

Background: The canonical regulation of CFTR is mediated by serine/threonine phosphorylation.
Results: GPCR stimulation of CFTR mutants lacking PKA/PKC sites revealed alternate pathways which involve tyrosine kinases.
Conclusion: Pyk2 and Src family tyrosine kinases mediate part of CFTR stimulation by the M3 muscarinic receptor.
Significance: This provides the first evidence that physiological secretagogues can activate CFTR through tyrosine phosphorylation.
Keywords: CFTR, G Protein-coupled Receptors (GPCR), PP2A, Src, Tyrosine Protein Kinase (Tyrosine Kinase), Muscarinic 3 Receptor
Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride (Cl−) channel, which plays an important role in physiological anion and fluid secretion, and is defective in several diseases. Although its activation by PKA and PKC has been studied extensively, its regulation by receptors is less well understood. To study signaling involved in CFTR activation, we measured whole-cell Cl− currents in BHK cells cotransfected with GPCRs and CFTR. In cells expressing the M3 muscarinic acetylcholine receptor, the agonist carbachol (Cch) caused strong activation of CFTR through two pathways; the canonical PKA-dependent mechanism and a second mechanism that involves tyrosine phosphorylation. The role of PKA was suggested by partial inhibition of cholinergic stimulation by the specific PKA inhibitor Rp-cAMPS. The role of tyrosine kinases was suggested by Cch stimulation of 15SA-CFTR and 9CA-CFTR, mutants that lack 15 PKA or 9 PKC consensus sequences and are unresponsive to PKA or PKC stimulation, respectively. Moreover the residual Cch response was sensitive to inhibitors of the Pyk2 and Src tyrosine kinase family. Our results suggest that tyrosine phosphorylation acts on CFTR directly and through inhibition of the phosphatase PP2A. Results suggest that PKA and tyrosine kinases contribute to CFTR regulation by GPCRs that are expressed at the apical membrane of intestinal and airway epithelia.
Introduction
Apical anion conductance is the rate-limiting step during transepithelial salt and fluid secretion. Secretagogues that elevate cAMP increase the activity of cystic fibrosis transmembrane conductance regulator (CFTR),3 an anion channel encoded by the gene mutated in cystic fibrosis (CF) (1) and CF-related diseases (2). CFTR function is also abnormal in other diseases; for example, it is reduced in the airways of people with chronic obstructive pulmonary disease (3) and hyperstimulated in the intestinal epithelium in cholera and some other secretory diarrheas (4, 5). CFTR-mediated secretion across the renal tubule drives the expansion of cysts in polycystic kidney disease (6). Studies of CFTR regulation and anion secretion are thus critical for understanding several disorders of epithelial ion transport.
CFTR is unique among ATP-Binding Cassette (ABC) transporters in possessing a regulatory domain (RD) with numerous phosphorylation sites (1). The open probability of the channel pore increases when the RD is phosphorylated by cAMP-dependent protein kinase A (PKA) and protein kinase C (PKC), and is reduced when dephosphorylated by PP2A and PP2C (7–9). RD has 9 dibasic and 5 monobasic consensus sequences for phosphorylation by PKA and 9 potential PKC sites in the RD and distal region of NBD1. According to the current paradigm CFTR is activated by peptide hormones and other secretatogogues such as PGE2 and epinephrine through phosphorylation by PKA. Protein kinase C (PKC) causes weak activation when added alone (7, 10, 11) and potentiates the response to PKA (12, 13). During metabolic stress, phosphorylation of the RD by 5′ adenosine monophosphate-activated protein kinase (AMPK) inhibits channel activity (14). Other kinases including Src (15), cysteine kinase 2 (16), spleen tyrosine kinase (Syk (16), and proline-rich tyrosine kinase (Pyk2 (17)) can also stimulate CFTR channel activity and/or trafficking; however their physiological roles are not known.
G protein-coupled receptors are important upstream regulators of CFTR. VIP, secretin, PGE2, epinephrine, and other secretagogues bind to Gαs-coupled basolateral receptors and lead to stimulation of adenylyl cyclase, elevation of cAMP, and PKA phosphorylation of the RD. This signaling cascade may occur locally in a sub-plasma membrane compartment when the GPCR is in the apical membrane, and may also be modulated by inhibitory GPCRs such as the type 2 lysophosphatidic acid receptor, which is coupled to Gαi, Gαq, and Gα12/13 and suppresses CFTR-mediated intestinal secretion (5).
Acetylcholine is a potent secretagogue in airway submucosal glands (18) where it acts primarily through the M3 muscarinic receptor (M3R) to mobilize Ca2+ from intracellular stores via the classical Gαq/11-phospholipase C-PKC/IP3 pathway. IP3 binding to its cognate receptor on the endoplasmic reticulum releases Ca2+, which activates TMEM16A and K+ channels and releases inositol 1,4,5-trisphosphate receptor-binding protein (IRBIT), which may stimulate multiple transporters and channels involved in secretion, most notably the sodium bicarbonate cotransporter (19). Although muscarinic responses are mediated through Ca2+ signaling, it has been noted that carbachol (Cch) responses are reduced in glands from CF human (20), ferret (21), and pig airways (22) compared with non-CF controls, especially after correction of the secretion rates for CF gland hypertrophy. This suggests part of the muscarinic secretory response may be mediated by CFTR. Further, using permeabilized sweat ducts, Reddy and Quinton showed that CFTR conductance can be activated by the G protein agonist GTPγS independently of cAMP (23), raising the possibility of alternative signaling pathways.
The purpose of this study was to explore CFTR regulation by the M3 muscarinic receptor (M3R), a prototypical Gαq/11-coupled receptor in CF-affected airway submucosal gland and intestinal epithelial cells. We found that the M3R can activate CFTR through Ca2+-dependent elevation of cAMP and conventional PKA phosphorylation, through inhibition of PP2A, and through direct tyrosine phosphorylation of CFTR by a pathway that includes the proline-rich tyrosine kinase Pyk2 and a Src family kinase.
EXPERIMENTAL PROCEDURES
Cell Culture
Untransfected baby hamster kidney (BHK) cells and BHK cells stably expressing either wild-type CFTR, a mutant lacking all 15 PKA consensus sequences (15SA-CFTR) (23), or a mutant lacking all 9 PKC consensus sequences (9CA-CFTR) (13) were plated on plastic coverslips at low density 2–3 days before patch-clamp experiments. The following day, cells were transfected with a pcDNA3-M3R plasmid using the Gene Juice (Millipore) reagent. The large T antigen and pMax-GFP were co-transfected with M3R to allow replication of the pcDNA3 plasmid and to identify transfected cells, respectively. Cells were cultured at 37 °C in 5% CO2.
Patch Clamp
Currents were measured using the broken-patch, whole-cell patch configuration. The holding potential was −40 mV and current/voltage (I-V) relationships were determined by pulsing from −40 mV to test voltages between −100 and +100 mV in 20 mV increments. Pipette capacitance was compensated electronically in the cell-attached mode. Voltage-clamp signals were recorded using an analog/digital interface (Digidata 1440; Axon Instruments, Inc., Burlingame, CA) and analyzed using pCLAMP version 10 (Axon Instruments). The external bath solution contained (in mm) 145 NaCl, 4 CsCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 tetradecyl sulfate (TES) (titrated with NaOH to pH 7.4) and had an osmolarity of 315 ± 5 mOsmol. The intrapipette solution contained (in mm) 113 l-aspartic acid, 113 CsOH, 27 CsCl, 1 NaCl, 1 MgCl2, 1 ethyleneglycoltetraacetic acid, 1 TES, and 3 MgATP (titrated with CsOH to pH 7.2) and had an osmolality of 285 ± 5 mOsmol. The calculated equilibrium potential for Cl− (ECl−) was −42 mV. Experiments were performed at room temperature (20–25 °C). Results are expressed as the means ± S.E. of n observations. Data were compared using the Student's t test with GraphPad Prism version 5.0 (GraphPad Software) and differences were considered significant when p < 0.05.
Biotinylation and Pulldown of CFTR on Streptavidin Beads
Surface expression of wild type and mutant CFTR channels was studied by streptavidin pull down after treating cells with sulfo-NHS-SS-biotin as described previously (24). Blots were also probed with α-tubulin antibody. To assess tyrosine phosphorylation on CFTR, BHK cells stably expressing WT-CFTR transiently co-transfected with v-Src were lysed, incubated with agarose protein G beads with or without M3A7 antibody (gift of J. Riordan), subjected to SDS-PAGE, transferred to PVDF membranes, immunoblotted using anti-phosphotyrosine antibody (4G10, used at 1:500 dilution, Millipore), exposed to secondary antibody conjugated to HRP (horseradish peroxidase; used at 1:5000) for 45 min, and visualized by enhanced chemiluminescence (Amersham Biosciences). Blots were then stripped and reprobed with anti-CFTR monoclonal antibody 23C5 (generated in collaboration with the laboratory of D.Y. Thomas, McGill University).
Measurement of Cytosolic Ca2+ Levels
BHK cells transfected with M3R and GFP plasmid were rinsed with standard external solution (130 mm NaCl, 5.4 mm KCl, 2.5 mm CaCl2, 0.8 mm MgCl2, 10 mm HEPES, and 10 mm d-glucose, adjusted to pH 7.4 with NaOH) and incubated for 45 min at 37 °C in the same solution supplemented with 5 μm of the acetoxy-methyl ester form of Fura-2 (Fura-2AM). Non-M3R and M3R-expressing cells were selected by monitoring GFP fluorescence using a UV lamp, then Fura-2 was excited alternately at 340 and 380 nm. Emitted fluorescence was measured at 510 nm and the 340/380 ratio was calculated. Experiments were conducted at room temperature (22–25 °C). To control the cell responses, after addition of Cch, 10 μm cyclopiazonic acid (CPA) was added to deplete Ca2+ endoplasmic reticulum store and activate Ca2+ influx.
Intracellular cAMP Assay
BHK-CFTR-wt cells transiently transfected with M3R were washed three times in PBS and incubated with forskolin, carbachol, or carbachol + BAPTA-AM in the same bath solution used for patch-clamp experiments. After 7 min of exposure at room temperature, lysates from 5 × 104 cells were harvested and assessed for level of intracellular cAMP using the enzyme immunoassay Parameter® kit (R&D Systems) according to the manufacturer's instructions. Extracellular cAMP was not measured, thus no correction was applied for cAMP released from the cells. Data represent two independent experiments performed in duplicate.
Chemicals
Chemicals were from Sigma. Stock solutions: 10 mm Fsk and 10 mm CFTRinh-172 were prepared in DMSO; 10 mm carbachol stock was prepared in H2O. Stock solutions of all inhibitors tested were prepared in DMSO at a 10× concentration.
Statistics
Results are expressed as the means ± S.E. of n observations. Data were compared using the Student's t test. Differences were considered statistically significant at p < 0.05. All statistical tests were performed using GraphPad Prism version 5.0 (GraphPad Software).
RESULTS
Cch Activates CFTR through the M3R
To investigate CFTR regulation by the muscarinic type 3 receptor we studied control BHK cells stably expressing CFTR-wt with, and without, transient co-transfection with the M3 receptor. First we monitored the effect of 10 μm Cch on whole cell Cl− current in control BHK cells expressing CFTR without M3R (Fig. 1, A and D). Cch failed to activate Cl− conductance under these conditions, although Fsk did stimulate a large non-rectifying Cl− conductance as expected (Fig. 1, A and D, dashed gray line). The current density at +40 mV during Cch exposure was 3.4 ± 0.33 pA/pF (n = 4) versus 71.5 ± 20 pA/pF (n = 9) during Fsk stimulation. This current was CFTR-dependent because BHK cells that transiently expressed M3R without CFTR did not respond to either Fsk or Cch (Fig. 1, B and E). However a large voltage- and time-independent Cl− current did appear when CFTR-expressing cells that had been transiently transfected with M3R were exposed to 10 μm Cch. The response to Cch was virtually abolished by the CFTR inhibitor CFTRinh172 (Fig. 1, C and D). Taken together, these results indicate that Cch activation of the muscarinic M3 receptor can cause robust stimulation of CFTR. Interestingly, the current density induced by Cch was 38% larger than during forskolin treatment (Fig. 1E, Cch 108.1 ± 13.2 pA/pF (n = 11) at +40 mV versus forskolin 78 ± 13.5 pA/pF (n = 13)), and adding Fsk in the continued presence of Cch did not cause a further increase (at +40mV: 102 ± 19 pA/pF (n = 5) data not shown). Strong activation of CFTR by M3R, which is generally coupled to Gαq/11/PLC/Ca2+/PKC signaling rather than the Gαs/adenylyl cyclase/cAMP/PKA pathway was unexpected; therefore we decided to dissect the signaling pathways involved.
FIGURE 1.

The cholinergic agonist Cch stimulates CFTR whole cell Cl− current through the M3R. BHK cells transfected with wild-type CFTR alone (CFTR-wt; A, D), M3R alone (B, E) or both CFTR-wt and M3R (C, F). A–C, representative traces of whole cell Cl− currents elicited by stepping from a holding potential of −40 mV to test potentials ranging from −100 to +100 mV in 20 mV increments under basal (unstimulated) conditions or in the presence of 10 μm Fsk or 10 μm Cch as indicated. Dashed lines indicate the zero current level. D–F, corresponding current-voltage (I-V) relationships normalized to cell capacitance (■, basal; □, Cch 10 μm; Δ, 10 μm Cch + 10 μm CFTR-Inh172; gray dotted line, 10 μm Fsk). Error bars show S.E. for n = 4 - 13 cells.
Carbachol Stimulation of CFTR via the PKA Pathway
The M3 receptor usually activates phospholipase C, which produces IP3 and diacylglycerol and leads to Ca2+ and activation of PKC. This was confirmed by loading cells with FURA-2AM and monitoring cytosolic Ca2+ as the fluorescence ratio when excited at 340 and 380 nm (Fig. 2A). Carbachol caused a rapid increase in the fluorescence ratio in cells transfected with the M3 receptor whereas those lacking the M3 receptor did not respond to Cch, although they showed a similar small Ca2+ response to the Ca2+-ATPase inhibitor CPA (Fig. 2A, upper panel). Cch-induced mobilization of Ca2+ was abolished in cells preloaded with the Ca2+ buffer BAPTA-AM, and in the presence of atropine, a specific muscarinic antagonist (Fig. 2A, lower panel).
FIGURE 2.
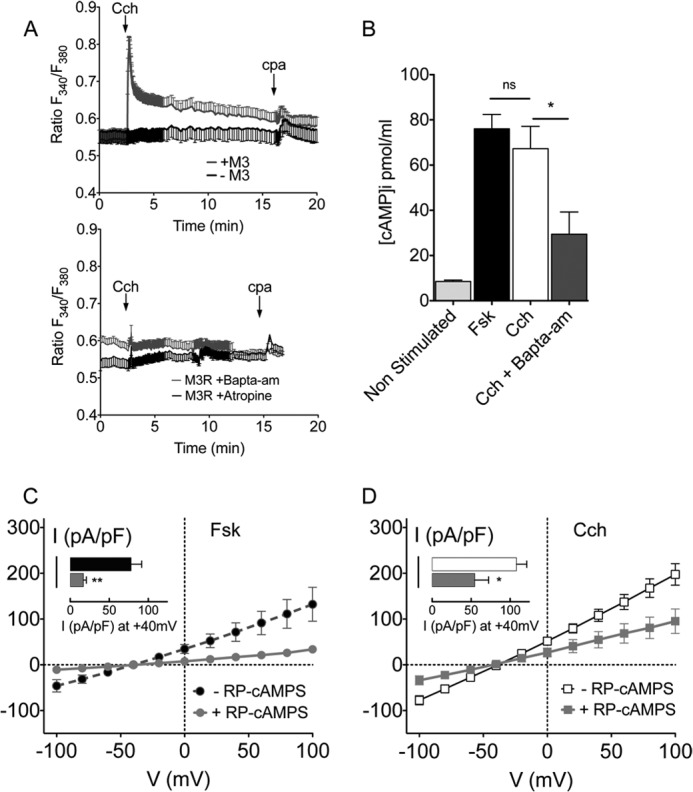
The cAMP/PKA pathway mediates part of the Cch stimulation of CFTR. A, time course of variations in the cytosolic Ca2+ level during treatment with Cch and the Ca2+-ATPase inhibitor cyclopiazonic acid (CPA). Upper panel: Cch-induced Ca2+ mobilization in control cells (black) and cells expressing the M3R (gray). Lower panel: Ca2+ response after 2 h of incubation with 10 μm BAPTA-AM (gray) or acute application of 10 μm atropine (black). B, intracellular cAMP levels (pmol/ml) in unstimulated condition or after 7 min of incubation with10 μm Fsk, 10 μm Cch or 10 μm Cch + 10 μm BAPTA-AM (incubated for 2 h at 37 °C). Results are the mean of two independent experiments with each set of data made in duplicate. cAMP concentration was determined using standard curves generated for each experiment. Error bars show the S.E. ns: non-significant difference, *, p < 0.05. C and D, I-V relationships of the Cl− current density recorded in presence of 10 μm Fsk (C) or Cch (D) after a 30 min incubation with DMSO (vehicle control) or 20 μm Rp-CAMPS as indicated. Histograms compare the current densities at +40 mV. Error bars show S.E. *, p < 0.05; **, p < 0.01.
Since Cch raises [cAMP] in some cells by activating Ca2+/calmodulin-dependent adenylate cyclases or through coupling to Gαi (25, 26), we measured cAMP in BHK-CFTR-wt cells during exposure to 10 μm Cch (Fig. 2B). Intracellular [cAMP] was 13.6 ± 2.9 pmol/ml under basal conditions and increased to 61.3 ± 10.3 pmol/ml after 7 min Cch stimulation. The cAMP response to Cch was comparable to that caused by Fsk stimulation (76.0 ± 6.3 pmol/ml). Pretreating cells with 10 μm BAPTA-AM for 2 h to chelate intracellular Ca2+ inhibited this cAMP response to 29.5 ± 9.8 pmol/ml. Taken together, these results suggest that part of the stimulation of CFTR by the M3R is mediated by a Ca2+-induced elevation of cAMP and subsequent activation of PKA.
The role of PKA in the activation of CFTR through by Cch was confirmed functionally using Rp-cAMPS (Rp-Cyclic 3′,5′-hydrogen phosphorothioate adenosine triethylammonium), a cell-permeant cAMP analog which is a potent PKA inhibitor (Fig. 2, C and D). Cells were pre-incubated with 20 μm Rp-CAMPS for 30 min, and 20 μm Rp-cAMPS was also included in the pipette solution while recording whole cell Cl− currents. Rp-CAMPS inhibited the Cl− currents elicited by both Fsk and Cch, however the Fsk response was inhibited more than the Cch response. The Fsk-induced current was 16.9 ± 3.8 pA/pF at +40mV (n = 9; Fig. 2C) in the presence of Rp-cAMPS, which corresponds to ∼80% inhibition of the control CFTR current. By contrast, the Cch-induced current during Rp-cAMPS exposure was 54.7 ± 17.7 pA/pF at +40mV (n = 5; Fig. 2D), an inhibition of ∼50% relative to controls. This suggests that other signaling mechanisms besides PKA contribute to the muscarinic activation of CFTR.
Carbachol Stimulates CFTR through PKA and Non-PKA Signaling Pathways
To explore PKA-independent regulation of CFTR without using inhibitors that might have confounding effects on other pathways, we studied the activation of 15SA-CFTR (S422A/S660A/S670A/S686A/T690A/S700A/S712A/S737A/S753A/S768A/T787A/T788A/S790A/S795A/S813A). This mutant lacks potential PKA sites on the R domain and was not responsive to PKA in previous single channel studies (27). Whole cell Cl− current did not increase when cells expressing 15SA-CFTR were exposed to Fsk, confirming that this mutant is indeed unresponsive to PKA (Fig. 3, C and E, dashed gray line). The lack of current does not reflect misprocessing because immunoblots of cell lysates and pulldowns of 15SA-CFTR on streptavidin beads after surface biotinylation revealed wild-type levels of mature protein (i.e. band C glycoform) and localization at the cell surface (Fig. 3A). Some α-tubulin was detected in the pulldowns along with CFTR (Fig. 3, A and B), however we showed previously that a small fraction of the cells (0.4–0.7% of the total population) are leaky to sulfo-NHS-SS-biotin and other molecules having Mr = 607–857, and this is sufficient to explain the intracellular labeling observed (24). Despite being unresponsive to forskolin, Cl− currents were stimulated by Cch by more than 4-fold (from 4.9 ± 1.9 pA/pF to 20.3 ± 5 pA/pF at +40 mV; Fig. 3E,), and were time- and voltage-independent and abolished by CFTRinh172 (Fig. 3, C and E, n = 5). These results indicate that 15SA-CFTR can be activated by this muscarinic agonist through a non-PKA mechanism. Based on the inhibition by Rp-cAMPS in Fig. 2D, the PKA-independent pathway accounts for about half of the Cch stimulation of wild-type CFTR.
FIGURE 3.
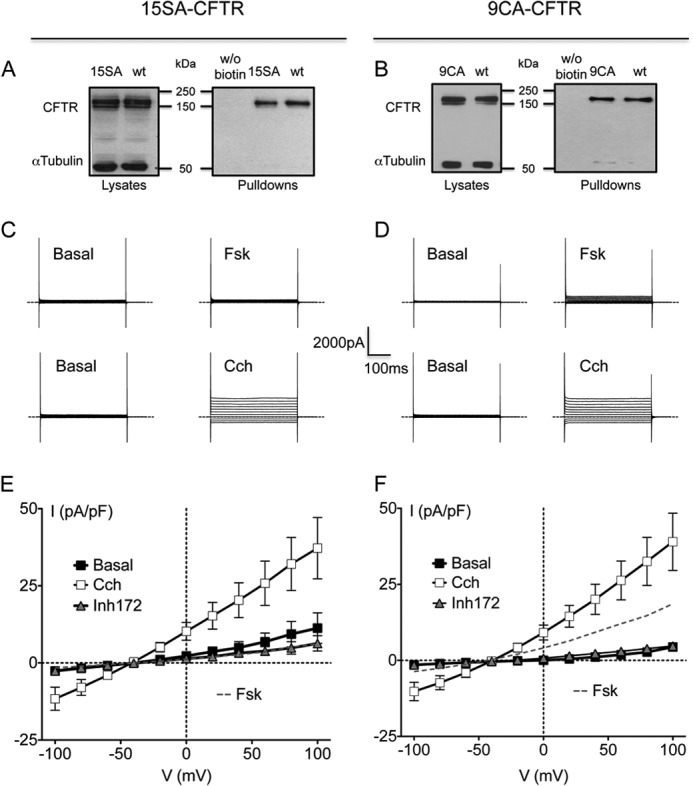
Cch regulates CFTR through cAMP/PKA-independent mechanisms. A and B, total and cell surface expression of wt-CFTR, 15SA-CFTR (A) and 9CA-CFTR (B) proteins. Left panels show expression in cell lysates (25 μg of total protein loaded). Right panels show representative immunoblots after surface biotinylation and pulldown on streptavidin beads showing CFTR at the cell surface (150 μg of total protein used for pulldowns). α-Tubulin was used as loading control when blotting cell lysates but could appeared in pulldowns due to labeling in a small population of sulfo-NHS-biotin permeable cells. C and D, representative traces of whole cell Cl− current in BHK cells expressing 15SA-CFTR (C) or 9CA-CFTR (D), elicited by stepping from holding potential of −40 mV to a series test potentials from −100 to +100 mV in 20-mV increments under basal conditions or in the presence of 10 μm Fsk or 10 μm Cch as indicated. Dashed lines indicate the zero current level. E and F, corresponding I-V relationships for 15SA-CFTR (E) and 9CA-CFTR (F) normalized to cell capacitance (■, basal; □, Cch 10 μm; Δ, 10 μm Cch + 10 μm CFTR-Inh172; dashed gray line, 10 μm Fsk). Error bars represent S.E. for n = 5–7 cells.
CFTR channels are stimulated by acute PKC exposure (13) and Gαq/11 activates PKC, therefore we examined the role of PKC in the CFTR response to Cch. To study PKC regulation without using inhibitors that could perturb other signaling pathways, we used BHK cells expressing 9CA-CFTR, a mutant that lacks all 9 PKC consensus sites in the RD and NBD1 regulatory extension (T582A/T604A/S641A/T682/S686A/S707A/S790A/T791A/S809A) (13). 9CA-CFTR is not activated by PKC and has greatly diminished responses to PKA, presumably because it lacks the potentiation caused by PKC (12). Fsk elicited relatively small currents of 9.3 ± 4.2 pA/pF (n = 5) at +40 mV (Fig. 3, D and F, dashed gray line) in agreement with previous studies. On the other hand Cch evoked much larger responses (20.1 ± 4.6 pA/pF (n = 7) at +40 pA/pF; Fig. 3F, open squares), comparable to those obtained with 15SA-CFTR cells. Again, immunoblots of the cell lysates and biotinylation/streptavidin pull-down experiments did not reveal any gross differences in either the biosynthesis or surface expression of 9CA-CFTR when compared with wt-CFTR (Fig. 3B). These results with 15SA-CFTR and 9CA-CFTR suggest that Cch regulates CFTR, at least in part, through a PKA and PKC-independent mechanism.
PKA-independent Regulation of CFTR by GPCRs Is Mediated by Protein Tyrosine Kinases
Src family kinases associate with GPCRs including muscarinic receptors (28) and mediate tyrosine phosphorylation on downstream targets during GPCR activation (29). Since Gαq/11 can activate the calcium-dependent tyrosine kinase Pyk2 (30, 31), which forms a complex with Src and increases its activity, we wondered if these tyrosine kinases play a role in the muscarinic stimulation of CFTR.
To investigate the role of tyrosine kinases we tested the Pyk2-specific inhibitor Tyrphostin A9 and the Src Family tyrosine Kinases (SFK) inhibitor “Src Inhibitor-1” (SrcInh1) for their effects on the carbachol response (Fig. 4). Neither inhibitor altered the Fsk-stimulated CFTR current as expected, however both inhibited the Cch-induced CFTR current significantly (Fig. 4A). Carbachol stimulation was reduced by 55% in the presence of 1 μm TyrphostinA9 (at +40mV current density was 58.94 ± 15.4 pA/pF), and by 65% after pre-incubation with 10 μm SrcInh1 (41.46 ± 13.2 pA/pF at +40mV). Both tyrosine kinase inhibitors had more dramatic effects on 15SA-CFTR than on wild-type channels (Fig. 4B). With Tyrphostin A9 or SrcInh1 present, Cl− currents during Cch exposure were similar to those under basal (unstimulated) conditions. 15SA-CFTR cells exhibited a basal current of 4.9 ± 1.9 pA/pF at +40mV whereas those exposed to Cch in combination with Tyrphostin A9 or SrcInh1 had current densities of 2.0 ± 0.4 pA/pF and 4.4 ± 1.9 pA/pF, respectively. Similar results were obtained when Cch was tested on 9CA-CFTR in the presence of Tyrophostin A9 or SrcInh1 (Fig. 4C).
FIGURE 4.

Non-PKA regulation of CFTR is mediated by Pyk2 and a Src family kinase. Upper panel: current-voltage relationships for wt-CFTR (A), 15SA-CFTR (B), and 9CA-CFTR (C) in presence of Cch after incubation with DMSO (control) or tyrosine kinase inhibitors as indicated, normalized to cell capacitance. Cells were incubated overnight with the SFK inhibitor Src Inhibitor-1 (10 μm). The Pyk2 inhibitor TyrphostinA9 (1 μm) was added to cells 30 min before recording whole-cell currents. Lower panel: comparison of the corresponding current density obtained at +40 mV. Results obtained in presence of Fsk instead of Cch are also shown. Error bars show S.E. (n = 4–13 cells). ns; no significant difference from untreated cells; *, p < 0.05; **, p < 0.01.
Inhibiting the Pyk2/Src pathway reduced CFTR activation by Cch; therefore we examined if these kinases could act by directly phosphorylating CFTR or through modulation of serine/threonine phosphorylation. Src could affect many signaling mechanisms including those that involve serine/threonine phosphorylation; e.g. through inhibition of the serine/threonine protein phosphatase PP2A, which is part of the CFTR interactome (9) and regulates its channel activity (32). To test the possible role of PP2A, we recorded whole cell current responses to Cch in the presence of Calyculin A, a potent and selective PP2A inhibitor, reasoning that any modulation that involves PP2A (e.g. through inhibition by Src) would be lost. Experiments were performed with and without SrcInh1. Preincubating cells with 5 nm Calyculin A alone did not affect the stimulation of wt- and 15SA-CFTR currents by Cch, suggesting that PP2A was already inactive and/or plays little role under these conditions (Fig. 5A). However, when cells were treated with both Calyculin A and SrcInh1, the current response of CFTR-wt cells to Cch stimulation was reduced by 30% and became similar to that observed during Fsk stimulation (at +40mV with Calyculin A + Src Inhibitor-1: 76.49 ± 12.2 pA/pF). This decrease was smaller than with Src Inhibitor-1 alone (Fig. 4A), indicating there is normally an up-regulation of PP2A by SrcInh1 alone (and hence a large decrease in current), which is mitigated by inhibiting PP2A with calyculin A. The up-regulation of PP2A by SrcInh1 is expected since SFKs are known to phosphorylate the catalytic subunit of PP2A at residue Y307 and inhibit its phosphatase activity (33).
FIGURE 5.
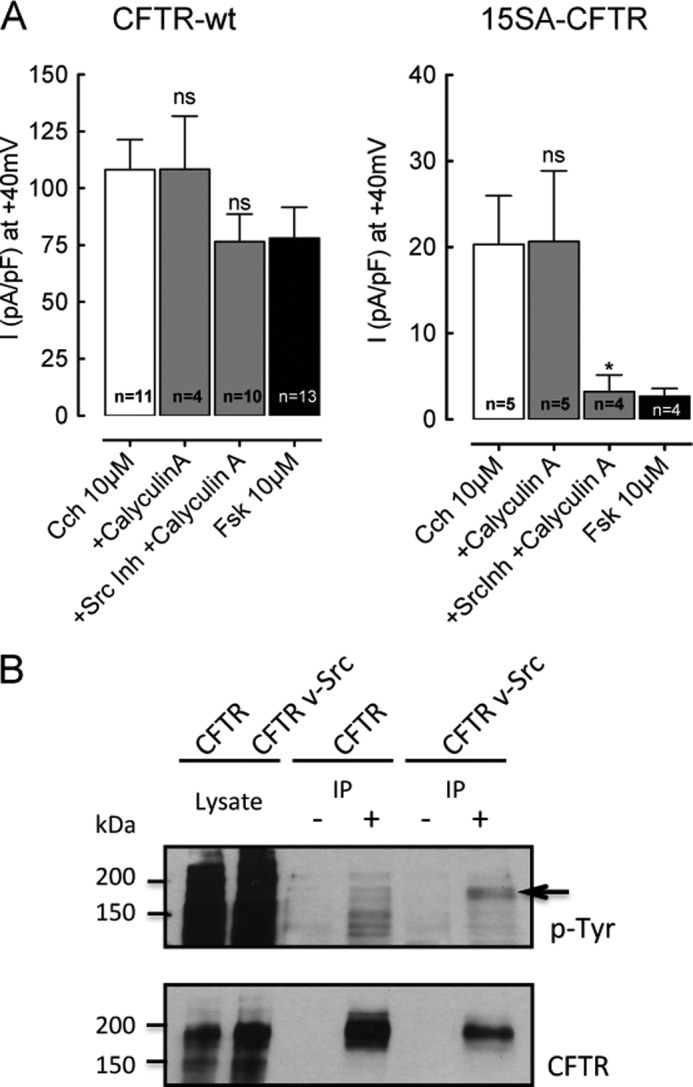
Src family kinase mediates Cch stimulation of CFTR through at least two mechanisms. A, Cl− current density at +40 mV for BHK cells expressing CFTR-wt (left) or 15SA-CFTR (right), in presence of 10 μm Fsk or Cch after incubation with DMSO (vehicle control), the PP2A inhibitor CalyculinA, or CalyculinA + Src Inhibitor-1 as indicated. Cells were incubated with 10 μm Src Inhibitor-1 overnight and treated with 5 nm CalyculinA for 30 min. ns, no significant difference from Cch control; *, p < 0.05. Error bars show S.E. for number of cells indicated. B, Western blot of BHK-CFTR-wt cells transfected with v-Src and pretreated with the tyrosine phosphatase inhibitors dephostatin (10 μm) and orthovanadate (50 μm) in serum-free medium. CFTR was immunoprecipitated, and the immunoblot was probed with anti-phosphotyrosine antibody (upper panel), then stripped and reblotted using anti-CFTR antibody 23C5 (lower panel). Lanes marked (−) are non-immune controls for nonspecific CFTR binding to the beads, and (+) indicate IP performed under identical conditions with M3A7. The phosphotyrosine (P-Tyr, indicated by arrow) and CFTR band (lower blot) are superimposable. Representative of three experiments.
Remarkably, in 15SA-CFTR cells the Cch-stimulated Cl− current was abolished when PP2A and SFKs were both inhibited, leaving a residual current (3.21 ± 1.9 pA/pF +40mV), which was comparable to to that under basal conditions in the absence of Cch. Thus, in addition to stimulating CFTR through inhibition of PP2A, Src also increases 15SA currents through another (PP2A-independent) mechanism, which could involve direct tyrosine phosphorylation of CFTR. To test tyrosine phosphorylation of CFTR, BHK CFTR-wt cells were transiently transfected with constitutively active v-Src and treated with tyrosine phosphatase inhibitors (10 μm dephostatin, 50 μm orthovanadate) in serum-free medium. CFTR was then immunoprecipitated using M3A7 antibody and subjected to SDS-PAGE. Immunoblots were probed using the anti-phosphotyrosine monoclonal antibody 4G10, stripped, and re-probed with the anti-CFTR monoclonal antibody 23C5. A clear phosphotyrosine band was detected on CFTR in v-Src transfected cells, but not in the untransfected control cells lacking v-Src despite higher CFTR expression in the control cells (Fig. 5B). Since constitutive Src activity apparently leads to CFTR phosphorylation, this phosphorylation might account for the activation of 15SA-CFTR by Cch in Fig. 5A (right panel) and why SrcInh1 abolishes the response (same figure). Taken together, these results suggest that tyrosine kinases exert a dual stimulatory effect as mentioned above: one mediated by inhibition of PP2A and the other due to direct phosphorylation of the channel. Both regulatory mechanisms operate on CFTR-wt whereas only the direct tyrosine phosphorylation pathway controls 15SA-CFTR, which lacks serine/threonine phosphorylation at PKA sites and therefore is less sensitive to PP2A.
DISCUSSION
We have found that carbachol activation of the M3 muscarinic receptor can stimulate the CFTR channel, and we have identified two distinct signaling pathways that mediate this regulation. Carbachol is a potent secretagogue in many epithelia which is thought to stimulate Ca2+-activated Cl− channels (CaCC) rather than CFTR (e.g. (34)). Nevertheless reduced carbachol responses have been observed in CF airway glands, raising the possibility that CFTR mediates part of the muscarinic secretory response. In a heterologous model cell line devoid of endogenous muscarinic receptors, CFTR and detectable CaCCs we found that Cch can stimulate Cl− currents, but only when the cells are transfected with both M3R and CFTR. Cells expressing CFTR alone did not respond to carbachol, and carbachol did not stimulate endogenous Cl− channels when added to cells that were transfected with M3R alone (Fig. 1). The lack of endogenous CaCC activity in M3R-transfected BHK cells was unexpected but not unprecedented, as many cell lines do not express TMEM16A including HEK 293, COS7, 9HTEo-, and others4, and other TMEM16 family members may respond very differently to Ca2+ elevation (e.g. TMEM16F (35)). The whole cell Cl− currents activated by carbachol were time and voltage-independent and did not display significant rectification (Fig. 1C), hallmarks of the CFTR channel (10, 36–38). Cch-induced Cl− current was also abolished by CFTRinh172, a CFTR inhibitor that does not act on Ca2+-activated Cl− channels (39).
Considering the robust activation of CFTR by Cch (stronger than forskolin), we studied the signaling pathways involved. M3R is a G-protein coupled receptor (class I) which binds muscarinic agonists such as Cch and activates G proteins of the Gαq/11 family (40). When stimulated, Gαq/11 activates phospholipase C (PLC), which in turn catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 elevates intracellular Ca2+ by activating IP3 receptors, whereas DAG activates some PKC isoforms. This classical signaling pathway regulates a multitude of cellular processes including ion channels and transporters.
Although CFTR channels are weakly stimulated by acute exposure to PKC (7), we confirmed that such activation is unlikely to explain the robust Cch-induced currents by studying 9CA-CFTR. This mutant lacks the PKC consensus sequences on the R domain and distal NBD1, yet still responded to Cch stimulation. PKC and Ca2+ can also activate the PKA pathway through stimulation of a Ca2+-dependent adenylate cyclase and cAMP synthesis (41). Indeed, cAMP measurements in Fig. 2 confirmed that Cch elevates [cAMP] in transfected BHK cells to levels which are comparable to those during forskolin stimulation. This cAMP response was Ca2+-dependent, further suggesting the involvement of a Ca2+/calmodulin-activated adenylyl cyclase (41). Further evidence for the involvement of PKA came from partial inhibition of the Cch response by the PKA-selective inhibitor Rp-cAMPS. There is synergism between Ca2+ and cAMP in the regulation of fluid secretion (42), and a recent study has highlighted the role of Gαq/11 stimulation and IRBIT (IP3 Receptor-Binding protein released with IP3) in stimulating CFTR and the anion exchanger slc26a (43). In BHK cells, Ca2+ release and cAMP production induced by M3R activation may cause IRBIT dissociation from the IP3 receptor and release it from the endoplasmic reticulum. Further experiments are underway to clarify the role of IRBIT in the regulation of CFTR by muscarinic receptors.
Stimulation of CFTR mutants lacking PKA or PKC phosphorylation consensus sites (15SA-CFTR and 9CA-CFTR, Fig. 3) and the significant residual current response that remains after exposure to PKA inhibitor led us to examine alternative pathways. PKC and Ca2+ activate the proline-rich tyrosine kinase Pyk2, which interacts with, and activates SFKs, therefore we examined the role of Pyk2-Src in the CFTR response to Cch. Inhibiting Pyk2 or SFKs consistently reduced the Cch-stimulated WT-CFTR current and abolished the Cch-stimulated 15SA-CFTR current, indicating a PKA-independent signaling pathway. This provides the first evidence for physiological regulation of CFTR through tyrosine phosphorylation, although it has been known for many years that adding purified Src to excised membrane patches alters CFTR gating (15). A recent study suggested that the antipsychotic drug Spiperone can activate both CFTR and CaCC (44) through Pyk2 (17). Our finding that Cch stimulation of 15SA-CFTR is abolished by inhibiting either tyrosine kinase suggests they mediate part of the M3 receptor activation of CFTR and act in a common pathway.
To determine if Src regulates CFTR directly or through an accessory protein we examined phosphotyrosine on CFTR in cells expressing constitutively active v-Src, while Liang and colleagues presented evidence for an involvement of Pyk2 (17). These kinases may act on CFTR as a heteromeric complex as they are known to interact and undergo reciprocal phosphorylation and activation. Tyrosine phosphorylation also impinges on the regulation of CFTR by PKA and PKC, which depends on the balance between these serine-threonine kinases and opposing phosphatases such as PP2A, which interacts with CFTR and down-regulates open probability (33). Inhibiting PP2A with calyculin A did not enhance the responses of wt and 15SA-CFTR to Cch, probably because the phosphatase was already inactive. However there was strong inhibition when Src was inhibited (Fig. 4A) and the current was partially restored when both Src and PP2A were inhibited (Fig. 5A). Together these findings suggest that the SFK/Pyk2 complex acts directly through phosphorylation of CFTR, and indirectly through inhibition of PP2A, which reduces the dephosphorylation of CFTR thereby enhancing its phosphorylation and activation by PKA + PKC. A scheme summarizing these pathways is shown in Fig. 6.
FIGURE 6.
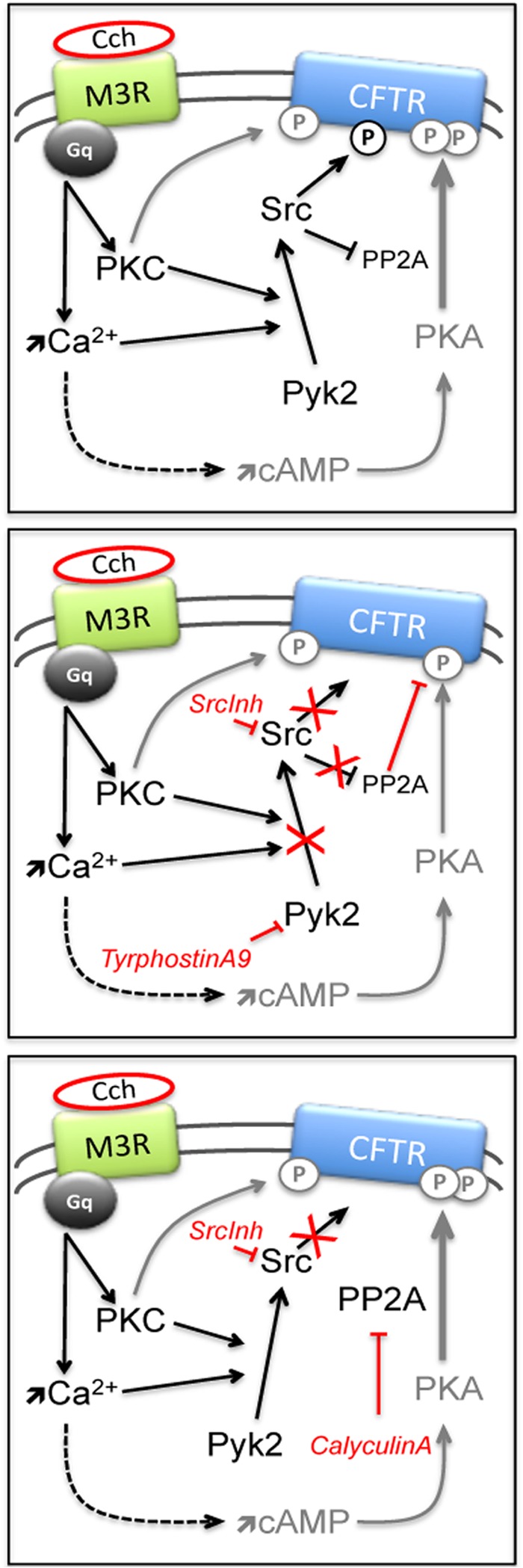
Scheme for CFTR stimulation by Gαq/11-coupled GPCR. Pathways mediating Cch/M3R stimulation of wt-CFTR (top panel) and the effects of inhibiting tyrosine kinases (middle) or SFK + PP2A (bottom panel). Gray “P” on CFTR represents serine/threonine phosphorylation, black “P” indicates tyrosine phosphorylation. Arrows symbolize activation and bars inhibition.
In this study we have shown strong activation of CFTR by a Gαq/11-coupled GPCR that is not usually thought to regulate this Cl− channel. We have found that the activation occurs through multiple mechanisms; Ca2+ mobilization stimulates Ca2+-activated adenylyl cyclase and the canonical PKA pathway, while tyrosine kinases provide alternative mechanisms of CFTR stimulation. The present results suggest that the Pyk2/SFK pathway phosphorylates CFTR directly and also acts indirectly by inhibiting PP2A. Further studies should identify the phosphotyrosines and extend these findings to other GPCRs, especially those expressed in the apical membrane of epithelia where they may form regulatory complexes with CFTR.
Acknowledgment
We thank Dr. Luis Galietta from the Gaslini Institute in Genova for helpful discussions.
This research was supported by the Canada Foundation for Innovation and Canadian Institutes for Health Research.
L. Galietta, personal communication.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- PKA/C
- protein kinase A/C
- SFK
- Src family kinases
- Fsk
- forskolin
- Cch
- carbachol
- GPCR
- G protein-coupled receptor
- RD
- regulatory domain
- IRBIT
- inositol 1,4,5-trisphosphate receptor-binding protein
- M3R
- muscarinic type 3 receptor
- CACC
- Ca2+-activated Cl− channel
- GTPγS
- guanosine 5'-O-[γ-thio]triphosphate.
REFERENCES
- 1. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 2. Kerem E. (2006) Atypical CF and CF related diseases. Paediatr Respir Rev 7, S144–S146 [DOI] [PubMed] [Google Scholar]
- 3. Cantin A. M., Hanrahan J. W., Bilodeau G., Ellis L., Dupuis A., Liao J., Zielenski J., Durie P. (2006) Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am. J. Respir. Crit. Care Med. 173, 1139–1144 [DOI] [PubMed] [Google Scholar]
- 4. Field M., Fromm D., al-Awqati Q., Greenough W. B. (1972) Effect of cholera enterotoxin on ion transport across isolated ileal mucosa. J. Clin. Invest. 51, 796–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li C., Dandridge K. S., Di A., Marrs K. L., Harris E. L., Roy K., Jackson J. S., Makarova N. V., Fujiwara Y., Farrar P. L., Nelson D. J., Tigyi G. J., Naren A. P. (2005) Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J. Exp. Med. 202, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li H., Yang W., Mendes F., Amaral M. D., Sheppard D. N. (2012) Impact of the cystic fibrosis mutation F508del-CFTR on renal cyst formation and growth. Am. J. Physiol. Renal Physiol. 303, F1176–E1186 [DOI] [PubMed] [Google Scholar]
- 7. Berger H. A., Travis S. M., Welsh M. J. (1993) Regulation of the cystic fibrosis transmembrane conductance regulator Cl- channel by specific protein kinases and protein phosphatases. J. Biol. Chem. 268, 2037–2047 [PubMed] [Google Scholar]
- 8. Luo J., Pato M. D., Riordan J. R., Hanrahan J. W. (1998) Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am. J. Physiol. 274, C1397–C1410 [DOI] [PubMed] [Google Scholar]
- 9. Thelin W. R., Kesimer M., Tarran R., Kreda S. M., Grubb B. R., Sheehan J. K., Stutts M. J., Milgram S. L. (2005) The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J. Biol. Chem. 280, 41512–41520 [DOI] [PubMed] [Google Scholar]
- 10. Tabcharani J. A., Chang X. B., Riordan J. R., Hanrahan J. W. (1991) Phosphorylation-regulated Cl- channel in CHO cells stably expressing the cystic fibrosis gene. Nature 352, 628–631 [DOI] [PubMed] [Google Scholar]
- 11. Anderson M. P., Rich D. P., Gregory R. J., Smith A. E., Welsh M. J. (1991) Generation of cAMP-activated chloride currents by expression of CFTR. Science 251, 679–682 [DOI] [PubMed] [Google Scholar]
- 12. Jia Y., Mathews C. J., Hanrahan J. W. (1997) Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J. Biol. Chem. 272, 4978–4984 [DOI] [PubMed] [Google Scholar]
- 13. Chappe V., Hinkson D. A., Zhu T., Chang X.-B., Riordan J. R., Hanrahan J. W. (2003) Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J. Physiol. 548, 39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hallows K. R., McCane J. E., Kemp B. E., Witters L. A., Foskett J. K. (2003) Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J. Biol. Chem. 278, 998–1004 [DOI] [PubMed] [Google Scholar]
- 15. Fischer H., Machen T. E. (1996) The tyrosine kinase p60c-src regulates the fast gate of the cystic fibrosis transmembrane conductance regulator chloride channel. Biophys. J. 71, 3073–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luz S., Kongsuphol P., Mendes A. I., Romeiras F., Sousa M., Schreiber R., Matos P., Jordan P., Mehta A., Amaral M. D., Kunzelmann K., Farinha C. M. (2011) Contribution of casein kinase 2 and spleen tyrosine kinase to CFTR trafficking and protein kinase A-induced activity. Mol. Cell. Biol. 31, 4392–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang L., Woodward O. M., Chen Z., Cotter R., Guggino W. B. (2011) A novel role of protein tyrosine kinase2 in mediating chloride secretion in human airway epithelial cells. PLoS ONE 6, e21991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corrales R. J., Nadel J. A., Widdicombe J. H. (1984) Source of the fluid component of secretions from tracheal submucosal glands in cats. J Appl Physiol 56, 1076–1082 [DOI] [PubMed] [Google Scholar]
- 19. Park S., Hong J. H., Ohana E., Muallem S. (2012) The WNK/SPAK and IRBIT/PP1 pathways in epithelial fluid and electrolyte transport. Physiology 27, 291–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salinas D., Haggie P. M., Thiagarajah J. R., Song Y., Rosbe K., Finkbeiner W. E., Nielson D. W., Verkman A. S. (2005) Submucosal gland dysfunction as a primary defect in cystic fibrosis. FASEB J. 19, 431–433 [DOI] [PubMed] [Google Scholar]
- 21. Sun X., Sui H., Fisher J. T., Yan Z., Liu X., Cho H.-J., Joo N. S., Zhang Y., Zhou W., Yi Y., Kinyon J. M., Lei-Butters D. C., Griffin M. A., Naumann P., Luo M., Ascher J., Wang K., Frana T., Wine J. J., Meyerholz D. K., Engelhardt J. F. (2010) Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Invest. 120, 3149–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joo N. S., Cho H.-J., Khansaheb M., Wine J. J. (2010) Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J. Clin. Invest. 120, 3161–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reddy M. M., Quinton P. M. (2001) cAMP-independent phosphorylation activation of CFTR by G proteins in native human sweat duct. Am. J. Physiol., Cell Physiol. 280, C604–C613 [DOI] [PubMed] [Google Scholar]
- 24. Luo Y., McDonald K., Hanrahan J. W. (2009) Trafficking of immature DeltaF508-CFTR to the plasma membrane and its detection by biotinylation. Biochem. J. 419, 211–219 [DOI] [PubMed] [Google Scholar]
- 25. Suidan H. S., Murrell R. D., Tolkovsky A. M. (1991) Carbachol and bradykinin elevate cyclic AMP and rapidly deplete ATP in cultured rat sympathetic neurons. Cell Regul. 2, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirst R. A., Lambert D. G. (1993) Carbachol stimulated cAMP formation in SH-SY5Y cells is dependent on both extracellular and intracellular calcium. Biochem. Soc. Trans. 21, 431S. [DOI] [PubMed] [Google Scholar]
- 27. Seibert F. S., Chang X. B., Aleksandrov A. A., Clarke D. M., Hanrahan J. W., Riordan J. R. (1999) Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochim. Biophys. Acta 1461, 275–283 [DOI] [PubMed] [Google Scholar]
- 28. Watcharasit P., Tucholski J., Jope R. S. (2001) Src family kinase involvement in muscarinic receptor-induced tyrosine phosphorylation in differentiated SH-SY5Y cells. Neurochem. Res. 26, 809–816 [DOI] [PubMed] [Google Scholar]
- 29. Luttrell D. K., Luttrell L. M. (2004) Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 23, 7969–7978 [DOI] [PubMed] [Google Scholar]
- 30. Brinson A. E., Harding T., Diliberto P. A., He Y., Li X., Hunter D., Herman B., Earp H. S., Graves L. M. (1998) Regulation of a calcium-dependent tyrosine kinase in vascular smooth muscle cells by angiotensin II and platelet-derived growth factor. Dependence on calcium and the actin cytoskeleton. J. Biol. Chem. 273, 1711–1718 [DOI] [PubMed] [Google Scholar]
- 31. Li X., Hunter D., Morris J., Haskill J. S., Earp H. S. (1998) A calcium-dependent tyrosine kinase splice variant in human monocytes. Activation by a two-stage process involving adherence and a subsequent intracellular signal. J. Biol. Chem. 273, 9361–9364 [DOI] [PubMed] [Google Scholar]
- 32. Gadsby D. C., Nairn A. C. (1999) Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 79, S77-S107 [DOI] [PubMed] [Google Scholar]
- 33. Chen J., Martin B. L., Brautigan D. L. (1992) Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 34. Schultheiss G., Siefjediers A., Diener M. (2005) Muscarinic receptor stimulation activates a Ca(2+)-dependent Cl(−) conductance in rat distal colon. J. Membr. Biol. 204, 117–127 [DOI] [PubMed] [Google Scholar]
- 35. Grubb S., Poulsen K. A., Juul C. A., Kyed T., Klausen T. K., Larsen E. H., Hoffmann E. K. (2013) TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J. Gen. Physiol. 141, 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berger H. A., Anderson M. P., Gregory R. J., Thompson S., Howard P. W., Maurer R. A., Mulligan R., Smith A. E., Welsh M. J. (1991) Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J. Clin. Invest. 88, 1422–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kartner N., Hanrahan J. W., Jensen T. J., Naismith A. L., Sun S. Z., Ackerley C. A., Reyes E. F., Tsui L. C., Rommens J. M., Bear C. E., Riordan J. R. (1991) Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell 64, 681–691 [DOI] [PubMed] [Google Scholar]
- 38. Bear C. E., Li C. H., Kartner N., Bridges R. J., Jensen T. J., Ramjeesingh M., Riordan J. R. (1992) Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68, 809–818 [DOI] [PubMed] [Google Scholar]
- 39. Ma T., Thiagarajah J. R., Yang H., Sonawane N. D., Folli C., Galietta L. J. V., Verkman A. S. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin – induced intestinal fluid secretion. J. Clin. Investig. 110, 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wess J. (1996) Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 10, 69–99 [DOI] [PubMed] [Google Scholar]
- 41. Halls M. L., Cooper D. M. F. (2011) Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb. Perspect. Biol. 3, a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi J. Y., Joo N. S., Krouse M. E., Wu J. V., Robbins R. C., Ianowski J. P., Hanrahan J. W., Wine J. J. (2007) Synergistic airway gland mucus secretion in response to vasoactive intestinal peptide and carbachol is lost in cystic fibrosis. J. Clin. Invest. 117, 3118–3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park S., Shcheynikov N., Hong J. H., Zheng C., Suh S. H., Kawaai K., Ando H., Mizutani A., Abe T., Kiyonari H., Seki G., Yule D., Mikoshiba K., Muallem S. (2013) Irbit Mediates Synergy Between Ca2+ and cAMP Signaling Pathways During Epithelial Transport in Mice. Gastroenterology 13, 00461–00467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang L., MacDonald K., Schwiebert E. M., Zeitlin P. L., Guggino W. B. (2009) Spiperone, identified through compound screening, activates calcium-dependent chloride secretion in the airway. Am. J. Physiol., Cell Physiol. 296, C131–C141 [DOI] [PMC free article] [PubMed] [Google Scholar]