

Background: Glioblastoma is characterized by heightened cell invasion and therapeutic resistance.
Results: The Src homology 3 domain-containing GEF (SGEF) promotes fibroblast growth factor-inducible 14 (Fn14) proinvasive signaling in glioblastoma via TNF receptor-associated factor 2 (TRAF2).
Conclusion: SGEF is an important regulator of glioblastoma cell invasion downstream of Fn14.
Significance: Therapy directed at mediators of invasion may confer increased chemotherapeutic and radiologic susceptibility in glioblastoma.
Keywords: Cell Invasion, Cell Signaling, Glioblastoma, Guanine Nucleotide Exchange Factor (GEF), Rho GTPases
Abstract
Glioblastoma (GB) is the highest grade of primary adult brain tumors, characterized by a poorly defined and highly invasive cell population. Importantly, these invading cells are attributed with having a decreased sensitivity to radiation and chemotherapy. TNF-like weak inducer of apoptosis (TWEAK)-Fn14 ligand-receptor signaling is one mechanism in GB that promotes cell invasiveness and survival and is dependent upon the activity of multiple Rho GTPases, including Rac1. Here we report that Src homology 3 domain-containing guanine nucleotide exchange factor (SGEF), a RhoG-specific guanine nucleotide exchange factor, is overexpressed in GB tumors and promotes TWEAK-Fn14-mediated glioma invasion. Importantly, levels of SGEF expression in GB tumors inversely correlate with patient survival. SGEF mRNA expression is increased in GB cells at the invasive rim relative to those in the tumor core, and knockdown of SGEF expression by shRNA decreases glioma cell migration in vitro and invasion ex vivo. Furthermore, we showed that, upon TWEAK stimulation, SGEF is recruited to the Fn14 cytoplasmic tail via TRAF2. Mutation of the Fn14-TRAF domain site or depletion of TNF receptor-associated factor 2 (TRAF2) expression by siRNA oligonucleotides blocked SGEF recruitment to Fn14 and inhibited SGEF activity and subsequent GB cell migration. We also showed that knockdown of either SGEF or RhoG diminished TWEAK activation of Rac1 and subsequent lamellipodia formation. Together, these results indicate that SGEF-RhoG is an important downstream regulator of TWEAK-Fn14-driven GB cell migration and invasion.
Introduction
One of the key hallmarks of glioblastoma is the heightened proclivity to invade normal brain tissue. Despite attempts at treatment with surgical resection, radiation, and chemotherapy with the alkylating agent temozolomide, patient survival rates are less than 10% at 5 years (1). Resistance to these current treatments is inevitable, with subsequent tumor recurrence and death. Recurrent tumors develop most commonly within 2 cm from the primary excised tumor, although single or grouped tumor cells have been observed frequently outside of this distance as well as in the contralateral hemisphere (2), and the possibility exists that cell invasion is an early acquired phenotype in GB2 tumor formation (3). Importantly, therapy directed at mediators of invasion has been shown to increase chemotherapeutic sensitivity (4, 5), and, thus, efforts are being made to identify and characterize the potential drivers of cell migration.
The Rho GTPase family is comprised of critical mediators of cell migration and invasion via cytoskeletal reorganization, most notably including Rac1, Cdc42, and RhoA (6–8). Rho family GTPases enable downstream signaling when in their activated GTP-bound state catalyzed by guanine nucleotide exchange factor (GEF)-mediated GTP loading. Conversely, stimulation of Rho GTPase intrinsic GTP-to-GDP hydrolysis by GTPase-activating proteins facilitates Rho GTPase inactivation (6). To allow activation of Rho GTPases, posttranslational prenylation modifications are attached to the carboxyl-terminal cysteine residue, the impact of which is to allow interactions with phospholipid membranes and signaling complexes (9, 10). A third category of Rho GTPase family regulators are the guanine nucleotide dissociation inhibitors, which sequester Rho GTPases in their inactive conformation in the cytosol, sequestering their hydrophobic tail (11).
Rac1 can be activated by multiple growth factors and cytokines. One mechanism of Rac1 activation in GB is via the tumor necrosis factor receptor superfamily 12A (TNFRSF12A) member known as Fn14, which has been highly characterized in GB for its role in promoting cell migration, invasion, and survival (12–14). Ligand-mediated activation of the receptor by TWEAK, the TNF-like weak inducer of apoptosis, specifically promotes Cdc42-dependent activation of Rac1 with subsequent robust lamellipodia formation (15). Interestingly, although Rac1 mRNA levels are ubiquitous in expression among many tissue types (16, 17), Rac1 protein localizes in situ to the plasma membrane in a subset of GB tumors, suggesting that heightened or constitutive Rac1 activity in this cancer promotes a malignant phenotype (18).
In several cancers, including glioblastoma, Rac1 activation has been shown to be mediated by small Rho GTPase RhoG (19–22). Notably, a role for RhoG in promoting GB migration and invasion has been described recently (22). RhoG protein levels are elevated in GB (22), and RhoG has been reported to stimulate lamellipodia formation and confer downstream activation of Rac1 with a subsequent increase in cell migration (19, 20, 22, 23). Mechanisms of RhoG activation and signaling in GB, however, have yet to be characterized.
Here we show that the Src homology 3 domain-containing GEF (SGEF/ARHGEF26), an exchange factor for RhoG, is overexpressed in high-grade brain tumors and correlates with poor patient survival. We report that SGEF activity is activated upon TWEAK stimulation and that SGEF promotes TWEAK-induced RhoG-dependent Rac1 activation as well as TWEAK-stimulated lamellipodia formation and migration and invasion of GB cells. The Fn14 cytoplasmic tail contains a single TNF receptor-associated factor (TRAF) consensus motif that has been shown to bind TRAFs 1, 2, 3, and 5, the recruitment of which is critical to Fn14 downstream signaling (24). Analysis of the SGEF protein sequence indicates five potential TRAF2 binding sites. We demonstrate that SGEF coimmunoprecipitates with Fn14 and that this interaction and the promotion of downstream signaling are dependent upon the presence of an intact TRAF binding domain on Fn14 and the recruitment of TRAF2.
EXPERIMENTAL PROCEDURES
Cell Culture Conditions
The human astrocytoma cell lines U87, U118, and T98G, as well as HEK293 cells (ATCC) were maintained in DMEM (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen) at 37 °C with 5% CO2. For all assays with TWEAK treatment, cells were cultured in reduced serum (0.5% fetal bovine serum) for 16 h before stimulation with recombinant TWEAK at 100 ng/ml in DMEM + 0.1% bovine serum albumin for the indicated time.
Antibodies, Plasmids, Reagents, and Western Blot Analysis
A polyclonal SGEF antibody was purchased from Sigma (St. Louis, MO). A monoclonal RhoG antibody and a monoclonal tubulin antibody were purchased from Millipore (Billerica, MA). Monoclonal anti-myc and a polyclonal antibody to TRAF2 were purchased from Cell Signaling Technologies (Beverly, MA). Anti-HA polyclonal antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Human recombinant TWEAK was purchased from PeproTech (Rock Hill, NJ). Human placental laminin was obtained from Sigma. Lipofectamine RNAiMax was purchased from Invitrogen. The plasmid pCMV-GST-ELMO-NT was obtained from Dr. Hironori Katoh (Kyoto University), pGEX4T-1-RhoG(15A) was obtained from Dr. Keith Burridge (University of North Carolina Chapel Hill), and pCMV-SGEF-myc was obtained from Dr. Thomas Samson (University of North Carolina Chapel Hill). The HA epitope-tagged wild-type Fn14 (Fn14wt) was constructed by amplifying the Fn14 coding sequence by polymerase chain reaction and ligating the product in-frame upstream of a 3× HA epitope in pcDNA3. The Fn14 variant designated Fn14TRAFaa containing a mutation of the TRAF binding domain (PIEE → PIAA) was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and the Fn14wt-HA plasmid as a template.
For immunoblotting, cells were lysed in 2× SDS sample buffer (0.25 m Tris-HCl (pH 6.8), 10% SDS, 25% glycerol) containing 10 μg/ml aprotinin, 10 μg/ml leupeptin, 20 mm NaF, 2 mm sodium orthovanadate, and 1 mm phenylmethylsulfonyl fluoride. Protein concentrations were determined using the BCA assay (Pierce) with BSA as a standard. Thirty micrograms of total cellular protein were loaded per lane and separated by SDS-PAGE. After transfer at 4 °C, the nitrocellulose (Invitrogen) was blocked with either 5% nonfat milk or 5% BSA in Tris-buffered saline (pH 8.0) containing 0.1% Tween 20 prior to addition of primary antibodies and followed with peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG. Protein bands were detected using SuperSignal West Dura chemiluminescent substrate (Thermo Scientific) with a UVP BioSpectrum 500 imaging system (Upland, CA).
Expression Profile Dataset of SGEF in Nonneoplastic Brain and Human Gliomas
SGEF gene expression was mined in the publicly available NCBI Gene Expression Omnibus dataset GSE4290 containing 195 clinically annotated brain tumor specimens. Expression values were filtered, and principal component analysis to investigate the relationship between samples was performed as described previously (14). Box plots for SGEF expression in each survival cluster derived from principal component analysis were graphed, and the significance between the two populations was tested with a two-sample Student's t test assuming unequal variances as described previously (14).
Immunohistochemical Analysis of SGEF Protein
A glioma invasion tissue microarray and immunohistochemistry protocol used to examine SGEF expression in glioblastoma tumor samples has been described previously (25). A scoring system of 0, negative; 1–2, moderate; and 3, strong was used to grade the staining.
Laser Capture Microdissection, RNA Isolation, and Quantitative RT-PCR
Laser capture microdissection of tumor core and invasive cells was done, and total RNA was isolated as described previously (26). cDNA was synthesized from 500 ng of total RNA in a 20-μl reaction volume using the SuperScript III First-Strand Synthesis SuperMix kit (Invitrogen) for 50 min at 50 °C, followed by 85 °C for 5 min. Quantitative PCR analysis of SGEF (sense, 5′-TGC TGA AAG GAC AAG GAA CA-3′ and antisense, 5′-GTA GTT TTG ATA CAG GAC AGC ATT-3′) and histone H3.3 (sense, 5′-CCA CTG AAC TTC TGA TTC GC-3′ and antisense, 5′-GCG TGC TAG CTG GAT GTC TT-3′) mRNA levels was conducted using SYBR Green (Roche) fluorescence for detection of amplification after each cycle with LightCycler analysis software and quantified as described previously (12).
Lentiviral Production
Lentiviral vectors containing SGEF shRNA were obtained from Open Biosystems (Fisher Scientific, Pittsburgh, PA). The SGEF shRNA constructs were packaged into vesicular stomatitis virus G pseudotyped lentiviral particles following transfection of 293T cells using the pPACKH1 packaging plasmid mix (Open Biosystems) according to the instructions of the manufacturer. For lentiviral transduction of target cells, lentiviral-containing supernatants were collected from packaging cells 48 and 72 h after transfection, concentrated by PEG precipitation, and added to subconfluent cultures of cells with 8 μg/ml polybrene for 4–6 h. Forty-eight hours after infection, infected cells were enriched by selection with 2 μg/ml puromycin for 2 weeks. Confirmation of knockdown was performed by Western blot analysis.
Radial Cell Migration Assay
Cell migration was quantified as described previously (27). Glioma cells stably expressing shRNA targeting SGEF, a control empty vector, or cells that were transfected with siRNA targeting luciferase or TRAF2 24 h post-transfection were plated onto 10-well glass slides precoated with 10 μg/ml laminin. Cells were cultured in reduced serum (0.5% FBS) for an additional 16 h prior to TWEAK addition, and the migration rate was assessed over 24 h.
Organotypic Brain Slice Invasion Assay
Preparation and culture of brain slices was carried out as described previously (28), with minor modification. Glioma cells (U87 and U118) stably expressing GFP were placed bilaterally onto the putamen of 400-μm-thick slices of freshly isolated 4- to 6-week-old murine brains. Glioma cell invasion into the brain slices was quantified using the LSM 5 confocal microscope, and depth of invasion (z axis stacks) was calculated as described previously (28).
RhoG and Rac Activity Assay and Nucleotide-free GEF Pull-downs
Activity assays for Rac1 were done according to the protocol of the manufacturer (Pierce). Lysates were harvested, and equal concentrations of lysates were assessed for Rac activity. RhoG activity was measured as described previously using a GST-ELMO-NT fusion protein (22). Affinity pull-downs of active SGEF bound to RhoG were performed using a nucleotide-free RhoG mutant (G15A) expressed and purified as described (29). Recombinant RhoG G15A-GST protein and GST-ELMO-NT (a GST fusion protein containing the N-terminal RhoG-binding domain of ELMO2, amino acids 1–362) were produced in Escherichia coli (BL21) cells. Cells were lysed in B-PER lysis buffer (Pierce) containing protease inhibitors and purified with glutathione-Sepharose beads (GE Healthcare). U87 and U118 cells were grown in 10-cm dishes in reduced serum for 16 h before treatment with TWEAK for the indicated times. Subsequently, equal amounts of total GST fusion protein were incubated with total cellular protein lysate (1 mg), and precipitated lysates were resolved with SDS-PAGE.
Immunofluorescence
For immunofluorescence, glioma cells were transfected with plasmids encoding Fn14wt-HA and SGEF-myc using effectene according to the protocol of the manufacturer (Qiagen). After 24 h, cells were plated onto 10-well glass slides precoated with 10 μg/ml laminin. Twenty-four hours later, cells were fixed in 4% formaldehyde/PBS, permeabilized with 0.1% Triton X-100 dissolved in PBS, and incubated with anti-HA and/or anti-myc antibodies, and Alexa Fluor phalloidin (Molecular Probes) to stain for F-actin. Slides were mounted with ProLong Gold antifade reagent with DAPI (Molecular Probes). Images were collected using a Zeiss LSM 510 microscope equipped with a ×63 objective, ZEN 2009 image analysis software, and Adobe Photoshop CS3.
Quantification of Lamellipodia Formation
T98G glioma cells were stably transduced with an empty lentivirus vector expressing GFP alone or with a recombinant lentivirus expressing a shRNA targeting SGEF. For transient knockdown experiments, T98G glioma cells were transfected with siRNA targeting luciferase or RhoG. 24 h after siRNA transfection, all cells were plated onto 10-well glass slides precoated with 10 μg/ml laminin. Twenty-four hours later, cells were cultured in reduced serum (0.5% FBS) for an additional 16 h prior to TWEAK stimulation for 5 min. Subsequently, cells were fixed in 4% formaldehyde/PBS, permeabilized with 0.1% Triton X-100 dissolved in PBS, and incubated with Alexa Fluor phalloidin (Molecular Probes) to stain for F-actin. Slides were mounted with ProLong Gold antifade reagent with DAPI (Molecular Probes). Images were collected using a Zeiss LSM 510 microscope equipped with a ×63 objective, ZEN 2009 image analysis software, and Adobe Photoshop CS3. For each experimental condition, at least 12 images were taken randomly. Lamellipodia were traced using Image J software. For each cell, the fraction of the cell perimeter that displayed lamellipodia was calculated.
Immunoprecipitation
For immunoprecipitation, cells were transfected as indicated with 1 μg of Fn14 WT-HA, Fn14 TRAFaa-HA, or SGEF-myc plasmid DNA. After 48 h, cells were lysed on ice for 10 min in a buffer containing 10 mm Tris-HCl (pH 7.4), 0.5% Nonidet P-40, 150 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, 1 mm EDTA, 2 mm sodium orthovanadate, 20 mm sodium fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. Equivalent amounts of protein (500 μg) were precleared and immunoprecipitated from the lysates using either TRAF2, Myc, or HA antibodies, as indicated, or a control isotype-matched antibody and then washed with lysis buffer followed by TX-100 buffer (10 mm HEPES (pH 7.4), 150 mm NaCl, 2 mm EDTA, 2 mm EGTA, 20 mm sodium fluoride, and 0.5% Triton X-100). Samples were then resuspended in 2× SDS sample buffer and boiled in the presence of 2-mercaptoethanol (Sigma), separated by SDS-PAGE, transferred to nitrocellulose for 1 h at 4 °C, and then proteins were detected using SuperSignal West Dura chemiluminescent substrate (Thermo Scientific).
Small Interfering RNA Transfection
Small interfering RNA (siRNA) oligonucleotides specific for GL2 luciferase were described previously (18). siRNA target sequences for TRAF2 were as follows: TRAF2-1, 5′-CTG GAC CAA GAC AAG ATT GAA and TRAF2-2, 5′-CTG CTG CGG AGC AGA CGT GAA). Transient transfection of siRNA was performed using Lipofectamine RNAiMax. Cells were plated at 70% confluence in DMEM + 10% FBS without antibiotics and were transfected within 8 h of plating. The siRNA and Lipofectamine were diluted in Opti-MEM (Invitrogen). After 5 min, the mixtures were combined and incubated for 20 min at room temperature to enable complex formation. siRNA oligonucleotides were transfected at 50 nm, and no cell toxicity was observed. Maximum inhibition of protein levels was achieved 48–72 h post-transfection.
Statistical Analysis
Statistical analyses were done using the two-sample Student's t test. p < 0.05 was considered significant.
RESULTS
SGEF Is Overexpressed in High-grade Gliomas, and Expression Correlates with Poor Patient Survival
To examine the expression pattern of SGEF in high-grade astrocytoma, we used a panel of publicly available grade three anaplastic astrocytoma and grade four glioblastoma patient tissue samples versus non-neoplastic brain (NCBI Gene Expression Omnibus data set GSE4290). We found that SGEF mRNA is significantly elevated in high-grade brain tumors relative to non-neoplastic control tissue (Fig. 1A, p < 0.001). Next, we assessed the levels of SGEF mRNA on this GB database, where a principle component analysis has been used to discern possible relationships between subgroups of samples and in which Kaplan-Meier survival curves were developed for each principal component cluster (14). One cluster had a median survival time of 401 days (short-term survival), and the other cluster had a median survival time of 952 days (long-term survival). An analysis of SGEF mRNA expression showed that GB patients in the short-term survival cluster had a significantly higher expression of SGEF than GB patients in the long-term survival cluster (Fig. 1B, p < 0.001). These data suggest that increased SGEF expression correlates with poor patient outcome.
FIGURE 1.

Expression profiling of SGEF in non-neoplastic brain and brain tumor samples. A, box and whisker plots of SGEF mRNA expression levels from the NCBI Gene Expression Omnibus GSE4290 (NB, non-neoplastic brain; AA, anaplastic astrocytoma). The significance between groups was tested with a two-sample Student's t test assuming unequal variances. B, principal component analysis of brain tumors from the NCBI Gene Expression Omnibus GSE4290 correlating SGEF expression level in long-term (LT) (median = 952 days) and short-term (ST) (median = 401 days) survival groups. The significance between groups was tested with a two-sample Student's t test assuming unequal variances. C, paraffin sections of an invasive glioma tissue microarray were immunostained with a polyclonal antibody against SGEF. a, representative SGEF immunohistochemistry micrograph of reactive tissue adjacent to the tumor. V, vessel; arrows, normal glial cells; arrowheads, reactive astrocytes in the tumor rim. b, representative SGEF immunohistochemistry micrograph of the tumor core. D, quantitative RT-PCR analysis of SGEF mRNA expression from patient tumor samples laser capture-microdissected for either the core (C) or rim (R) tumor population.
To determine the levels of SGEF protein expression in GB tissue, we analyzed 43 tumors via immunohistochemistry for SGEF. Overall, the majority of GB tumor tissue displayed elevated staining intensity (76.7%, moderate and 4.7%, strong, Fig. 1C, b), whereas minimum to no SGEF staining was detected in non-neoplastic cells, including endothelial, neuronal, and glial cells (Fig. 1C, a). To assess protein levels of SGEF in the tumor rim versus the tumor core, four patient GB tissues were demarcated by a board-certified pathologist as tumor core versus rim, and respective cells from each population were obtained via laser capture microdissection. In three of the four cases, the levels of SGEF mRNA were elevated 2- to 10-fold in cells from the tumor rim versus cells from the tumor core (Fig. 1D). Collectively, these data show that elevated SGEF message and protein expression is found in high-grade gliomas and is enriched in the invasive cell population, and that elevated SGEF expression correlates with poor clinical outcome.
SGEF Is Important for TWEAK-Fn14-induced Cell Migration and Invasion
We have shown previously that the TWEAK-Fn14 ligand-receptor signaling axis promotes the activation of Rac1 in GB cells, leading to increased cell migration and invasion (14, 15). To determine the role that SGEF may play in GB cell migration, we stably transduced U87 and U118 glioma cells with lentiviruses expressing either control empty vector or SGEF-targeting shRNA. Two independent shRNA constructs targeting SGEF were used, and the level of SGEF knockdown in both cell lines was greater than 90% (Fig. 2A). Knockdown of SGEF expression in both U87 and U118 cells abrogated TWEAK-induced cell migration (Fig. 2B, p < 0.01) but did not change the basal cell migration (data not shown). We next investigated whether knockdown of SGEF affects glioma invasion in an ex vivo mouse brain model. In both U87 and U118 cells, knockdown of SGEF diminished the depth of invasion into the brain slices (Fig. 2C, p < 0.01). These data suggest that SGEF functions within the TWEAK-Fn14 signaling pathway to enhance glioma cell migration in vitro and promote invasion ex vivo.
FIGURE 2.
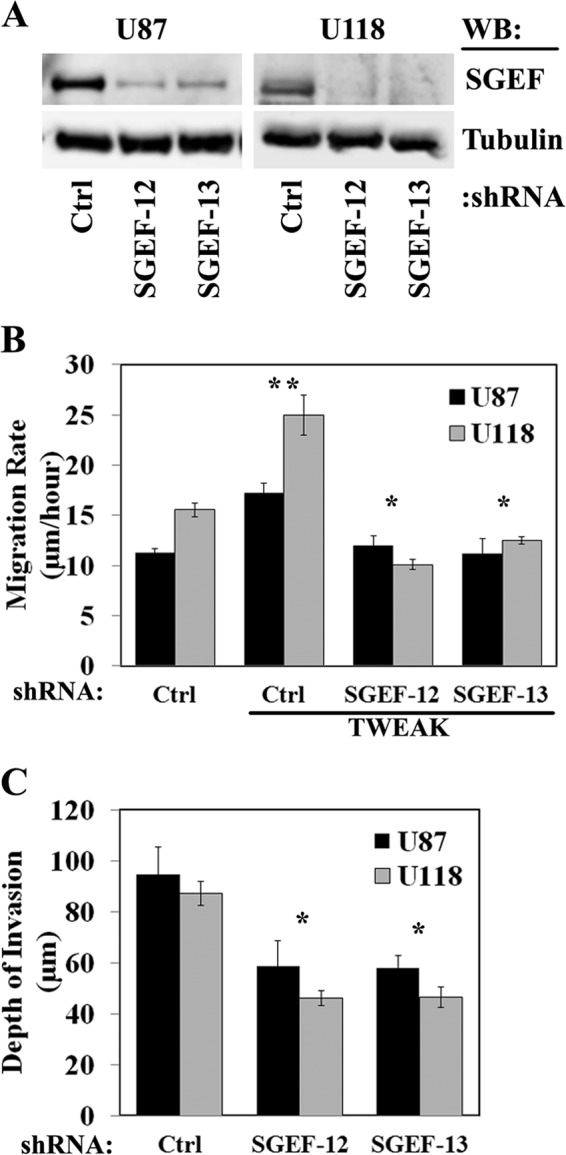
Knockdown of SGEF expression suppresses glioma cell migration and invasion. A, U87 or U118 cells were stably transduced with two independent lentivirus-encoding shRNAs targeting SGEF (SGEF-12 and SGEF-13) or an empty vector (Ctrl), and cell lysates were immunoblotted (WB) for SGEF. B, U87 and U118 glioma cells stably transduced with shRNAs targeting either SGEF or an empty vector were seeded onto 10-well glass slides precoated with 10 μg/ml human laminin. Cells were either left untreated or TWEAK-stimulated, and cell migration was assessed over 24 h. Data represent the average of 10 replicates. * and **, p < 0.01. C, U87 and U118 glioma cells stably transduced with shRNA targeting either SGEF or empty vector were implanted into the bilateral putamen of murine organotypic brain slices and observed at 48 h. The depth of invasion was calculated from z-axis images collected by confocal laser scanning microscopy. Data represent the average of nine replicates. *, p < 0.01.
SGEF Activates RhoG Downstream of the TWEAK-Fn14 Signaling Axis
Our data show that SGEF is overexpressed in glioblastoma and is important in TWEAK-induced glioma cell migration. We therefore assessed whether SGEF is activated by TWEAK. U87 glioma cells were treated with TWEAK, and lysates were analyzed for levels of active SGEF bound to RhoG G15A, a nucleotide-free mutant of RhoG with a high affinity for active GEFs (29). Upon TWEAK stimulation, a rapid increase in the level of active SGEF was detected within 2 min and subsequently diminished after 2 min (Fig. 3, A and B). SGEF is known to catalyze guanine nucleotide exchange on RhoG (30), and RhoG has been shown to be overexpressed and activated by both EGF and HGF in glioblastoma cells (22). We therefore assessed whether SGEF plays a role in activating RhoG downstream of the Fn14 receptor. Because we were unable to consistently detect significant activity of RhoG in U87 cells, potentially because of lower levels of RhoG expression (data not shown), we utilized U118 cells to examine the kinetics of RhoG activation upon TWEAK stimulation. Similar to SGEF activation levels, RhoG activity was sharply increased within 2 min of TWEAK activation in U118 cells, an effect that diminished after 10 min (Fig. 3C). To confirm whether the increase in active RhoG upon TWEAK stimulation was dependent upon SGEF activity, we assessed RhoG activation in U118 glioma cells with stable knockdown of SGEF protein. Knockdown of SGEF abrogated TWEAK-stimulated RhoG activity (Fig. 3D). Together, these data demonstrate that SGEF activates RhoG downstream of TWEAK and Fn14.
FIGURE 3.
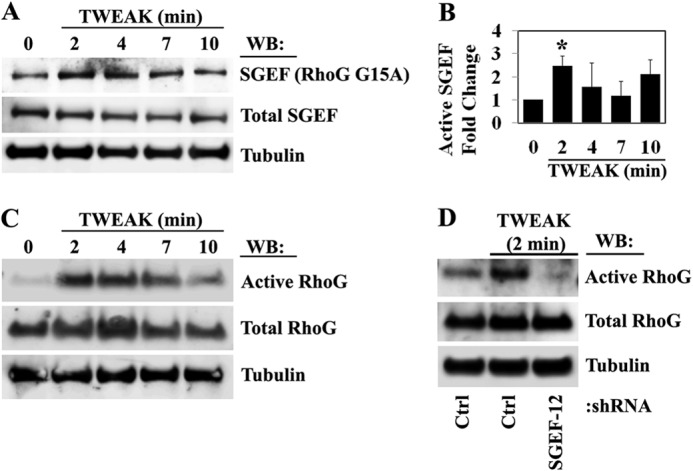
TWEAK-Fn14 signaling activates SGEF and the SGEF dependent activation of RhoG. A, U87 glioma cells were cultured in reduced serum (0.5% FBS) overnight and then treated with TWEAK for the indicated times. SGEF activation in control and treated cell lysates was assessed using RhoG G15A-GST constructs. B, the mean ± S.E. of three independent replicates of SGEF activation were quantified as levels of active SGEF-bound RhoG G15A-GST relative to total SGEF. *, p < 0.05. WB, Western blot. C, U118 glioma cells were cultured in reduced serum (0.5% FBS) overnight and then treated with TWEAK for the indicated times. RhoG activation in control and treated cell lysates was assessed using GST-ELMO-NT constructs. D, U118 glioma cells stably transduced with an empty lentivirus vector (Ctrl) or shRNA targeting SGEF (SGEF-12) were cultured overnight in reduced serum (0.5% FBS) and either left untreated or TWEAK-treated for 2 min. Levels of active RhoG in control and treated cell lysates were assessed using GST-ELMO-NT. C and D, data are representative of two independent experiments.
TWEAK-Fn14-stimulated Activation of Rac1 Is Dependent upon SGEF and RhoG Function
SGEF has been shown to exchange for RhoG (30). Because RhoG is known to directly bind ELMO as a downstream effector in its activated state and the ELMO-Dock180 GEF complex has been shown to confer guanine nucleotide exchange directly for Rac1 downstream of RhoG activation (20, 21, 23), we sought to determine whether TWEAK-induced activation of SGEF and RhoG modulated downstream Rac1 activation. TWEAK induction of Rac1 activity was diminished in U118 and U87 cells with stable knockdown of SGEF expression compared with control cells (Fig. 4A). Furthermore, transfection of U87 glioma cells with siRNA targeting RhoG (Fig. 4B) abrogated TWEAK-induced Rac1 activity in a similar fashion to SGEF depletion (C). Thus, increased Rac1 activity induced by TWEAK-Fn14 signaling is dependent upon SGEF and RhoG activity.
FIGURE 4.

TWEAK-Fn14 activation of Rac1 is dependent upon SGEF and RhoG. A, U87 and U118 glioma cells stably expressing either control empty vector (Ctrl) or shRNA targeting SGEF (SGEF-12) were cultured overnight in reduced serum medium (0.5% FBS). Cells were left untreated or treated with TWEAK for 5 min. Cells were lysed and assessed for levels of active Rac1. WB, Western blot. B, U87 glioma cells were transfected with one of two independent siRNA oligonucleotides targeting RhoG (RhoG-1 and RhoG-2), with siRNA targeting non-mammalian luciferase (Ctrl), or left untreated (NT). After 72 h, cell lysates were immunoblotted with the indicated antibodies. C, U87 glioma cells were transfected with siRNA targeting non-mammalian luciferase (Ctrl) or with a siRNA oligonucleotide targeting RhoG (RhoG-2). 48 h after transfection, cells were cultured an additional 16 h overnight in reduced-serum medium (0.5% FBS) prior to addition of TWEAK treatment for 5 min in the indicated samples. Cells were lysed and assessed for levels of Rac1 activity. Data are representative of two independent experiments.
Depletion of SGEF and RhoG Suppresses TWEAK-induced Lamellipodia Formation
Activated RhoG and Rac1 are known to promote actin cytoskeletal rearrangement, resulting in lamellipodia formation (31). To determine the role of SGEF and RhoG in the regulation of lamellipodia, we treated T98G glioma cells, which display robust lamellipodia formation, with TWEAK and stained cells for filamentous actin using phalloidin. The addition of TWEAK to T98G cells transduced with a control shRNA or transfected with a control siRNA robustly induced lamellipodia formation (Fig. 5A, b and e, arrows; p < 0.01) relative to untreated cells (a and d). In contrast, stable knockdown of SGEF or knockdown of RhoG by transient transfection with siRNA (Fig. 5D) abrogated lamellipodia formation following TWEAK stimulation (A, c and f, B, and C; p < 0.01). Therefore, both SGEF and RhoG play an important role in the promotion of TWEAK-Fn14-induced actin cytoskeleton rearrangement, lamellipodia formation, and invasive behavior.
FIGURE 5.

TWEAK-induced lamellipodia formation in glioma cells requires the function of SGEF and RhoG. T98G glioma cells were stably transduced with lentivirus encoding shRNA targeting SGEF (SGEF-12) or an empty vector (Ctrl). Additionally, T98G glioma cells were transfected with siRNA targeting either control non-mammalian luciferase (ctrl), or RhoG (RhoG-2). After 24 h of siRNA transfection, all cells were seeded onto 10-well glass slides precoated with 10 μg/ml human laminin. Cells were further grown for 24 h and then cultured in reduced-serum medium (0.5% FBS) for 16 h. Cells were either left untreated or treated with TWEAK (5 min) and stained for filamentous actin using Alexa Fluor-phalloidin. A, representative images of empty vector control non-treated cells (a), TWEAK-treated cells (b), or TWEAK-treated cells with knockdown of SGEF (c). Additionally, representative images of control luciferase-transfected non-treated (d) or TWEAK-treated (e) cells as well as TWEAK-treated cells after siRNA depletion of RhoG (f) are shown. Arrows indicate lamellipodia. B, quantification of lamellipodia formation in T98G with and without SGEF knockdown. a.u., arbitrary unit. C, quantification of lamellipodia formation in control and RhoG knockdown T98G cells. Data represent the average of at least 12 cells/condition. * and **, p < 0.01. D, immunoblot analysis (WB) of T98G cells stably transduced with lentivirus empty vector (Ctrl) or lentivirus encoding shRNA targeting SGEF (SGEF-12) (upper panel) or T98G cells transfected with siRNA targeting either luciferase (Ctrl) or RhoG (RhoG-2) (lower panel).
SGEF Binding to the Fn14 Cytoplasmic Tail Requires an Intact TRAF Domain
To determine whether SGEF binds to the Fn14 cytoplasmic tail, we transfected HEK293 cells with both Fn14 wild-type-expressing (Fn14WT-HA) and SGEF-expressing (SGEF-myc) plasmids and performed coimmunoprecipitation. Cellular lysates immunoprecipitated with anti-myc antibody confirmed the presence of Fn14 as assessed by HA immunoblot analysis (data not shown), indicating that SGEF binds in complex with Fn14.
Fn14 downstream signaling, including the activation of NF-κB, has been shown to depend on an intact TRAF binding site (13, 24). Using a functional site prediction analysis algorithm, we observed that SGEF contains five TRAF2 binding consensus motifs of the pattern (P/S/A/T/)X(Q/E)E. The five TRAF2 consensus sites in SGEF include TPEE (aa 157–160), PSQE (aa 221–224), AGEE (aa 292–295), SDEE (aa 392–395), and SQEE (aa 434–437), all of which are located N-terminally to the GEF canonical Dbl homology-pleckstrin homology domains (Fig. 6A). To determine whether the TRAF binding site on Fn14 is required for SGEF recruitment to its cytoplasmic tail, we mutated the TRAF binding domain of Fn14 (Fn14TRAFaa-HA) and assessed SGEF binding by coimmunoprecipitation. As expected, the Fn14 wild type was present in the anti-TRAF2 immunoprecipitate. However, mutation of the TRAF2 binding site in the Fn14TRAFaa construct prevented its coimmunoprecipitation with TRAF2 (Fig. 6C). Furthermore, immunoprecipitation of TRAF2 in cells cotransfected with both the Fn14 wild type and SGEF indicated that both Fn14 and SGEF coimmunoprecipitate with TRAF2 (Fig. 6D). Interestingly, SGEF did not coimmunoprecipitate with the Fn14TRAFaa mutant, suggesting that the TRAF binding domain of Fn14 is required for SGEF binding (Fig. 6E). The reverse immunoprecipitation for HA also demonstrated that SGEF robustly coimmunoprecipitated with Fn14WT but did not readily coimmunoprecipitate with the Fn14TRAFaa mutant (Fig. 6F). Therefore, taken together, recruitment of SGEF to Fn14 requires an intact TRAF binding domain.
FIGURE 6.

SGEF and Fn14 interaction is dependent upon a functional TRAF domain. A, the SGEF protein contains five TRAF2 binding consensus motifs of the pattern (P/S/A/T/)X(Q/E)E, including TPEE (aa 157–160), PSQE (aa 221–224), AGEE (aa 292–295), SDEE (aa 392–395), and SQEE (aa 434–437). NLS, nuclear localization sequence. B, whole cell lysates of HEK293 cells transiently transfected with plasmids encoding Fn14wt-HA, Fn14TRAFaa-HA, or SGEF-myc were immunoblotted (WB) as indicated. C, whole cell lysates of HEK293 cells transiently transfected with plasmids encoding Fn14wt-HA or Fn14TRAFaa were collected and precleared, followed by immunoprecipitation (IP) using antibodies as indicated for anti-TRAF2 or control immunoprecipitation (Ctrl) and resolution via SDS-PAGE analysis using an anti-HA antibody. D, whole cell lysates of HEK293 cells transiently transfected with plasmids encoding Fn14wt-HA or SGEF-myc were collected and precleared, followed by immunoprecipitation using antibodies as indicated for anti-TRAF2 or control immunoprecipitation (Ctrl) and resolution via SDS-PAGE analysis using anti-myc and anti-HA antibodies. E and F, HEK293 cells transiently transfected with plasmids encoding Fn14wt-HA, Fn14TRAFaa, or SGEF-myc were collected and precleared, followed by immunoprecipitation using antibodies as indicated for anti-myc (E), anti-HA (F), or control immunoprecipitation (Ctrl) and resolution via SDS-PAGE analysis using an anti-HA (E) or anti-myc (F) antibody. G, U87 glioma cells were cultured for 16 h in reduced-serum (0.5% FBS) medium. Cells were either left untreated or treated with TWEAK for 2 min, after which whole cell lysates were precleared, followed by immunoprecipitation using antibodies indicated for anti-TRAF2 or control immunoprecipitation (Ctrl) and resolution via SDS-PAGE analysis using anti-SGEF and anti-Fn14 antibodies.
To further probe the role of TRAF2 in the Fn14-SGEF complex, we performed immunoprecipitation of endogenous TRAF2 in U87 cells. We found that both Fn14 and SGEF bound to TRAF2 (Fig. 6G). Interestingly, Fn14 binding to TRAF2 is dependent upon TWEAK activation, whereas SGEF binding to TRAF2 is constitutive.
TWEAK-Fn14-induced Activation of SGEF and Stimulation of Migration Is Dependent upon TRAF2
We next sought to determine the requirement of TRAF2 in SGEF activation downstream of TWEAK-Fn14 signaling on the basis of the predicted TRAF2 binding sites in the SGEF protein (Fig. 6A). Using two independent siRNA oligonucleotides targeting TRAF2 (Fig. 7A), we assessed TWEAK stimulation of SGEF activity in U87 glioma cells following knockdown of TRAF2. In control cells, TWEAK stimulation increased the amount of SGEF bound to the RhoG G15A nucleotide-free construct relative to unstimulated cells. Conversely, knockdown of TRAF2 decreased the amount of SGEF bound to RhoG G15A under TWEAK treatment to base-line levels (Fig. 7B). Therefore, TRAF2 is necessary for the TWEAK-induced increase in SGEF activity. Furthermore, targeted depletion of TRAF2 protein significantly abrogated TWEAK-induced glioma cell migration in both U87 and U118 cells (Fig. 7C), supporting the role of TRAF2 recruitment in the TWEAK-Fn14-stimulated migratory phenotype of GB.
FIGURE 7.

TWEAK-induced activation of SGEF and stimulation of migration require TRAF2 function. A, U87 and U118 glioma cells were transfected with one of two independent siRNA oligonucleotides targeting TRAF2 (TRAF2-1 and TRAF2-2) or with siRNA targeting non-mammalian luciferase (Ctrl). After 72 h, cells were lysed, and lysates were immunoblotted (WB) as indicated. B, U87 glioma cells were transiently transfected with siRNA targeting non-mammalian luciferase (Ctrl) or TRAF2 (TRAF2-1) for 48 h. Cells were then serum-reduced (0.5% FBS) for an additional 16 h and treated with TWEAK for 2 min. SGEF activation in cell lysates was assessed using RhoG G15A-GST constructs. C, U87 and U118 cells were transiently transfected with siRNA targeting non-mammalian luciferase (Ctrl) or two independent siRNA oligonucleotides targeting TRAF2 (TRAF2-1 and TRAF2-2). After 24 h, cells were plated on glass slides precoated with 10 μg/ml laminin and cultured an additional 16 h in reduced-serum (0.5% FBS) medium. Cells were either left untreated or treated with TWEAK, and glioma cell migration was assessed over 24 h. Data represent the average of 10 replicates. *, p < 0.01.
DISCUSSION
In this study, we report the importance of SGEF and RhoG signaling downstream of TWEAK-Fn14 in promoting glioblastoma cell invasion. Levels of SGEF mRNA and protein expression are elevated significantly in high-grade brain tumors relative to non-neoplastic brain tissue, and elevated expression in GB samples correlates with a poorer patient prognosis. Our data are consistent with data in the Human Protein Atlas, a publicly available portal with protein expression profiles across human tumors, where SGEF protein staining is stronger in both glioma and liver cancer tissues relative to normal organ tissue staining. Furthermore, SGEF expression is elevated in cells found at the invasive edge or rim of GB tumor specimens relative to tumor core cells. SGEF has been shown to have the ability to confer cell invasiveness during human papillomavirus-mediated transformation and to direct actin cytoskeleton remodeling following infection with Salmonella and in leukocyte transendothelial migration (30, 32–34). We have reported previously that glioma cells with the increased capacity for migration have a decreased expression of proapoptotic genes and are, coincidently, less sensitive to cytotoxic therapy-induced apoptosis (13, 14, 26, 35, 36), arguing for a more detailed characterization and new therapeutic strategies for the invasive cell population. Moreover, we demonstrated that knockdown of SGEF protein inhibits TWEAK-Fn14-stimulated cell migration and invasion. Therefore, elevated SGEF signaling may contribute to the invasive behavior of GB cells, and these data highlight the potential for targeted therapy development.
In glioblastoma, the Fn14 receptor is overexpressed significantly, and glioma cells with an increased migratory capacity display elevated levels of Fn14 (12). Our data demonstrate that TWEAK activates SGEF, followed by RhoG and subsequent Rac1 activation in glioblastoma cells. Moreover, loss of SGEF or RhoG protein inhibits TWEAK-Fn14-induced lamellipodia formation, which is consistent with the ability of SGEF to promote TWEAK stimulation of glioma cell migration and downstream Rac1 activation. These data corroborate our previous finding that TWEAK-Fn14-increased cell migration is dependent on Rac1 activation (14). RhoG signaling has been described to occur in both Rac1-dependent and Rac1-independent manners, with these two GTPases sharing overlapping signal transduction pathways (23, 31, 37, 38). Our data support a role for RhoG upstream of Rac1 that is dependent upon TWEAK-Fn14 stimulation of SGEF activity.
Several studies have linked the ability of RhoG to confer downstream activation of Rac1 with a subsequent increase in cell migration via nucleotide exchange by the bipartite GEF engulfment and motility dedicator of cytokinesis 180 (ELMO-Dock180) (19, 20, 23). The Dock180 superfamily of proteins is an unconventional family of Rho GTPase-specific GEFs containing a conserved “Dock homology region 2” (DHR-2) or “Docker” domain as opposed to the characteristic GEF Dbl homology domain for catalytic nucleotide exchange (39, 40) and requires the interaction with the pleckstrin homology domain of ELMO. ELMO is a direct effector of RhoG (23). It is possible that the dependence of TWEAK-Fn14-induced Rac1 activity on the presence of SGEF and RhoG may utilize the ELMO-Dock180 GEF to facilitate guanine nucleotide exchange for Rac1. This possibility is the focus of future studies. Furthermore, SGEF has also been shown to confer weak nucleotide exchange for Cdc42 (30). Our previous report showed that Ect2-Cdc42 is important in TWEAK-Fn14 activation of Rac1 (15). It is possible that SGEF functions in a compensatory pathway to Ect2 to activate Cdc42 and, subsequently, Rac1 downstream of TWEAK stimulation, which warrants further investigation.
We have shown previously that immunoprecipitates of Fn14 contain Rac1 and that this interaction is dependent upon the presence of a functional Fn14 cytoplasmic tail. The deletion of the TRAF binding site from the cytoplasmic domain results in the loss of Fn14-Rac1 coimmunoprecipitation (14). Here we have assessed the mechanism by which SGEF is recruited to the Fn14 cytoplasmic domain. Using predicted site analysis, we observed that SGEF contains five TRAF2 consensus binding sites, suggesting that SGEF may interact directly with TRAF2. Precisely which predicted TRAF2 binding site(s) on SGEF is/are required for TRAF2 interaction will be the focus of future investigation. We also demonstrated that SGEF activity downstream of TWEAK-Fn14 requires the presence of a functional TRAF binding site in the Fn14 cytoplasmic domain. In addition, loss of TRAF2 decreases TWEAK-stimulated SGEF activation. Taken together, these results indicate a requirement for TRAF2 upstream of SGEF in the TWEAK-Fn14 signaling axis.
Signaling through TRAF2, but not TRAF1 or TRAF3, is known to promote NF-κB activity, and TRAF2 is also responsible for promoting JNK/SAPK activity, inflammation, cell migration, and chemo- and radioresistance of cancer cells (41–47). Moreover, specific depletion of TRAF2 in GB has been shown to inhibit growth and confer radiosensitization to tumor cells (48). Similarly, signaling through Fn14 by TWEAK in glioma results in increased resistance to cytotoxic therapy-induced apoptosis and enhanced survival via TRAF recruitment and Rac1-dependent NF-κB activation (13, 14, 24, 36). Furthermore, RhoG has been shown to promote the activation of NF-κB (49). Thus, the TRAF2-SGEF complex may be necessary in the TWEAK-Fn14-TRAF signaling axis, resulting in increased levels of NF-κB activity.
Activated Rho GTPases are localized at the plasma membrane in close proximity with signaling complexes (50, 51). We found that the SGEF and Fn14 proteins colocalize to areas of induced membrane ruffling and that SGEF coimmunoprecipitated with Fn14 from cells coexpressing SGEF and Fn14 (data not shown). Notably, transfection of the SGEF-myc plasmid alone into HEK293 cells resulted in robust membrane ruffling(data not shown). Thus, SGEF binds to the Fn14 receptor complex and colocalizes with Fn14 at the leading edge of cells. Interestingly, although levels of active SGEF are dependent upon the presence of TRAF2, we found that the forced expression of SGEF alongside the expression of Fn14TRAFaa is still found predominantly at the cell edge (data not shown). Other studies have also provided evidence that the overexpression of SGEF alone can induce membrane ruffling in fibroblasts and colocalization with filamentous actin (30), thus further supporting the role of SGEF in modulating cytoskeletal dynamics.
New treatment strategies are needed to cure glioblastoma. Patient prognosis remains poor, and actively invading cells survive current therapeutic regimens. Importantly, therapy directed at mediators of invasion has been shown to increase chemotherapeutic sensitivity (4, 5). Our data support a role for SGEF and RhoG activation and signaling downstream of the TWEAK-Fn14 axis to confer increased Rac1 activity and promote GB cell migration and invasion (Fig. 8). Together, these data provide a rationale for the targeting of this signaling axis as an adjuvant therapy in glioblastoma to limit dispersion of malignant cells and increase susceptibility to traditional radiation and chemotherapies. The role of SGEF in other disease processes is also emerging. SGEF has been shown recently to contribute to the formation of atherosclerosis by promoting endothelial docking structures for leukocytes at areas of inflammation (52). Thus, SGEF presents a therapeutic target for atherosclerosis, and, in this study, we validated SGEF as a target for glioblastoma.
FIGURE 8.

Schematic of TWEAK-Fn14 signaling via SGEF and RhoG to drive glioma migration/invasion. TWEAK ligand binding to the Fn14 receptor results in the activation of RhoG by the SGEF guanine nucleotide exchange factor. SGEF-RhoG signaling results in the activation of Rac1 via additional guanine nucleotide exchange, promoting cytoskeletal reorganization and glioma cell migration and invasion.
Acknowledgments
We would like to thank Dr. Hironori Katoh (Kyoto University) for the pCMV-GST-ELMO-NT plasmid and Drs. Keith Burridge and Thomas Samson (University of North Carolina Chapel Hill) for the pGEX4T-1-RhoG(15A) and pCMV-SGEF-myc plasmids, respectively. We also thank Molly Kupfer for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA130940 (to N. L. T.). This work was also supported by an Achievement Reward for College Scientists Foundation Eller scholarship and a Science Foundation Arizona fellowship (to S. P. F. E.).
- GB
- Glioblastoma
- GEF
- guanine nucleotide exchange factor
- TWEAK
- TNF-like weak inducer of apoptosis
- SGEF
- Src homology 3 domain-containing guanine nucleotide exchange factor
- TRAF
- TNF receptor-associated factor
- aa
- amino acids
- ELMO
- engulfment and cell motility.
REFERENCES
- 1. Stupp R., Hegi M. E., Mason W. P., van den Bent M. J., Taphoorn M. J., Janzer R. C., Ludwin S. K., Allgeier A., Fisher B., Belanger K., Hau P., Brandes A. A., Gijtenbeek J., Marosi C., Vecht C. J., Mokhtari K., Wesseling P., Villa S., Eisenhauer E., Gorlia T., Weller M., Lacombe D., Cairncross J. G., Mirimanoff R. O. (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study. 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 [DOI] [PubMed] [Google Scholar]
- 2. Burger P. C., Heinz E. R., Shibata T., Kleihues P. (1988) Topographic anatomy and CT correlations in the untreated glioblastoma multiforme. J. Neurosurg. 68, 698–704 [DOI] [PubMed] [Google Scholar]
- 3. Sampetrean O., Saga I., Nakanishi M., Sugihara E., Fukaya R., Onishi N., Osuka S., Akahata M., Kai K., Sugimoto H., Hirao A., Saya H. (2011) Invasion precedes tumor mass formation in a malignant brain tumor model of genetically modified neural stem cells. Neoplasia 13, 784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munson J. M., Fried L., Rowson S. A., Bonner M. Y., Karumbaiah L., Diaz B., Courtneidge S. A., Knaus U. G., Brat D. J., Arbiser J. L., Bellamkonda R. V. (2012) Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 4, 127–136 [DOI] [PubMed] [Google Scholar]
- 5. Acharyya S., Oskarsson T., Vanharanta S., Malladi S., Kim J., Morris P. G., Manova-Todorova K., Leversha M., Hogg N., Seshan V. E., Norton L., Brogi E., Massagué J. (2012) A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nobes C. D., Hall A. (1995) Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 7. Ridley A. J., Paterson H. F., Johnston C. L., Diekmann D., Hall A. (1992) The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410 [DOI] [PubMed] [Google Scholar]
- 8. Ridley A. J., Hall A. (1992) The small GTP-binding protein Rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399 [DOI] [PubMed] [Google Scholar]
- 9. Ando S., Kaibuchi K., Sasaki T., Hiraoka K., Nishiyama T., Mizuno T., Asada M., Nunoi H., Matsuda I., Matsuura Y. (1992) Post-translational processing of Rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J. Biol. Chem. 267, 25709–25713 [PubMed] [Google Scholar]
- 10. Seabra M. C. (1998) Membrane association and targeting of prenylated Ras-like GTPases. Cell. Signal. 10, 167–172 [DOI] [PubMed] [Google Scholar]
- 11. Olofsson B. (1999) Rho guanine dissociation inhibitors. Pivotal molecules in cellular signalling. Cell. Signal. 11, 545–554 [DOI] [PubMed] [Google Scholar]
- 12. Tran N. L., McDonough W. S., Donohue P. J., Winkles J. A., Berens T. J., Ross K. R., Hoelzinger D. B., Beaudry C., Coons S. W., Berens M. E. (2003) The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am. J. Pathol. 162, 1313–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tran N. L., McDonough W. S., Savitch B. A., Sawyer T. F., Winkles J. A., Berens M. E. (2005) The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFκB pathway activation and BCL-XL/BCL-W expression. J. Biol. Chem. 280, 3483–3492 [DOI] [PubMed] [Google Scholar]
- 14. Tran N. L., McDonough W. S., Savitch B. A., Fortin S. P., Winkles J. A., Symons M., Nakada M., Cunliffe H. E., Hostetter G., Hoelzinger D. B., Rennert J. L., Michaelson J. S., Burkly L. C., Lipinski C. A., Loftus J. C., Mariani L., Berens M. E. (2006) Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-κB and correlate with poor patient outcome. Cancer Res. 66, 9535–9542 [DOI] [PubMed] [Google Scholar]
- 15. Fortin S. P., Ennis M. J., Schumacher C. A., Zylstra-Diegel C. R., Williams B. O., Ross J. T., Winkles J. A., Loftus J. C., Symons M. H., Tran N. L. (2012) Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol. Cancer Res. 10, 958–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Didsbury J., Weber R. F., Bokoch G. M., Evans T., Snyderman R. (1989) Rac, a novel Ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 264, 16378–16382 [PubMed] [Google Scholar]
- 17. Bosco E. E., Mulloy J. C., Zheng Y. (2009) Rac1 GTPase. A “Rac” of all trades. Cell Mol. Life Sci. 66, 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salhia B., Tran N. L., Chan A., Wolf A., Nakada M., Rutka F., Ennis M., McDonough W. S., Berens M. E., Symons M., Rutka J. T. (2008) The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am. J. Pathol. 173, 1828–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiramoto K., Negishi M., Katoh H. (2006) Dock4 is regulated by RhoG and promotes Rac-dependent cell migration. Exp. Cell Res. 312, 4205–4216 [DOI] [PubMed] [Google Scholar]
- 20. Katoh H., Hiramoto K., Negishi M. (2006) Activation of Rac1 by RhoG regulates cell migration. J. Cell Sci. 119, 56–65 [DOI] [PubMed] [Google Scholar]
- 21. Jarzynka M. J., Hu B., Hui K. M., Bar-Joseph I., Gu W., Hirose T., Haney L. B., Ravichandran K. S., Nishikawa R., Cheng S. Y. (2007) ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res. 67, 7203–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kwiatkowska A., Didier S., Fortin S., Chuang Y., White T., Berens M. E., Rushing E., Eschbacher J., Tran N. L., Chan A., Symons M. (2012) The small GTPase RhoG mediates glioblastoma cell invasion. Mol. Cancer 11, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Katoh H., Negishi M. (2003) RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature 424, 461–464 [DOI] [PubMed] [Google Scholar]
- 24. Brown S. A., Richards C. M., Hanscom H. N., Feng S. L., Winkles J. A. (2003) The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-κB activation. Biochem. J. 371, 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fortin S. P., Ennis M. J., Savitch B. A., Carpentieri D., McDonough W. S., Winkles J. A., Loftus J. C., Kingsley C., Hostetter G., Tran N. L. (2009) Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol. Cancer Res. 7, 1871–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoelzinger D. B., Mariani L., Weis J., Woyke T., Berens T. J., McDonough W. S., Sloan A., Coons S. W., Berens M. E. (2005) Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 7, 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berens M. E., Rief M. D., Loo M. A., Giese A. (1994) The role of extracellular matrix in human astrocytoma migration and proliferation studied in a microliter scale assay. Clin. Exp. Metastasis 12, 405–415 [DOI] [PubMed] [Google Scholar]
- 28. Nakada M., Niska J. A., Miyamori H., McDonough W. S., Wu J., Sato H., Berens M. E. (2004) The phosphorylation of EphB2 receptor regulates migration and invasion of human glioma cells. Cancer Res. 64, 3179–3185 [DOI] [PubMed] [Google Scholar]
- 29. García-Mata R., Wennerberg K., Arthur W. T., Noren N. K., Ellerbroek S. M., Burridge K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 [DOI] [PubMed] [Google Scholar]
- 30. Ellerbroek S. M., Wennerberg K., Arthur W. T., Dunty J. M., Bowman D. R., DeMali K. A., Der C., Burridge K. (2004) SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol. Biol. Cell 15, 3309–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prieto-Sánchez R. M., Bustelo X. R. (2003) Structural basis for the signaling specificity of RhoG and Rac1 GTPases. J. Biol. Chem. 278, 37916–37925 [DOI] [PubMed] [Google Scholar]
- 32. Krishna Subbaiah V., Massimi P., Boon S. S., Myers M. P., Sharek L., Garcia-Mata R., Banks L. (2012) The invasive capacity of HPV transformed cells requires the hDlg-dependent enhancement of SGEF/RhoG activity. PLoS Pathog. 8, e1002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patel J. C., Galán J. E. (2006) Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J. Cell Biol. 175, 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Buul J. D., Allingham M. J., Samson T., Meller J., Boulter E., García-Mata R., Burridge K. (2007) RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J. Cell Biol. 178, 1279–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mariani L., Beaudry C., McDonough W. S., Hoelzinger D. B., Demuth T., Ross K. R., Berens T., Coons S. W., Watts G., Trent J. M., Wei J. S., Giese A., Berens M. E. (2001) Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes. A cDNA microarray analysis. J. Neurooncol. 53, 161–176 [DOI] [PubMed] [Google Scholar]
- 36. Kislin K. L., McDonough W. S., Eschbacher J. M., Armstrong B. A., Berens M. E. (2009) NHERF-1: modulator of glioblastoma cell migration and invasion. Neoplasia 11, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wennerberg K., Ellerbroek S. M., Liu R. Y., Karnoub A. E., Burridge K., Der C. J. (2002) RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 277, 47810–47817 [DOI] [PubMed] [Google Scholar]
- 38. Gauthier-Rouvière C., Vignal E., Mériane M., Roux P., Montcourier P., Fort P. (1998) RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol. Biol. Cell 9, 1379–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Côté J. F., Vuori K. (2002) Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 115, 4901–4913 [DOI] [PubMed] [Google Scholar]
- 40. Brugnera E., Haney L., Grimsley C., Lu M., Walk S. F., Tosello-Trampont A. C., Macara I. G., Madhani H., Fink G. R., Ravichandran K. S. (2002) Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 4, 574–582 [DOI] [PubMed] [Google Scholar]
- 41. Yang H. J., Youn H., Seong K. M., Jin Y. W., Kim J., Youn B. (2013) Phosphorylation of ribosomal protein S3 and antiapoptotic TRAF2 protein mediates radioresistance in non-small cell lung cancer cells. J. Biol. Chem. 288, 2965–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin W. J., Su Y. W., Lu Y. C., Hao Z., Chio I. I., Chen N. J., Brüstle A., Li W. Y., Mak T. W. (2011) Crucial role for TNF receptor-associated factor 2 (TRAF2) in regulating NFκB2 signaling that contributes to autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 108, 18354–18359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jang K. W., Lee K. H., Kim S. H., Jin T., Choi E. Y., Jeon H. J., Kim E., Han Y. S., Chung J. H. (2011) Ubiquitin ligase CHIP induces TRAF2 proteasomal degradation and NF-κB inactivation to regulate breast cancer cell invasion. J. Cell Biochem. 112, 3612–3620 [DOI] [PubMed] [Google Scholar]
- 44. Natoli G., Costanzo A., Guido F., Moretti F., Bernardo A., Burgio V. L., Agresti C., Levrero M. (1998) Nuclear factor κB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 273, 31262–31272 [DOI] [PubMed] [Google Scholar]
- 45. Rothe M., Sarma V., Dixit V. M., Goeddel D. V. (1995) TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science 269, 1424–1427 [DOI] [PubMed] [Google Scholar]
- 46. Liu Z. G., Hsu H., Goeddel D. V., Karin M. (1996) Dissection of TNF receptor 1 effector functions. JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 87, 565–576 [DOI] [PubMed] [Google Scholar]
- 47. Natoli G., Costanzo A., Ianni A., Templeton D. J., Woodgett J. R., Balsano C., Levrero M. (1997) Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science 275, 200–203 [DOI] [PubMed] [Google Scholar]
- 48. Zheng M., Morgan-Lappe S. E., Yang J., Bockbrader K. M., Pamarthy D., Thomas D., Fesik S. W., Sun Y. (2008) Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res. 68, 7570–7578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murga C., Zohar M., Teramoto H., Gutkind J. S. (2002) Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-κB. Oncogene 21, 207–216 [DOI] [PubMed] [Google Scholar]
- 50. Ridley A. J. (2006) Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 16, 522–529 [DOI] [PubMed] [Google Scholar]
- 51. DerMardirossian C., Bokoch G. M. (2005) GDIs. Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 15, 356–363 [DOI] [PubMed] [Google Scholar]
- 52. Samson T., van Buul J. D., Kroon J., Welch C., Bakker E. N., Matlung H. L., van den Berg T. K., Sharek L., Doerschuk C., Hahn K., Burridge K. (2013) The guanine-nucleotide exchange factor SGEF plays a crucial role in the formation of atherosclerosis. PLoS ONE 8, e55202. [DOI] [PMC free article] [PubMed] [Google Scholar]