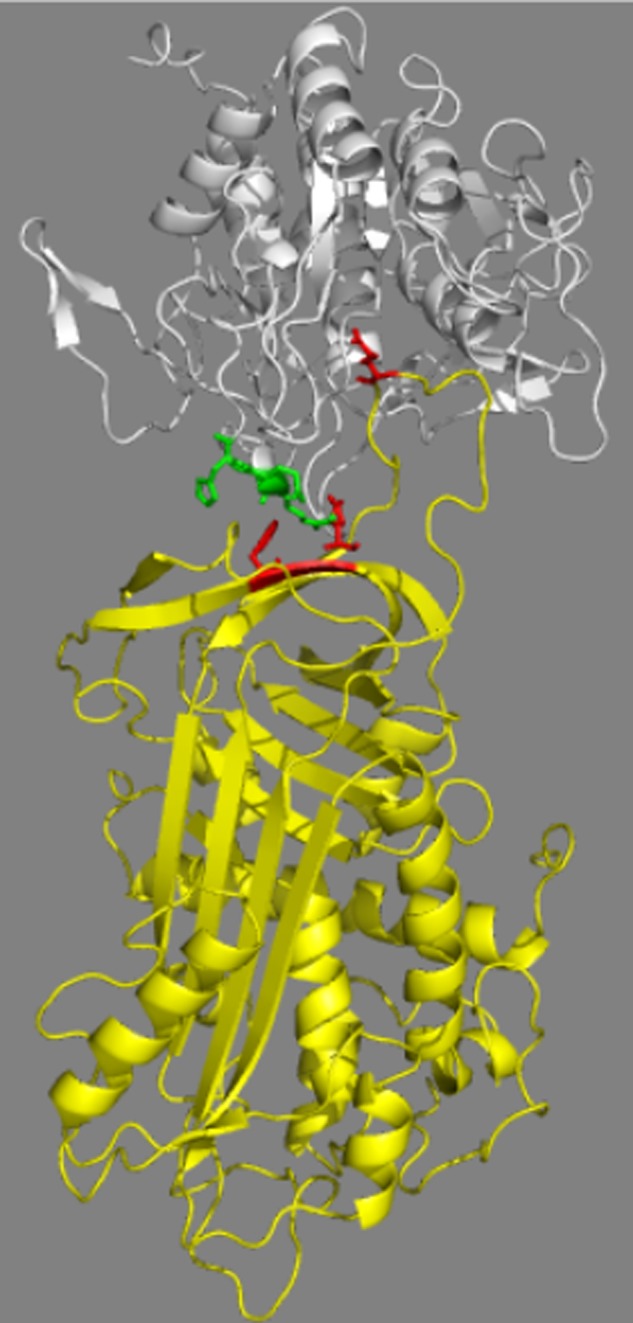
Molecular model of the serpin B8-furin Michaelis complex. The complex is depicted as ribbon structures with serpin B8 in yellow and the furin catalytic domain in white. Highlighted in stick are the furin(298–300) loop residues (green), serpin B8 residues Phe198 and Glu200 of strand 3 of β-sheet C (red), and the serpin B8 P1 residue (red). The complex shown was modeled based on the antithrombin-S195A factor Xa Michaelis complex structure but modeling based on two other Michaelis complex structures gave similar results (see “Experimental Procedures”). The proximity of the furin loop and the strand 3C exosite residues is evident. The figure was prepared using PyMol software.
To determine whether these strand 3C residues influenced serpin reactivity with furin, the homologous strand 3C residues in α1PDX, Lys222 and Leu224, were mutated to the serpin B8-like residues, Tyr and Glu, respectively. ka for reaction of furin with the α1PDX-YE s3C chimera was reduced by 5-fold and the inhibition stoichiometry was modestly increased relative to that with α1PDX (Table 1), indicating a marked effect on reactivity comparable with that of the serpin B8 RCL swap. To determine whether this change in reactivity resulted from interactions with the furin(298–300) loop, we measured ka and SI for reaction of the α1PDX-YE chimera with the four furin loop mutants (Fig. 9 and Table 3). The reactivity of α1PDX-YE with the furin loop variants was reduced in all cases from that of wild-type furin, similar to the reactivity changes of α1PDX with the furin variants except for the 298–300 loop variant. Whereas α1PDX-YE showed a 2-fold lower reactivity with the furin(298–300) loop mutant, α1PDX exhibited a 2-fold higher reactivity with the variant than furin (Figs. 6 and 9).
FIGURE 9.

Reactivity profiles of α1PDX exosite variants with furin loop variants. Reactivity of α1PDX-YE exosite variant with furin variable loop mutants and with furin(298–300) loop chimeras (left-hand panels). Reactivity of α1PDX-serpin B8 P6–P5′-YE dual chimera with the furin variable loop mutants and 298–300 loop chimeras (right-hand panels). ka values are from Tables 3 and 5. Reactivity profiles of the furin loop variants with α1PDX variants are expressed relative to wild-type furin reactivities.
To further probe whether the effects of the α1PDX strand 3C mutations on furin reactivity depended on the furin(298–300) loop sequence, we measured ka and SI for the reactions of α1PDX-YE with the panel of furin(298–300) loop mutants (Fig. 9 and Table 5). The reactivity profile of α1PDX-YE with the furin loop chimeras showed marked differences from that of α1PDX in the case of the PC1 and PC5 loop chimeras. These chimeras thus showed reactivity enhancements with α1PDX-YE relative to wild-type furin that were much greater (4.4–4.5-fold) than those observed with α1PDX (2-fold) (Fig. 7). Overall, the effects of the strand 3C mutations on α1PDX reactivity with each of the furin(298–300) loop variants varied over a wide range from 2.3-fold for the PC5 loop variant to 12-fold for the triple alanine variant, indicating a significant dependence of the reactivity changes on the 298–300 loop sequence.
Synergistic Effects of RCL and Strand 3C Determinants on α1PDX Reactivity with Furin
We next investigated the effects of combining the RCL and strand 3C mutations in α1PDX on furin reactivity (Fig. 9 and Table 5). The α1PDX-serpin B8 P6–P5′-YE variant exhibited a striking ∼730-fold loss in reactivity relative to α1PDX. This reactivity loss was notable in that it greatly exceeded the additive losses of the RCL and strand 3C mutations alone (i.e. 10- and 5-fold, respectively) (Fig. 10). The low reactivity of the α1PDX dual RCL/strand 3C chimera with furin was further reduced with all furin alanine loop variants except for the 298–300 loop variant (Fig. 9 and Table 5). Again, the α1PDX reactivity losses with each of the furin variants was greater than the additive losses expected from the individual mutations alone except for the 298–300 loop variant whose reactivity loss was additive (Fig. 10). Remarkably, the reactivity profile of the α1PDX dual RCL/strand 3C chimera with the panel of furin(298–300) loop chimeras differed substantially from that of α1PDX, the single RCL or strand 3C α1PDX chimeras, or the RXXR substrate (Fig. 9 and Table 5). The furin chimeras thus showed the most marked alterations in furin reactivity with the α1PDX dual chimera that ranged from ∼2-fold lower (PC2 and PACE4) to 17-fold higher (PC5) than furin. As a result, α1PDX reactivity losses with each of the furin(298–300) loop chimeras caused by the dual RCL and strand 3C mutations varied dramatically from ∼100–1600-fold, indicating a major dependence of the reactivity loss on the furin(298–300) loop sequence (Fig. 10).
FIGURE 10.
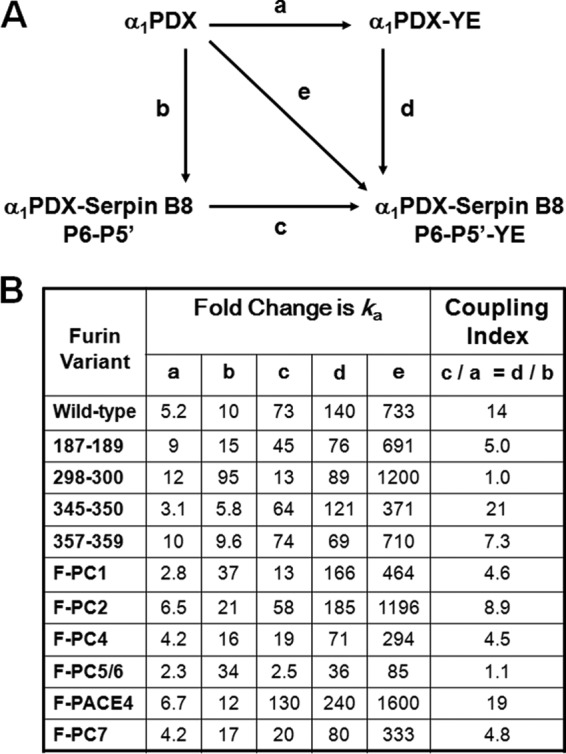
Coupling effects of RCL and strand 3C substitutions on α1PDX reactivity with furin. A, double mutation cycle for substitution of serpin B8 RCL and strand 3C sequences into α1PDX. Lower case letters indicate the relative change in ka resulting from the mutations indicated by the arrow. B, fold-changes in ka for reactions of α1PDX with furin and the indicated furin loop variants resulting from individual or combined RCL and s3C substitutions in α1PDX. The coupling index indicates the ratio of the relative changes in ka for one set of mutations in the presence or absence of the second set of mutations. A value of 1 indicates no coupling.
DISCUSSION
Furin and other members of the PC family of proteases play critical roles in the processing of a variety of proteins to their biologically active form (3–5). Because PCs have been implicated in the pathogenesis of numerous diseases, specific inhibitors of these enzymes could be of great potential value as research tools and therapeutics (6, 7). α1PDX was engineered to be a furin inhibitor by replacing the P1 and P4 RCL residues of α1PI with Arg, but the recognition of the consensus RXXR sequence by all PC family members makes this inhibitor unsuitable for selective targeting of individual PCs (14).
Our studies used a chimeric approach to investigate whether the natural PC inhibitor, serpin B8, possessed determinants that could transform α1PDX into a specific and selective inhibitor of PCs. Stable recombinant forms of serpin B8 were shown to inhibit furin at a physiologically significant rate through the P4–P1 RXXR recognition sequence, in agreement with previous reports (15) and the observation that serpin B8-furin complexes are detectable in platelets (22). A second RXXR sequence in the P1–P3′ RCL residues of serpin B8 was found to be unreactive with furin, although the P3′ Arg in this sequence contributed 3-fold to the specificity of serpin B8 for inhibiting furin.
α1PDX was surprisingly found to be a 5-fold faster inhibitor of furin than serpin B8. This difference in specificity was largely accounted for by the different RCL sequences of the two serpins. Whereas the P6–P1 sequence of serpin B8 enhanced α1PDX specificity for furin by 2-fold, the serpin B8 P1′–P5′ sequence reduced α1PDX specificity for furin by 20-fold. Substituting the full P6–P5′ serpin B8 sequence into α1PDX produced an additive net 10-fold reduction in specificity for furin, within 2-fold of that of serpin B8. These findings demonstrate a major impact of the primed RCL sequence on serpin reactivity with furin in addition to the established importance of the unprimed RXXR sequence. The primed sequence thus represents an important overlooked determinant for engineering specificity and selectivity in α1PDX for PC family members.
The determinants of serpin reactivity with furin were additionally probed in the protease. Four variable loops surrounding the active site were identified by sequence alignments of PC catalytic domains. All alanine mutations of these loops resulted in moderate reductions in kcat/Km for furin cleavage of RXXR peptide substrates and comparable reductions in ka for furin inhibition by serpin B8 and α1PDX except for the 298–300 loop mutations, which showed markedly different effects on ka for the two serpins. Mutations in this loop thus enhanced reactivity with α1PDX and depressed reactivity with serpin B8. Substituting the primed or full serpin B8 RCL but not the unprimed RCL sequence into α1PDX reversed the effect of the 298–300 loop mutation on furin reactivity to that observed with serpin B8. These findings suggest that interactions of the furin 298–300 loop with the primed RCL sequence of serpin B8 contribute positively to reactivity, whereas interactions of the furin loop with the primed sequence of α1PDX antagonize reactivity. The importance of furin(298–300) loop interactions with RCL primed residues in determining reactivity is in keeping with this loop residing close to the primed substrate binding pockets of the protease (29).
Further substitutions of the 298–300 loops of other PCs into furin similarly revealed effects on α1PDX reactivity that differed significantly from the effects on serpin B8 reactivity or RXXR peptide substrate reactivity. Again, substituting the serpin B8 RCL into α1PDX altered the effects of the 298–300 loop variants on α1PDX reactivity to resemble more closely those on serpin B8. The 298–300 loop sequences of PCs thus contribute to variable reactivities with serpin B8 and α1PDX through differential interactions with the primed RCL sequence. Such differential interactions may contribute to the selectivity of serpin B8 for different PCs and could be exploited for engineering selective PC inhibitors.
Alanine mutations of the three other furin loops also exhibited small differential effects on serpin B8 and α1PDX reactivity, but these effects were marginally different from the changes in RXXR substrate reactivity. These loops thus appear to mostly affect furin active-site interactions with the serpin P4–P1 RXXR recognition sequence. Nevertheless, the loops make important contributions to furin reactivity with serpins, the 187–189 loop being of particular importance given the significant negative effects of mutating this loop on furin reactivity. The variable sequences of these loops in PC family members suggests that they could contribute to variable reactivities of serpin B8 with PCs and that such variability might be exploited in engineering selectivity in α1PDX for different PCs.
Our finding that the furin(298–300) loop influences reactivity with serpins lead us to explore potential interactions of this loop with exosites on the serpin body. A model of the serpin B8-furin Michaelis complex revealed strand 3C residues of serpin B8 that were close to this loop. Substitution of serpin B8-like strand 3C residues into the homologous residues of α1PDX resulted in a loss of furin reactivity comparable with that caused by substituting the serpin B8 RCL into α1PDX. This reactivity loss appeared to involve the furin(298–300) loop sequence because substituting the sequences of other PC loops showed that the reactivity losses of the α1PDX-serpin B8 strand 3C chimera strongly depended on the furin loop sequence. The strand 3C exosites thus appear to perturb furin(298–300) loop interactions with the α1PDX primed RCL sequence that govern reactivity.
Combining the serpin B8 strand 3C substitutions with the serpin B8 RCL substitution in α1PDX resulted in an unexpectedly dramatic ∼700-fold loss in furin reactivity, i.e. greater than the loss expected from additive effects of the mutations. Notably, the four furin alanine loop mutants showed similar large losses in reactivity with the dual chimera that reflected synergistic effects of the individual serpin B8 substitutions in the chimera except for the 298–300 loop mutant whose reactivity losses reflected additive effects of the individual substitutions. This suggested that interactions among the furin(298–300) loop, the serpin primed RCL sequence, and the strand 3C exosite are strongly coupled and that this coupling is lost by mutating the furin loop to alanines (30, 31). This conclusion is supported by the finding that the extent of coupling was dependent on the furin(298–300) loop sequence with the wild-type sequence showing the largest coupling and the PC5 sequence showing no coupling. Notably, such differential coupling effects significantly enhanced the selectivity of the α1PDX dual chimera for the furin(298–300) loop mutants. The observation that the reactivity of the dual chimera with furin is comparable in order of magnitude to the kcat/Km values for RXXR substrate cleavage suggests that the reactivity of this variant with furin may be governed solely by the RXXR sequence in the serpin RCL (32). This could result from the strand 3C mutations abrogating the contribution of the primed RCL sequence to reactivity. However, such an abrogation must be specific to the α1PDX dual chimera because serpin B8 possesses the RCL and similar strand 3C determinants of this chimera but shows a ∼100-fold higher reactivity. The effects of the strand 3C exosite residues on serpin reactivity thus appear to markedly depend on their context in the two serpins with the negative effects of the exosite residues expressed only in α1PDX. Importantly, our findings demonstrate that serpin exosites can have a profound effect on furin reactivity as a result of their perturbing protease active-site interactions with the serpin RCL and these perturbing effects can be exploited to engineer specificity and selectivity for inhibiting PCs. Together, our findings suggest the potential for engineering specific and selective serpin inhibitors of PCs utilizing α1PDX as a scaffold by targeting RCL and exosite determinants of α1PDX that affect PC specificity.
Acknowledgments
We thank Saivenkat Vagvala and Danté Brown for excellent technical assistance with the studies and Dr. Peter Gettins of the University of Illinois at Chicago, Department of Biochemistry and Molecular Biology, for critical comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R37-HL39888.
- PC
- proprotein convertase
- α1PI
- α1-protease inhibitor
- α1PDX
- α1-protease inhibitor Portland variant with P1 and P4 residues replaced with Arg
- RCL
- reactive center loop
- SI
- stoichiometry of inhibition
- PDB
- Protein Data Bank.
REFERENCES
- 1. Molloy S. S., Bresnahan P. A., Leppla S. H., Klimpel K. R., Thomas G. (1992) Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 267, 16396–16402 [PubMed] [Google Scholar]
- 2. Walker J. A., Molloy S. S., Thomas G., Sakaguchi T., Yoshida T., Chambers T. M., Kawaoka Y. (1994) Sequence specificity of furin, a proprotein-processing endoprotease, for the hemagglutinin of a virulent avian influenza virus. J. Virol. 68, 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scamuffa N., Calvo F., Chrétien M., Seidah N. G., Khatib A. M. (2006) Proprotein convertases. Lessons from knockouts. FASEB J. 20, 1954–1963 [DOI] [PubMed] [Google Scholar]
- 4. Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. (2008) The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 40, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 5. Artenstein A. W., Opal S. M. (2011) Proprotein convertases in health and disease. N. Engl. J. Med. 365, 2507–2518 [DOI] [PubMed] [Google Scholar]
- 6. Seidah N. G., Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 7. Couture F., D'Anjou F., Day R. (2011) On the cutting edge of proprotein convertase pharmacology. From molecular concepts to clinical applications. Biomol. Concepts 2, 421–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fugère M., Day R. (2002) Inhibitors of the subtilase-like pro-protein convertases (SPCs). Curr. Pharm. Des. 8, 549–562 [DOI] [PubMed] [Google Scholar]
- 9. Basak A. (2005) Inhibitors of proprotein convertases. J. Mol. Med. 83, 844–855 [DOI] [PubMed] [Google Scholar]
- 10. Henrich S., Lindberg I., Bode W., Than M. E. (2005) Proprotein convertase models based on the crystal structures of furin and kexin. Explanation of their specificity. J. Mol. Biol. 345, 211–227 [DOI] [PubMed] [Google Scholar]
- 11. Komiyama T., Fuller R. S. (2000) Engineered eglin c variants inhibit yeast and human proprotein processing proteases, Kex2 and furin. Biochemistry. 39, 15156–15165 [DOI] [PubMed] [Google Scholar]
- 12. Lu W., Zhang W., Molloy S. S., Thomas G., Ryan K., Chiang Y., Anderson S., Laskowski M., Jr. (1993) Arg15-Lys17-Arg18 turkey ovomucoid third domain inhibits human furin. J. Biol. Chem. 268, 14583–14585 [PubMed] [Google Scholar]
- 13. Van Rompaey L., Ayoubi T., Van De Ven W., Marynen P. (1997) Inhibition of intracellular proteolytic processing of soluble proproteins by an engineered α2-macroglobulin containing a furin recognition sequence in the bait region. Biochem. J. 326, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anderson E. D., Thomas L., Hayflick J. S., Thomas G. (1993) Inhibition of HIV-1 gp160-dependent membrane fusion by a furin-directed α1-antitrypsin variant. J. Biol. Chem. 268, 24887–24891 [PubMed] [Google Scholar]
- 15. Dahlen J. R., Jean F., Thomas G., Foster D. C., Kisiel W. (1998) Inhibition of soluble recombinant furin by human proteinase inhibitor 8. J. Biol. Chem. 273, 1851–1854 [DOI] [PubMed] [Google Scholar]
- 16. Osterwalder T., Kuhnen A., Leiserson W. M., Kim Y. S., Keshishian H. (2004) Drosophila serpin 4 functions as a neuroserpin-like inhibitor of subtilisin-like proprotein convertases. J. Neurosci. 24, 5482–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richer M. J., Keays C. A., Waterhouse J., Minhas J., Hashimoto C., Jean F. (2004) The Spn4 gene of Drosophila encodes a potent furin-directed secretory pathway serpin. Proc. Natl. Acad. Sci. U.S.A. 101, 10560–10565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bentele C., Krüger O., Tödtmann U., Oley M., Ragg H. (2006) A proprotein convertase-inhibiting serpin with an endoplasmic reticulum targeting signal from Branchiostoma lanceolatum, a close relative of vertebrates. Biochem. J. 395, 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Silverman G. A., Bird P. I., Carrell R. W., Church F. C., Coughlin P. B., Gettins P. G., Irving J. A., Lomas D. A., Luke C. J., Moyer R. W., Pemberton P. A., Remold-O'Donnell E., Salvesen G. S., Travis J., Whisstock J. C. (2001) The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 276, 33293–33296 [DOI] [PubMed] [Google Scholar]
- 20. Gettins P. G., Olson S. T. (2009) Exosite determinants of serpin specificity. J. Biol. Chem. 284, 20441–20445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Strik M. C., Bladergroen B. A., Wouters D., Kisiel W., Hooijberg J. H., Verlaan A. R., Hordijk P. L., Schneider P., Hack C. E., Kummer J. A. (2002) Distribution of the human intracellular serpin protease inhibitor 8 in human tissues. J. Histochem. Cytochem. 50, 1443–1454 [DOI] [PubMed] [Google Scholar]
- 22. Leblond J., Laprise M. H., Gaudreau S., Grondin F., Kisiel W., Dubois C. M. (2006) The serpin proteinase inhibitor 8. An endogenous furin inhibitor released from human platelets. Thromb. Haemost. 95, 243–252 [DOI] [PubMed] [Google Scholar]
- 23. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 24. Izaguirre G., Rezaie A. R., Olson S. T. (2009) Engineering functional antithrombin exosites in α1-proteinase inhibitor that specifically promote the inhibition of factor Xa and factor IXa. J. Biol. Chem. 284, 1550–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kwon K. S., Lee S., Yu M. H. (1995) Refolding of α1-antitrypsin expressed as inclusion bodies in Escherichia coli. Characterization of aggregation. Biochim. Biophys. Acta 1247, 179–184 [DOI] [PubMed] [Google Scholar]
- 26. Pannell R., Johnson D., Travis J. (1974) Isolation and properties of human plasma α1-proteinase inhibitor. Biochemistry 13, 5439–5445 [DOI] [PubMed] [Google Scholar]
- 27. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 28. Dufour E. K., Denault J. B., Hopkins P. C., Leduc R. (1998) Serpin-like properties of α1-antitrypsin Portland towards furin convertase. FEBS Lett. 426, 41–46 [DOI] [PubMed] [Google Scholar]
- 29. Henrich S., Cameron A., Bourenkov G. P., Kiefersauer R., Huber R., Lindberg I., Bode W., Than M. E. (2003) The crystal structure of the proprotein processing furin explains its stringent specificity. Nat. Struct. Biol. 10, 520–526 [DOI] [PubMed] [Google Scholar]
- 30. Horovitz A. (1987) Non-additivity in protein-protein interactions. J. Mol. Biol. 196, 733–735 [DOI] [PubMed] [Google Scholar]
- 31. Di Cera E. (1998) Site-specific thermodynamics: understanding cooperativity in molecular recognition. Chem. Rev. 98, 1563–1592 [DOI] [PubMed] [Google Scholar]
- 32. Dementiev A., Simonovic M., Volz K., Gettins P. G. (2003) Canonical inhibitor-like interactions explain reactivity of α1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J. Biol. Chem. 278, 37881–37887 [DOI] [PubMed] [Google Scholar]