

Background: Calmodulin (CaM) is recruited into the death-inducing signaling complex in cholangiocarcinoma cells.
Results: CaM binds to FasDD in a 2:1 CaM:FasDD model. CaM antagonists abolish FasDD-CaM interactions.
Conclusion: Data offer a structural basis for Fas-CaM interactions and mechanisms of inhibition.
Significance: Elucidating the structural determinants of Fas-CaM interaction is critical to understanding the functional role of CaM in Fas-mediated apoptosis.
Keywords: Apoptosis, Calmodulin, Fas, NMR, Surface Plasmon Resonance (SPR), Cholangiocarcinoma
Abstract
The extrinsic apoptotic pathway is initiated by cell surface death receptors such as Fas. Engagement of Fas by Fas ligand triggers a conformational change that allows Fas to interact with adaptor protein Fas-associated death domain (FADD) via the death domain, which recruits downstream signaling proteins to form the death-inducing signaling complex (DISC). Previous studies have shown that calmodulin (CaM) is recruited into the DISC in cholangiocarcinoma cells, suggesting a novel role of CaM in Fas-mediated signaling. CaM antagonists induce apoptosis through a Fas-related mechanism in cholangiocarcinoma and other cancer cell lines possibly by inhibiting Fas-CaM interactions. The structural determinants of Fas-CaM interaction and the underlying molecular mechanisms of inhibition, however, are unknown. Here we employed NMR and biophysical techniques to elucidate these mechanisms. Our data show that CaM binds to the death domain of Fas (FasDD) with an apparent dissociation constant (Kd) of ∼2 μm and 2:1 CaM:FasDD stoichiometry. The interactions between FasDD and CaM are endothermic and entropically driven, suggesting that hydrophobic contacts are critical for binding. We also show that both the N- and C-terminal lobes of CaM are important for binding. NMR and surface plasmon resonance data show that three CaM antagonists (N-(6-aminohexyl)-5-chloro-1-naphthalene sulfonamide, tamoxifen, and trifluoperazine) greatly inhibit Fas-CaM interactions by blocking the Fas-binding site on CaM. Our findings provide the first structural evidence for Fas-CaM interactions and mechanism of inhibition and provide new insight into the molecular basis for a novel role of CaM in regulating Fas-mediated apoptosis.
Introduction
Apoptosis is a strictly regulated process by which abnormal cells are removed from the body without altering the immune system or generating an inflammatory response. Inappropriate apoptosis (enhanced or diminished) is linked to many human diseases including neurodegenerative and autoimmune disorders, AIDS, and many types of cancers (1). Cells from a wide variety of human malignancies show a decreased ability to undergo apoptosis in response to various stimuli, which may contribute to the clonal expansion of cancer cells (2). The apoptotic pathway is normally initiated by cell surface death receptors, which belong to a tumor necrosis factor (TNF) super family of receptors (3–5). The cytoplasmic regions of two major receptors, Fas (also called CD95/Apo1) and TNF-receptor-1, share a homologous cytoplasmic region of ∼80 amino acids called the death domain (DD)4 (5, 6). These receptors undergo a conformational change in response to their cognate ligands, allowing them to interact with adaptor proteins such as Fas-associated death domain (FADD), which also contains a homologous DD region (7, 8). Engagement of Fas by Fas ligand (FasL) initiates a cascade of interactions that lead to activation of specific proteases called caspases. Caspase activation can be achieved through two distinct but ultimately converging apoptotic pathways, extrinsic and intrinsic (9). In the extrinsic pathway, Fas-FasL interaction leads to activation of caspase 8 (by FADD) and formation of death-inducing signaling complex (DISC). Active caspase 8 then cleaves and activates caspases 3, 6, and 7, which target cellular substrates and ultimately lead to cell death (8–10). DISC formation and subsequent protein recruitment is a critical initial step in regulating Fas-mediated apoptosis.
There is compelling evidence that the DD of Fas (FasDD) interacts with various molecules, suggesting that Fas signaling is complex and regulated by multiple proteins (5). Previous studies have shown that calmodulin (CaM) is recruited into DISC in cholangiocarcinoma (11–17) and pancreatic cancer cells (18), suggesting a novel role of CaM in Fas signaling. It has been hypothesized that Fas-CaM interaction may affect Fas-FADD interaction and thus regulates DISC assembly and inhibits apoptosis in cholangiocarcinoma cells (14). Fas has been shown to interact with CaM in cholangiocarcinoma cells in a calcium-dependent manner; the amount of CaM recruited into DISC has increased upon Fas stimulation (11). Elevated levels of CaM have been linked to diseases characterized by pathological, unregulated cell growth, such as cancer (19). CaM is a ubiquitous and highly conserved calcium-binding protein expressed in all eukaryotic cells and is implicated in a variety of cellular functions (20–25). Binding of Ca2+ to CaM triggers major structural rearrangements in the N- and C-terminal lobes resulting in the opening of large binding pockets on the surface of each domain consisting of hydrophobic residues that are essentially buried in the apoprotein (21, 24, 25). The CaM protein has a “dumbbell-like” architecture with the N- and C-terminal lobes (CaM-N and CaM-C, respectively) connected by a flexible helix called the central linker. CaM-N and CaM-C each possess two helix-loop-helix motifs called “EF-hands” (26–28).
Based on mutagenesis studies, it was suggested that the CaM-binding site in FasDD is located between residues 215 and 238 (numbered 231–254 in that study) (13). The NMR structure of Fas shows that these residues form helices I and II (α1 and α2) (29). Mutations that may alter the structure and/or function of Fas have been detected in many types of cancers (30, 31). In particular, mutation of Val-238 to asparagine (V238N, named V254N in other studies (13, 32)) is analogous to the identified mutant allele of Fas in lpr-autoimmune mice that have a deficiency in Fas-mediated apoptosis (33). Although it has been shown that the Fas V254N mutant reduced CaM binding (13), it is not yet known whether residue Val-238 is directly involved in CaM binding or if it exerts an allosteric effect that affects Fas binding to CaM.
Several molecules with distinct chemical structures have been found to inhibit CaM-mediated processes (12, 14). N-(6-Aminohexyl)-5-chloro-1-naphthalene sulfonamide (W7), the anti-psychotic trifluoperazine (TFP), and the anti-estrogen tamoxifen (TMX) induce apoptosis through a Fas-related mechanism in cholangiocarcinoma and other cancer cell lines (12, 14, 34). CaM antagonists also inhibit tumor cell invasion in vitro (35) and metastasis in vivo (36), suggesting that these are promising chemotherapeutic agents for malignancies. The molecular mechanism by which CaM antagonists manipulate the Fas signaling pathway is still unclear.
In summary, the Fas-CaM interaction appears to be an inhibitory component of DISC and may play a vital role in obstruction of caspases activation. Elucidation of the structural determinants of Fas-CaM interaction and mechanism of inhibition will be critical to understanding the precise molecular mechanism of Fas-mediated apoptosis, which may help in the development of new anticancer therapeutic strategies.
Here we employ NMR and biophysical techniques to identify the structural determinants of FasDD-CaM interactions. We show that CaM binds directly to FasDD with a dissociation constant of ∼2 μm and 2:1 CaM:FasDD stoichiometry. Our NMR data show that both of the N- and C-terminal lobes of CaM are important for FasDD binding. In addition, we show that CaM antagonists block interaction with FasDD, providing a structural basis for their role in the activation of Fas-mediated apoptosis.
EXPERIMENTAL PROCEDURES
Sample Preparation
A plasmid encoding full-length (amino acids 1–148) Norvegicus rattus calmodulin was a kind gift from Dr. Madeline Shea (University of Iowa). The Rattus CaM protein sequence is identical to that of human CaM (Swiss-port code: P62158). Plasmids encoding for CaM-N (residues 1–80) and CaM-C (residues 76–148) were constructed using the pT7–7 vector. CaM protein expression and purification were conducted as described (37). CaM-N and CaM-C proteins were expressed as described for CaM (37) and purified by ion exchange (Q-column) and gel filtration chromatography methods. CaM samples were stored in a buffer containing 50 mm HEPES or Tris at pH 7.0, 100 mm NaCl, and 5 mm CaCl2.
A vector (pET28) harboring the FasDD gene encoding for residues 191–335 fused to a His6-SUMO tag on the N terminus was kindly provided by Dr. Jay McDonald (University of Alabama at Birmingham). Consistent with the NMR studies of FasDD, we used the numbering of FasDD amino acids as described (3, 29). Thus, we numbered residues 191–335 as 175–320. This plasmid was used to construct two new Fas plasmids encoding for residues 205–305 (Faswt). The first plasmid encodes for an N-terminal His6-SUMO tag fused to Faswt in a pET28 vector. The second plasmid contains Fas gene encoding for residues 205–305 cloned into the pET-11a vector at its NdeI and BamHI sites in-frame with the C-terminal His6 tag (His6-Fas). The His6-Fas clone was used to make Fas D244A mutant (Fas monomer (Fasm)) via QuikChange site-directed mutagenesis.
FasDD proteins were expressed in Escherichia coli BL21 (DE3) codon+ RIL cells. To make uniformly 15N- and 13C,15N-labeled Fas samples, cells were grown in 4 liters of LB media at 37 °C until the A600 was ∼0.6. Next, cells were spun down and washed with 1× M9 salt before transferring them to 1 liter of M9 minimal media containing 15NH4Cl and/or [13C]glucose as the sole sources to produce 15N- and/or 13C-labeled proteins, respectively. Cells were induced with isopropyl β-d-1-thiogalactopyranoside, grown at 37 °C for ∼12 h, spun down, and stored overnight at −80 °C. Cells were then lysed by sonication methods. The FasDD proteins were purified by nickel or cobalt affinity chromatography (Thermo Scientific) and gel filtration chromatography (GE Healthcare). For His6-SUMO-Faswt, the His6-SUMO tag was cleaved via SUMO protease, and the Faswt protein was subsequently purified by gel filtration chromatography. Fasm protein was stored in 50 mm HEPES or Tris (pH 7.0), 100 mm NaCl, and 5 mm CaCl2, whereas Faswt was stored in 50 mm MOPS (pH 6.5), 100 mm NaCl, and 5 mm CaCl2. Because of the high tendency of Faswt to form large and insoluble aggregates, the protein was kept at very low concentrations (∼10 μm). Protein molecular weights were confirmed by electrospray ionization mass spectrometry. Because of the zero extinction coefficient for CaM-N, protein concentrations were measured using bicinchoninic protein assay (Thermo Scientific). TFP, W7 and TMX were purchased from Sigma. TFP and W7 were dissolved in 20:80 DMSO:H2O at concentration of ∼20 mm and TMX was dissolved in 100% DMSO at concentrations of ∼0.1 mm.
Gel Filtration Assay
The mobility of Faswt, Fasm, and CaM and their complexes was analyzed by gel filtration assay. Briefly, 0.5–2 ml of CaM, Faswt, Fasm, or their complexes (∼20–400 μm) were run through a HiLoad 16/60 Superdex 75 column (GE Healthcare) in a buffer containing 50 mm MES or MOPS (pH 6.5–7.0), 100 mm NaCl, and 5 mm CaCl2. Protein fractions were analyzed by SDS-PAGE. Molecular weight calibration kits (GE Healthcare) were used to determine the approximate molecular weights of loaded proteins.
Analytical Ultracentrifugation Experiments
Sedimentation velocity measurements were collected on a Beckman XL-I Optima system equipped with a 4-hole An-60 rotor (Beckman Coulter). Protein samples were prepared in 50 mm HEPES, 100 mm NaCl, and 5 mm CaCl2. Loading concentrations were at 60 and 20 μm for CaM and Fasm, respectively. Three CaM-Fasm complexes have been prepared at 0.5:1, 1:1, and 2:1 CaM:Fasm stoichiometries with Fasm concentration set constant at 20 μm. Rotor speed was set at 40,000 rpm (20 °C), and scans were acquired at 280 nm. Partial specific volumes (v-bar) and molar extinction coefficients were calculated by using the program SEDNTERP, and buffer densities were measured pycnometrically. Sedimentation velocity data analysis were performed by using SEDFIT (38–41).
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted on protein samples in a buffer containing 50 mm HEPES (pH 7.0) or MOPS (pH 6.5), 100 mm NaCl, and 5 mm CaCl2. Thermodynamic parameters of CaM binding to Faswt and Fasm were determined using an Auto-iTC200 microcalorimeter (MicroCal Corp.). CaM at 410–430 μm was titrated into the cell sample containing 17 μm Fasm or Faswt. In the reciprocal ITC titrations, Fasm at 400 μm was titrated into the cell sample containing 80 μm CaM. Heat of reaction was measured at 35 °C for 19 injections. Heat of dilution was measured by titrating CaM or FasDD proteins into buffers under identical conditions. Data analysis was performed using the Microcal Origin package. Base-line corrections were performed by subtracting heat of dilution from the raw Fas-CaM titration data. Binding curves were analyzed, and dissociation constants (Kd) were determined by nonlinear least-square fitting of the base-line-corrected data. The formula used to fit the data as single set of identical sites is,
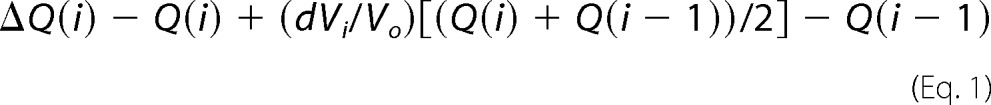 |
where ΔQ(i) is the heat released at ith injection, Q(i) is the total heat content of the solution, dVi is injection volume, and Vo is total volume.
NMR Spectroscopy
NMR data were collected at 35 °C on a Bruker Avance II (700 MHz 1H) spectrometer equipped with a cryogenic triple-resonance probe, processed with NMRPIPE (42), and analyzed with NMRVIEW (43). All NMR samples were prepared in a buffer containing 50 mm Tris-d11 (pH 7.0), 50 mm NaCl, and 5 mm CaCl2. Sample concentrations used for NMR titration data were at 100 μm. 1H, 13C, and 15N NMR chemical shifts for CaM have been reported (44,45). Fasm backbone signals were assigned using standard triple resonance (HNCA, HNCO, HNCOCA, HNCACB, HNCOCACB) and 15N-edited HSQC-NOESY and HSQC-TOCSY (two-dimensional total correlation spectroscopy) datasets collected on ∼0.5–0.8 mm samples (see Refs. 46 and 47 and citations therein).
Surface Plasmon Resonance (SPR) Experiments
SPR experiments were performed on a BIAcore 2000 system (GE Healthcare) at 20 °C using HBS running buffer (10 mm HEPES (pH 7.4), 150 mm NaCl, 0.005% surfactant P20, and 5 mm CaCl2). CaM was immobilized by amine-coupling chemistry on CM5 sensor chip (GE Healthcare) that had been activated with a 1:1 mixture of 0.1 m N-hydroxysuccinimide and 0.1 m 3-(N,N-dimethylamino)propyl-N-ethylcarbondiimide. Surfaces were blocked by the injection of 1 m ethanolamine for 7 min and regenerated by injecting HBS running buffer containing 10 mm EDTA for 1 min at a flow rate of 20 μl/min. Faswt or antagonist binding to CaM were evaluated at increased concentrations (0.5–20 μm) at a flow rate of 15 μl/min. Experiments were performed in duplicate. Results were analyzed using BIAevaluation software Version 3.2 (GE Healthcare), and the response curves were globally fit to a one-site Langmuir binding model.
RESULTS
Mutants Inhibit Fas Self-association
Structural studies of FasDD and characterization of its interactions with other proteins have been hampered by the high propensity to self-associate and form large aggregates in solution (8, 29). Previous studies have shown that solubility of FasDD is as low as ∼20 μm at pH 7 (29). Consequently, the NMR structure was determined at pH 4 (Fig. 1) (29). Mutagenesis studies have shown that self-association of FasDD is mediated by several charged residues localized in helix α3 (29). These include Asp-244, Glu-245, and Lys-247. Recent solution studies on FasDD and its interaction with FADD were conducted on a mutant (K247A) that inhibits FasDD aggregation (48). NMR data have also shown that D244A mutant inhibits FasDD self-association without altering the Fas structure (29, 49). For initial characterization of its binding to CaM, we have made a protein construct of wild type FasDD encompassing residues 205–305 (Faswt). Consistent with previous studies (29), solubility of Faswt was very low (∼20 μm). Because of these complications, we have made a D244A mutant (Fasm) and examined the effect of this mutant on CaM binding by gel filtration, ITC, and NMR methods.
FIGURE 1.
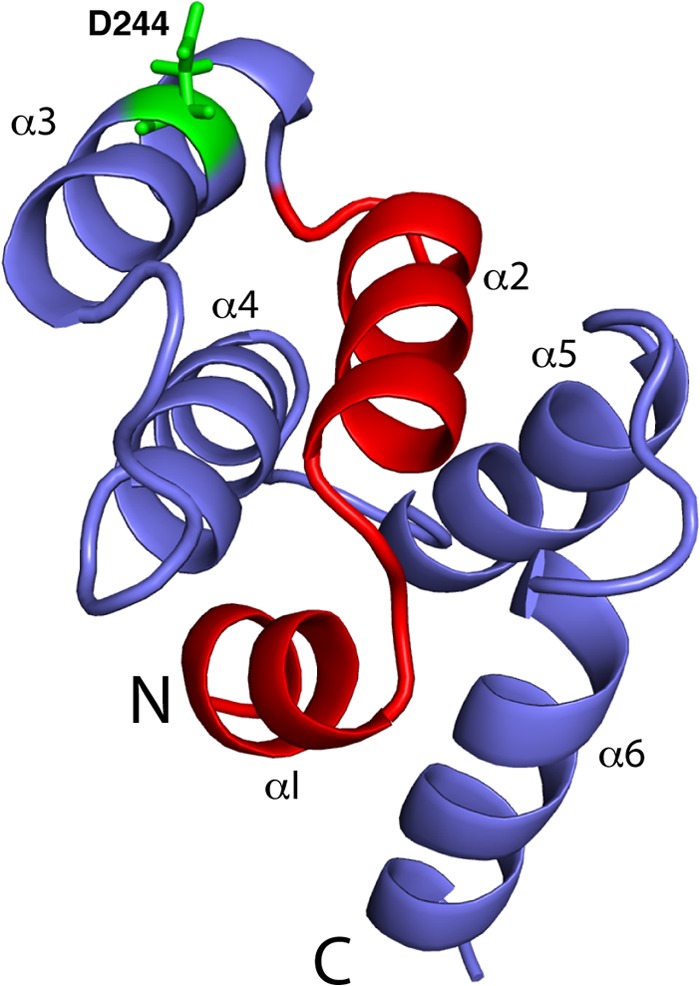
Schematic representation of the FasDD structure (PDB ID 1DDF) highlighting the originally proposed CaM binding region (red) (13). Residue Asp-244 is shown in green sticks.
Mobility Characteristics of FasDD-CaM Complexes as Determined by Gel Filtration Assay
We first analyzed the solution properties of Faswt, CaM, and Faswt-CaM complex by gel filtration mobility assay. Samples were run on a size exclusion column (Superdex 75) under similar buffer conditions. Faswt protein concentration was at ∼20 μm. As shown in Fig. 2A, the elution volumes of Faswt and CaM were at 76 and 65 ml, respectively. Interestingly, we found that the elution volume of the Faswt-CaM complex is dependent on the stoichiometry of the proteins. At a 1:1 molar ratio, the complex eluted at 63.5 ml, whereas a complex formed by mixing a slight excess of CaM (1.5:1 CaM:Faswt) eluted at 59 ml (Fig. 2A). Despite the presence of excess amount of CaM, a peak indicating free Faswt was also observed at 76 ml. At 2:1 CaM:Faswt, the elution volume of the complex had changed to 56 ml, suggesting formation of a larger molecular weight complex (Fig. 2A). A further increase of CaM concentration (2.5:1 CaM:Faswt) led to only a minor change of the elution volume to 55 ml (Fig. 2B, red). No further changes in the elution volume of the complex were observed at higher ratio, indicating complete formation of the complex at molar ratio 2–2.5:1 CaM:Faswt. The observation of a single peak for the complex with a variable elution volume may suggest an equilibrium between 1:1 and ternary CaM-Fas complexes.
FIGURE 2.

Gel filtration assay of CaM binding to FasDD using a HiLoad Superdex 75 (16/60) column. A, shown are chromatograms of CaM, Faswt, and their complex. Data show that complex formation is dependent on the molar ratio. B, shown are chromatograms of CaM, Faswt, Fasm, and their complexes. Elution volumes of CaM-Faswt and CaM-Fasm are identical, indicating that the Asp-244 mutation in FasDD does not affect binding to CaM. C, complex fractions were analyzed by 12% SDS-PAGE. Gels were stained with Coomassie Brilliant Blue. The band representing CaM is more intense than that of Faswt or Fasm, indicating that CaM is more abundant in both complexes. D, a low molecular weight calibration curve was used to determine the approximate molecular weight of CaM, Faswt, Fasm, and their complexes.
One possible explanation of the results described above is that Faswt self-associates and forms high order complexes. This has been shown previously for Faswt-FADD complexes (8,10). This scenario was examined by characterizing CaM interaction with Fasm. Gel filtration data show that the elution volumes of free Fasm and Faswt are identical (Fig. 2B), indicating that Faswt does not self-associate at 20 μm. A complex formed between CaM and Fasm at 2.5:1 also eluted at 55 ml, indicating that both Faswt and Fasm form similar complexes with CaM and that D244A mutation has no detectable effect on CaM binding. Fractions of the complexes were collected and analyzed by SDS-PAGE. As shown in Fig. 2C, two bands representing CaM and FasDD proteins were observed. The band representing the CaM protein is more intense than that of Faswt or Fasm, indicating that CaM is more abundant in both complexes. A gel filtration mobility assay with known protein standards revealed that the molecular mass of unbound Fasm or Faswt is ∼14 kDa, consistent with a monomer. The migration behavior of CaM (∼27 kDa) is attributed to its elongated dumbbell shape (45, 50). Both CaM:Faswt and CaM:Fasm complexes with 2.5:1 molar ratio migrated as ∼45–48-kDa complexes, an apparent molecular mass that is significantly higher than a 1:1 complex (Fig. 2D). At a 1:1 molar ratio, the calculated molecular mass of the CaM-Fasm complex was ∼35 kDa, an average value of the molecular masses of 1:1 and 2:1 CaM:Fasm complexes, suggesting an equilibrium between the 1:1 and 2:1 complexes. Altogether, our gel filtration results provide compelling evidence for direct binding of CaM to FasDD and indicate that formation of the complex is dependent on stoichiometry and that two molecules of CaM probably bind to FasDD. We also show that D244A mutation has no effect on the formation of the CaM-FasDD complex.
Sedimentation Studies of FasDD-CaM Interactions
Gel filtration results provided insights on the mobility of the Fasm and CaM proteins as well as their complex. Here, we examined the oligomerization and equilibrium properties and stoichiometry of binding using analytical ultracentrifugation. As shown in Fig. 3, sedimentation velocity profiles of Fasm and CaM exhibited a single sedimentation boundary. Analysis of the data using SEDFIT yielded peaks at 1.4 and 1.9 S for Fasm and CaM, respectively. Molecular weight distribution analysis gave 11 and 17 kDa for Fasm and CaM, respectively. The sedimentation velocity data obtained on 3 sample mixtures of the CaM-Fasm complex with variable molar ratios (0.5:1, 1:1, and 2:1 CaM:Fasm) are shown in Fig. 3. Sedimentation velocity profiles of the complex exhibited two sedimentation boundaries, of which one represented unbound Fasm at 1.4 S and a second represented the complex at 3 S. Upon increasing the ratio of CaM, the intensity of the peak representing the complex was increased, whereas the intensity of the peak denoting the unbound Fasm was decreased. The apparent molecular mass for the CaM-Fasm complex was calculated to be ∼42 kDa, consistent with the formation of a 2:1 CaM-Fasm complex. These results have been recapitulated for the CaM-Faswt complex (data not shown).
FIGURE 3.
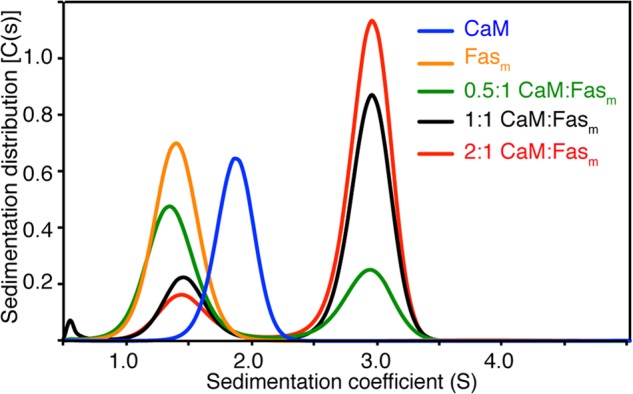
Sedimentation coefficient distributions, c(s), obtained from the sedimentation profiles for free Fasm and CaM and the Fasm-CaM complex at variable molar ratios. Molecular mass of the CaM-Fasm complex (∼42 kDa) is consistent with of 2:1 binding mode. Loading concentrations were at 60 and 20 μm for CaM and Fasm, respectively. Three CaM-Fasm complexes have been prepared at 0.5:1, 1:1, and 2:1 CaM:Fasm stoichiometries with Fasm concentration set constant at 20 μm. Rotor speed was set at 40,000 rpm (20 °C), and scans were acquired at 280 nm.
Thermodynamic Properties of CaM Binding to FasDD as Detected by ITC
The two hydrophobic regions on the N- and C-terminal lobes of CaM have been widely implicated in CaM cellular activity (24). Additional electrostatic interactions have also been shown to stabilize CaM-protein interactions (24, 51, 52). The ITC method is widely used to assess the relative contribution of hydrophobic versus electrostatic factors. ITC data provide values for Kd, stoichiometry (n), and enthalpy change (ΔH°). The Kd value is then used to calculate the change in Gibbs energy (ΔG°), which together with ΔH° allows the calculation of the entropic term TΔS°. The entropic and enthalpic components of the free energy reveal the nature of the forces that drive the binding reaction.
Thermodynamic parameters of CaM binding to Faswt and Fasm were assessed by ITC. The results allowed us to determine whether D244A mutation has any effect on the thermodynamic properties of CaM binding to FasDD. As shown in Fig. 4A, fitting of the ITC data to a single set of identical sites upon titration of CaM (410 μm) into Fasm (17 μm) yielded the following thermodynamic parameters: Kd = 1.7 ± 0.2 μm, n = 1.96, ΔH° = 11.7 ± 0.2 kcal/mol, and ΔS° = 64.4 cal/mol/degree. In the reciprocal experiment, fitting of the data obtained on Fasm (402 μm) titrated into CaM (80 μm) has yielded the following thermodynamic parameters: n = 0.47, Kd = 3.2 ± 0.1 μm, ΔH° = 18.0 ± 0.2 kcal/mol, and ΔS° = 85.3 cal/mol/degree (data not shown). Consistent with the sedimentation velocity and gel filtration data, the ITC data clearly show that stoichiometry of binding is 2, indicating that the two CaM molecules bind to FasDD. Similarly, the ITC data obtained upon titration of CaM (430 μm) into Faswt (17 μm) yielded the following parameters: n = 1.94, Kd = 2.3 ± 0.3 μm, ΔH° = 9.7 ± 0.3 kcal/mol, and ΔS° = 57.3 cal/mol/degree (Fig. 4B). Collectively, our ITC data show that (i) CaM binds to FasDD in a 2:1 model, (ii) binding affinity and stoichiometry are not affected by Asp-244 mutation, and (iii) Fas binding to CaM is entropically driven as indicated by the sign of the heat of enthalpy (endothermic).
FIGURE 4.
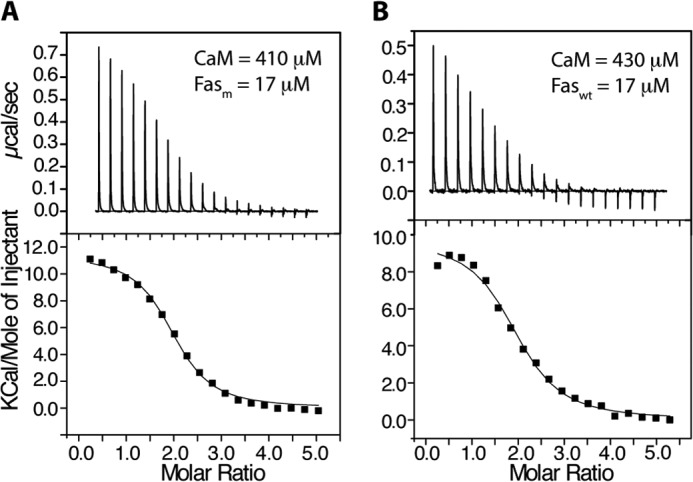
ITC data obtained upon titration of CaM (410 μm) into Fasm (17 μm) (A) and CaM (430 μm) into Faswt (17 μm) (B). Data-fitting afforded Kd values of 1.7 and 2.3 μm for left and right panels, respectively. As indicated by the molar ratio, the stoichiometry of binding is 2:1 CaM:FasDD.
CaM-Fasm Interactions as Detected by NMR Spectroscopy
The binding sites in CaM and FasDD can be identified through NMR resonance perturbations as detected in two-dimensional 1H,15N HSQC (heteronuclear single quantum coherence) spectra. These experiments allow for identification of residues that are involved in the interaction and/or accompanying conformational changes and provide an effective method for examining the folding of the protein. Because of the high propensity of Faswt to self-associate, which often complicates the NMR studies, and because mutation of Asp-244 does not affect CaM binding, we conducted our NMR experiments with the Fasm protein. Consistent with previous studies, mutation of Asp-244 to alanine has increased solubility of Fas by 103-fold to >2 mm. We assessed whether FasDD undergoes significant conformational changes upon substituting a single amino acid (D244A). Asp-244 is located at the center of α3 (Fig. 1). 1H, 13C, and 15N NMR signals of Fasm have been assigned using standard triple resonance methods (see “Experimental Procedures”). NMR chemical shifts of Fasm were very similar to those described for Fas K247A (48) except for residues in the proximity of the mutation site. No new intramolecular NOEs were detected that would be indicative of an altered protein conformation. Three-dimensional 15N-edited HSQC-NOESY data of Fasm confirmed that the helical character of α3 is retained (data not shown). These NMR observations indicate that D244A mutation inhibits FasDD self-association without altering the helical character of α3 or the overall structure of the protein.
To identify the binding interface, we first conducted NMR titrations on a uniformly 15N-labeled Fasm (100 μm) as a function of added unlabeled CaM (Fig. 5). The addition of substoichiometric amounts of unlabeled CaM to Fasm (0.5:1) led to a decrease in intensity for the majority of 1H,15N resonances (Fig. 5A, red). A steady decrease in intensity for the vast majority of the 1H,15N signals in the HSQC spectra was clearly observed with further addition of CaM (Fig. 5A, green). At 2.5:1 CaM:Fasm, the vast majority of 1H,15N resonances disappeared, but many new others were observed (Fig. 5B). No further changes in the HSQC spectrum were observed above 2.5:1 CaM:Fasm.
FIGURE 5.
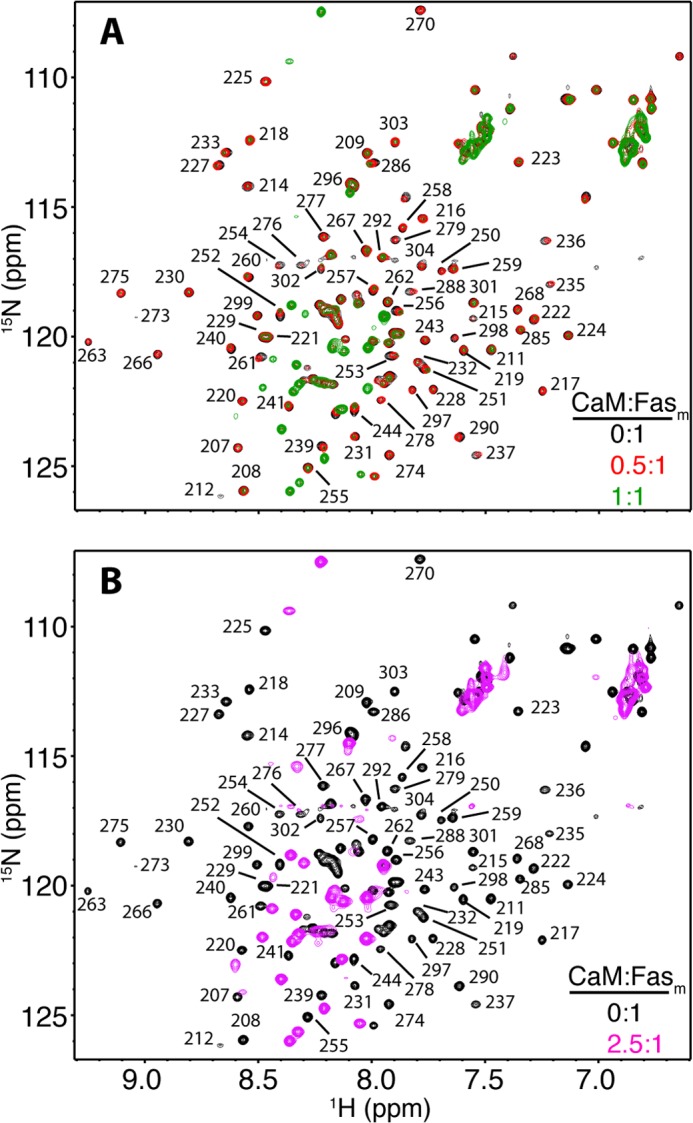
Overlay of two-dimensional 1H,15N HSQC spectra obtained for 15N-labeled Fasm upon titration with unlabeled CaM at CaM:Fasm = 0:1 (black), 0.5:1 (red), and 1:1 (green) (A), and CaM:Fasm = 0:1 (black) and 2.5:1 (magenta) (B).
Severe broadening and/or loss of NMR signals can be attributed to one or more of the following events; first, an intermediate chemical exchange between the free and bound states of Fasm. To determine if this was the case, we collected a set of two-dimensional HSQC data on a 15N-labeled Fasm bound to CaM at different temperatures (15–40 °C). No major improvement in signal intensity in the HSQC spectra was observed as a function of temperature. Second, the line-width (short transverse relaxation time, T2) of signals was increased as a result of increasing the molecular size of the CaM-Fasm complex. This is the mostly likely factor as the complex is considered large (∼45 kDa) and tumbles significantly slower than unbound proteins. Third, binding of CaM induces a conformational change, leading to alteration or unfolding of the tertiary structure of Fasm. In the two-dimensional HSQC spectrum of CaM:Fasm at 2.5:1 (Fig. 5B), about 40 of the new resonances appeared strong and are clustered in a narrow chemical shift range, which may suggest that a portion of the Fasm protein became unfolded upon binding to CaM. The alternative explanation is that these signals correspond to residues localized in highly dynamic regions or loops in the Fasm protein. The number of new amide resonances is significantly higher than the number of residues localized within flexible regions (25 residues) in the free FasDD protein (PDB ID 1DDF), which may suggest that CaM induced a conformational change in FasDD, rendering a portion of the protein more flexible.
Next, we sought to identify the FasDD binding region on CaM. We collected the reciprocal HSQC experiment by which a uniformly 15N-labeled sample of CaM was titrated with unlabeled Fasm (Fig. 6). NMR signal assignments of CaM are described elsewhere (44, 45). The addition of substoichiometric amounts of Fasm has led to dramatic changes in the HSQC spectra of CaM. A significant number of 1H,15N resonances have disappeared, and many others have become very broad. Changes in the HSQC spectra ceased at 0.5:1 Fasm:CaM, indicating complete complex formation at this molar ratio (Fig. 6). This result is consistent with formation of a 1:2 Fasm:CaM complex. As evident from the NMR titration data, 1H,15N resonances that exhibited substantial chemical shift changes correspond to residues spread throughout the CaM protein. These results suggest engagement of a wide interacting interface or induction of significant conformational changes in CaM upon binding to Fasm.
FIGURE 6.
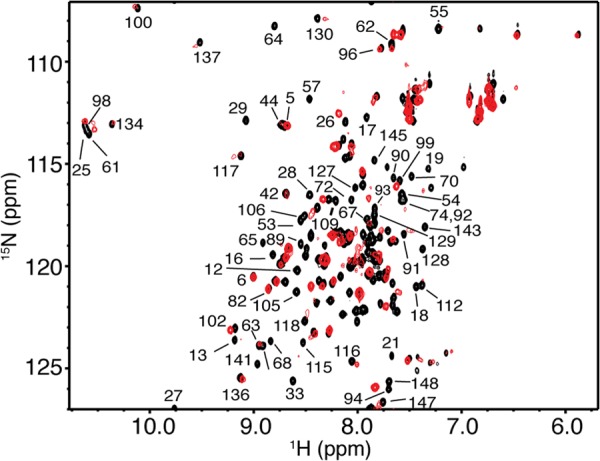
Overlay of two-dimensional 1H,15N HSQC spectra obtained for 15N-labeled CaM in the free state (black) and in complex with unlabeled Fasm at saturation (0.5:1 Fasm:CaM, red). No changes were observed in the HSQC spectrum with further addition of Fasm, indicating saturation at this molar ratio. Data are consistent with formation of a 2:1 CaM:Fasm complex.
Both N- and C-terminal Lobes of CaM Are Required for FasDD Binding
To identify which domains of CaM are critical for FasDD binding, we obtained ITC data on Fasm as titrated with the isolated N- and C-terminal domains of CaM (CaM-N and CaM-C, respectively). ITC data clearly show that the binding affinity is dramatically reduced upon titration of CaM-N or CaM-C into Fasm (Fig. 7). Although it was virtually impossible to fit the titration data of CaM-C due to the extremely weak binding, fitting of the CaM-N data yielded a Kd of 20 μm, which is 10-fold higher than that of full-length CaM. These results indicate that both N- and C-terminal lobes of CaM are important for FasDD binding.
FIGURE 7.
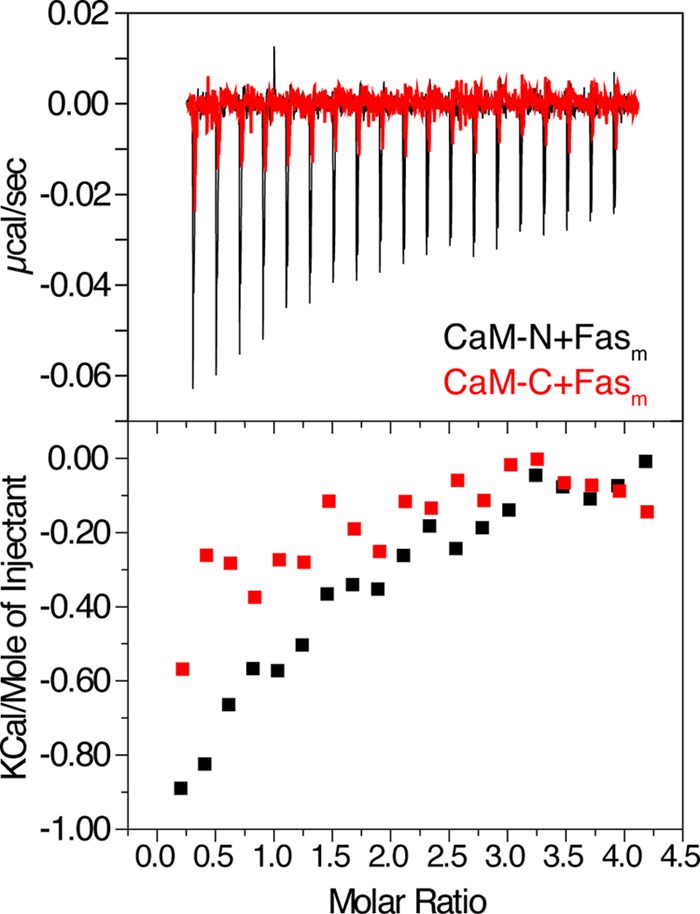
ITC data obtained upon titration of CaM-N (560 μm) and CaM-C (500 μm) into Fasm (19 μm). Data indicate that CaM-N and CaM-C bind FasDD much weaker than intact CaM.
CaM Antagonists Block Binding of FasDD
A variety of small organic molecules with distinct chemical structures interact directly with CaM and inhibit CaM-mediated processes (12). As described above, W7, TFP, and TMX were shown to induce apoptosis through a Fas-related mechanism in cholangiocarcinoma and other cancer cell lines (12, 14, 34). The molecular mechanism by which CaM antagonists manipulate the Fas signaling pathway is still unclear. Previous structural studies revealed that W7 and TFP bind to CaM with different modes. Whereas two W7 molecules bind to both hydrophobic pockets on the N and C termini of CaM (53), controversy still surrounds the mode of TFP binding to CaM. Three x-ray structures revealed that one, two, or four TFP molecules are capable of binding to CaM (54–56). NMR studies have also suggested that four TFP molecules can bind to CaM (57). The binding affinity of the first TFP molecule is, however, suggested to be significantly higher than the additional molecules (54).
One aim of the current study is to examine the effect of these antagonists on CaM-FasDD interactions. To do so, we employed NMR and SPR methods to assess whether antagonists block FasDD binding to CaM. First, we performed two-dimensional HSQC NMR titrations on a uniformly 15N-labeled sample of CaM as a function of added TFP. As shown in Fig. 8A, the vast majority of 1H,15N resonances exhibited substantial chemical shift changes upon the addition of TFP. Likewise, the majority of amide signals in the HSQC spectrum of CaM exhibited substantial chemical shift changes upon titration of W7 (Fig. 8B); these changes are similar to those reported previously (53), confirming that two molecules of W7 are packed in the N and C termini hydrophobic pockets of CaM. After saturating the CaM protein with TFP or W7, unlabeled Fasm was added at 1:1 molar ratio followed by acquisition of two-dimensional HSQC spectra (Fig. 8, C and D). Interestingly, no chemical shift changes were detected, indicating that TFP and W7 blocked binding of Fasm.
FIGURE 8.

Effect of antagonists on CaM-Fasm interaction as detected by two-dimensional NMR. A and B, HSQC spectra were obtained for 15N-labeled CaM in the free state (black) and in complex with TFP or W7 (red). C and D, HSQC spectra of CaM-TFP or CaM-W7 complexes in the absence (red) or presence (green) of Fasm at 1:1 ratio are shown. Notice the absence of any chemical shift changes upon the addition of Fasm (C and D), indicating that TFP and W7 inhibited CaM-Fasm interactions.
TMX, which was also shown to induce apoptosis through a Fas-related mechanism in cholangiocarcinoma cells, is poorly soluble, which precluded studying its effect on FasDD-CaM interactions by NMR. Thus, we resorted to SPR methods to assess whether TMX also disrupts binding of CaM to FasDD. In a control experiment, injection of Faswt over the CaM surface resulted in sensorgrams (Fig. 9) that could be globally fit to yield association (kon) and dissociation rates (koff) of 5.3 × 104 m−1 s−1 and 0.0477 s−1, respectively. Using these kinetic constants, the equilibrium dissociation constant (Kd = koff/kon) for the interaction between CaM and Faswt is 0.9 μm, which is in good agreement with the binding constant obtained from the ITC data. Despite the compelling evidence that CaM binds to FasDD in a 2:1 model, the SPR data were best fit to a one-site model. It has been shown that if the kon rates for both sites are within a factor of five, one-site fitting yields the best fit (58). Injection of either TMX or TFP at increased concentrations (5–20 μm) led to a significant decrease in the response units, indicating disruption of the CaM-Fas interactions. Altogether, consistent with previous studies (12, 14, 34), our data suggest that TFP, W7, and TMX probably induce apoptosis in cholangiocarcinoma cells by directly interfering with Fas-CaM interactions.
FIGURE 9.
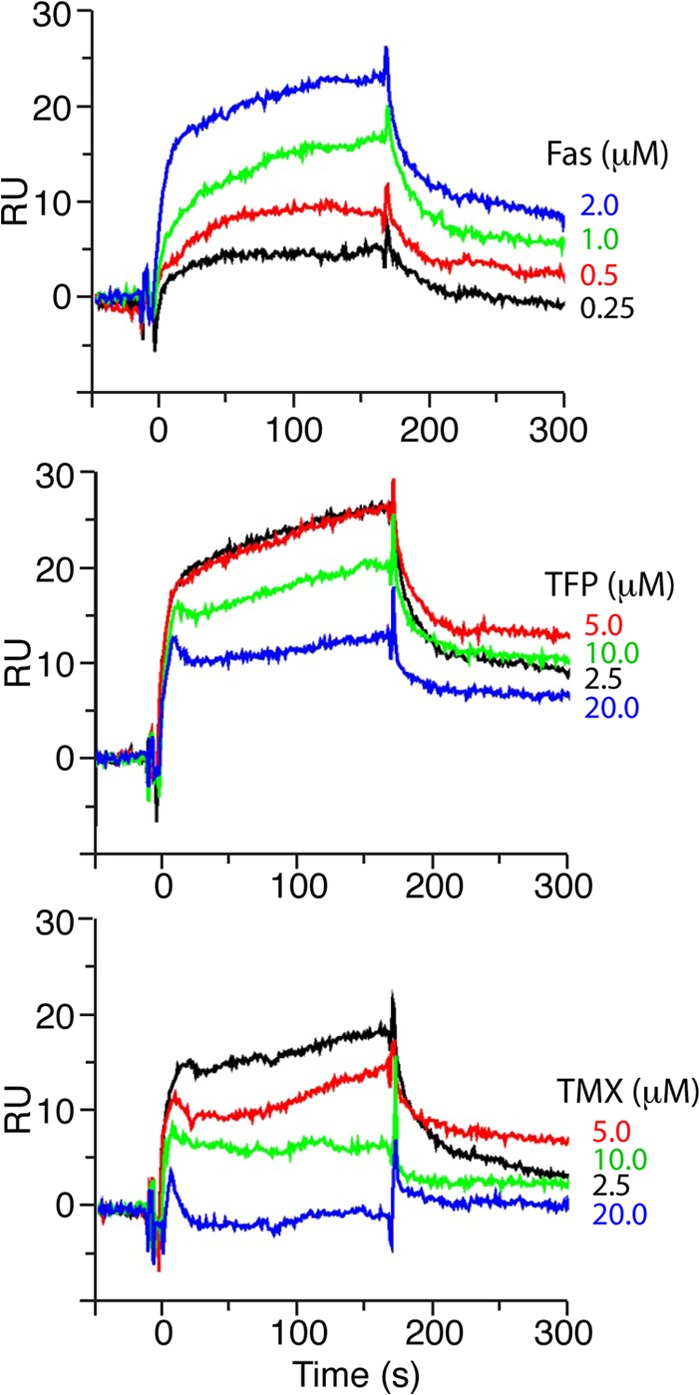
Effect of antagonists on CaM-Faswt interactions as detected by SPR methods. CaM was immobilized on CM5 sensor chip. Upper panel, shown is Faswt at increased concentrations (0.25–2 μm). The apparent Kd value for CaM binding to Faswt is 0.9 μm, a value that is similar to that obtained by ITC (1.7 μm). Middle and lower panels, Faswt (2 μm) with increased concentrations of TFP or TMX (2.5–20 μm) in HBS-P buffer containing 5 mm CaCl2 or 5 mm EGTA were injected for 3 min (20 or 15 μl/min, respectively). Experiments were collected in duplicate. Binding curves were obtained by subtraction of reference flow cells. RU, response units.
DISCUSSION
It is established that the Fas receptor undergoes major conformational changes upon binding to FasL, allowing its cytoplasmic DD to interact with the DD of FADD (5–8). Fas-FasL engagement initiates a cascade of downstream interactions that lead to activation of caspases. In the extrinsic apoptotic pathway, Fas-FasL interaction leads to formation of DISC, a prerequisite for activation of caspases 8, 3, 6, and 7 (8–10). There is growing evidence that CaM interacts with FasDD upon recruitment into DISC in cholangiocarcinoma cells, suggesting a novel role of CaM in Fas-mediated apoptosis (11–17). In this report we have sought to identify the molecular requirements for Fas-CaM interaction and the mechanism of inhibition using small molecule CaM antagonists. We have shown that (i) CaM binds to FasDD with an apparent Kd of ∼2 μm (0.9 μm by SPR methods) and 2:1 CaM:FasDD stoichiometry, (ii) CaM interacts with FasDD D244A mutant in an identical manner to WT FasDD, (iii) CaM appears to induce a conformational change in FasDD, (iv) the interaction between CaM and FasDD is entropically driven, (v) both the N- and C-terminal lobes of CaM are important for FasDD binding, and (vi) CaM antagonists block and interfere with binding of FasDD.
Typically, the CaM binding domain of target proteins is a short peptide (15–20 residues) that is hydrophobic-basic in nature and has the propensity to form an α-helix (22). In many of the classical CaM binding targets, hydrophobic residues usually occupy conserved positions at 1-5-10 or 1-8-14, which point to one face of the helix (22). Although these patterns are typically found in many CaM-binding proteins, other unclassified examples were also identified (22). Perhaps one of the most surprising results is the finding that two CaM molecules bind to FasDD. Analysis of the protein sequence using a web-based tool provides scores from 0 to 9 based on multiple criteria including hydropathy, α-helical propensity, residue weight, residue charge, hydrophobic residue content, helical class, and occurrence of particular residues (59). The most likely binding site in a protein sequence is given a score of repeated 9s. Analysis of the FasDD protein sequence using this method yielded the highest score (8–9) for residues 224–238 (MTLSQVKGFVRKNGV), which form helix α2 and a short loop. This region contains a classical 1-5-10 motif (underlined residues). A second potential CaM-binding site with a repeated score of 7 has also been detected for residues 283–297 (KKANLCTLAEKIQTI), which are localized in α6 and a preceding loop. This region does not contain any of the known CaM binding motifs. Based on mutagenesis studies, previous studies suggested that the CaM-binding site in FasDD is located within helices α1 and α2 and the connecting loop (Fig. 1) (13, 60). Co-immunoprecipitation studies conducted on truncated Fas constructs also suggested a single CaM binding domain (13). However, NMR, ITC, gel filtration, and analytical ultracentrifugation data described above indicate a 2:1 CaM:FasDD model. The contradiction between the results described here and the previous mutagenesis and co-immunoprecipitation data is probably a consequence of using different methods. One possible explanation for the 2:1 model is the induction of CaM dimerization upon binding to FasDD. Non-covalent dimerization of CaM has been observed in many cases (61–63). FasDD-induced CaM dimerization, however, is ruled out based on the HSQC NMR data obtained for CaM, which revealed only one set of 1H,15N resonances upon titration of Fasm (Fig. 6). Thus, the most likely scenario is that CaM binds to two regions on FasDD. The precise binding mode of CaM to FasDD is under investigation.
Mutations that may alter the structure and/or function of Fas have been detected in many types of cancer (30, 31). In particular, mutation of valine 238 to asparagine (V238N, named V254N in other studies; Refs. 13 and 32) is analogous to the identified mutant allele of Fas in lpr-autoimmune mice that have a deficiency in Fas-mediated apoptosis (33). Immunoprecipitation data have shown that Fas V238N mutant has a lower binding affinity to CaM (13). Val-238, which is located at the end of helix α2, is an essential hydrophobic residue in the 1-5-10 motif and is probably involved in binding to CaM. In a previous NMR study, it was reported that V238N mutation caused significant chemical shift changes in 1H,15N resonances, suggesting a significant change in the FasDD structure (29). Although it has yet to be elucidated whether the effect of V238N mutant on CaM-Fas interactions is direct or allosteric, analysis of the FasDD protein sequence with V238N mutation using the web-based tool yielded a repeated score of 4 for residues 224–237 (MTLSQVKGFVRKNG) and a score of 9 for residues 283–297 (KKANLCTLAEKIQTI). These results may suggest that CaM may still bind to Fas V238N but with either a lower affinity or an altered stoichiometry. Studies are currently focused on understanding, at the structural level, the effect of V238N mutation on FasDD structure and CaM binding properties.
The CaM protein adopts a collapsed structure when bound to peptides with a 1-5-10 motif (24). Previous molecular modeling studies suggested a collapsed structure for CaM when bound to helix α2 of FasDD (13). As shown in the three-dimensional structure of FasDD (Fig. 1), helix α2 is packed against helices α3, α4, and α5. The accessibility of helix α2 of FasDD to generate a collapsed structure of CaM probably requires protein unfolding. This unfolding event also releases helix α6 and make it accessible to CaM. The ability of CaM to disrupt protein targets has been recently observed in studies of the interactions between CaM and the human immunodeficiency virus type-1 matrix protein (37, 45, 64). CaM-induced unfolding of protein targets is not unusual. Partial unfolding is considered critical for the biological function of target proteins as in enzymatically driven cleavage reactions (65).
Recent structural analysis of the FasDD and CaM proteins has revealed interesting and unexpected findings (66). Despite the absence of sequence similarity, FasDD and CaM share significant structural features. During a signaling event and upon binding to target proteins, CaM undergoes a conformational change involving bending of the central helix to form a closed form (21, 22). On the other hand, FasDD undergoes a transition from a closed state to open state when it is bound to FADD (8). Thus, at the resting state CaM and FasDD are in open and closed conformations, respectively. It was hypothesized that both proteins possess a “designed mobility” or instability (66). This molecular effect is governed by a capability of the intermolecular interactions to dominate in a complex and disrupt the original structure, causing a structural transformation that leads to a signaling event that is frequently associated with a hinge motion. Thus, binding of CaM to FasDD may involve a conformational switch upon recruitment to DISC.
The current findings provide new insights into understanding the precise molecular mechanism of CaM-Fas interaction and will help in identification of the functional role of CaM in Fas-mediated apoptosis. CaM is known as a primary transducer of Ca2+-dependent signals. Elevated levels of CaM have been linked to diseases characterized by pathological, unregulated cell growth such as cancer (19). CaM antagonists have long been known as anti-proliferative agents (67) and have been identified as inhibitors of tumor cell invasion in vitro (35) and metastasis in vivo (36). We have shown that CaM antagonists (TFP, W7, and TMX) inhibit binding of FasDD, providing a potential molecular basis for the role of these antagonists in inducing Fas-mediated apoptosis in cholangiocarcinoma and Jurkat cells (12, 14, 34). Previous structural studies have shown that binding of TFP and W7 not only block the hydrophobic lobe(s) that are important for protein/peptide binding but also induce structural changes in CaM and likely alter the binding interface. Combined with interferon (IFN-γ), CaM antagonists have been also been shown to induce apoptosis in Fas-low apoptosis-resistant cholangiocarcinoma cells (12). CaM antagonists can also inhibit interactions with other DISC components such as FLICE-like inhibitory protein (FLIP). Overexpression of FLIP lacking the CaM binding region increased both spontaneous and Fas-stimulated apoptosis in cholangiocarcinoma cells, suggesting that FLIP-CaM interaction is important in mediating survival signals by FLIP (11, 14, 15, 60, 68).
In summary, we have provided new insights on the molecular determinants of Fas-CaM interactions and mechanism of inhibition. Molecular characterization of signaling pathways emanating from Fas is critical for identifying novel molecular targets that are crucial in switching between the death versus survival signals in response to the same ligand, which may help in the development of new anticancer therapeutic strategies.
Acknowledgments
We thank members of our laboratories and Dr. Jay McDonald (University of Alabama at Birmingham) for helpful discussions. We thank Diego Esposito and Paul Driscoll (Medical Research Council National Institute for Medical Research, United Kingdom) for providing NMR signal assignments of FasDD. We thank the University of Alabama Comprehensive Cancer Center X-ray core facility, funded by NCI, National Institutes of Health (NIH) Grant P30CA13148, which houses the Auto-ITC200, acquired through NIH Grant 1S10RR026478.
This work was supported in part by a pilot grant from the Comprehensive Cancer Center (University of Alabama at Birmingham).
- DD
- death domain
- FADD
- Fas-associated death domain
- CaM
- calmodulin
- DISC
- death-inducing signaling complex
- HSQC
- heteronuclear single quantum coherence
- ITC
- isothermal titration calorimetry
- SPR
- surface plasmon resonance
- W7
- N-(6-aminohexyl)-5-chloro-1-naphthalene sulfonamide
- TFP
- trifluoperazine
- TMX
- tamoxifen
- Fasm
- monomeric Fas
- FLIP
- FLICE-like inhibitory protein.
REFERENCES
- 1. Elmore S. (2007) Apoptosis. A review of programmed cell death. Toxicol Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soini Y., Pääkkö P., Lehto V. P. (1998) Histopathological evaluation of apoptosis in cancer. Am. J. Pathol. 153, 1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itoh N., Yonehara S., Ishii A., Yonehara M., Mizushima S., Sameshima M., Hase A., Seto Y., Nagata S. (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66, 233–243 [DOI] [PubMed] [Google Scholar]
- 4. Peter M. E., Scaffidi C., Medema J. P., Kischkel F., Krammer P. H. (1999) The death receptors. Results Probl. Cell Differ. 23, 25–63 [DOI] [PubMed] [Google Scholar]
- 5. Daniel P. T., Wieder T., Sturm I., Schulze-Osthoff K. (2001) The kiss of death. Promises and failures of death receptors and ligands in cancer therapy. Leukemia 15, 1022–1032 [DOI] [PubMed] [Google Scholar]
- 6. Marsters S. A., Sheridan J. P., Donahue C. J., Pitti R. M., Gray C. L., Goddard A. D., Bauer K. D., Ashkenazi A. (1996) Apo-3, a new member of the tumor necrosis factor receptor family, contains a death domain and activates apoptosis and NF-κB. Curr. Biol. 6, 1669–1676 [DOI] [PubMed] [Google Scholar]
- 7. Chinnaiyan A. M., O'Rourke K., Tewari M., Dixit V. M. (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81, 505–512 [DOI] [PubMed] [Google Scholar]
- 8. Scott F. L., Stec B., Pop C., Dobaczewska M. K., Lee J. J., Monosov E., Robinson H., Salvesen G. S., Schwarzenbacher R., Riedl S. J. (2009) The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 457, 1019–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bouillet P., O'Reilly L. A. (2009) CD95, BIM, and T cell homeostasis. Nat. Rev. Immunol. 9, 514–519 [DOI] [PubMed] [Google Scholar]
- 10. Wang L., Yang J. K., Kabaleeswaran V., Rice A. J., Cruz A. C., Park A. Y., Yin Q., Damko E., Jang S. B., Raunser S., Robinson C. V., Siegel R. M., Walz T., Wu H. (2010) The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat. Struct. Mol. Biol. 17, 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y., Pawar P., Pan G., Ma L., Liu H., McDonald J. M. (2008) Calmodulin binding to the Fas-mediated death-inducing signaling complex in cholangiocarcinoma cells. J. Cell. Biochem. 103, 788–799 [DOI] [PubMed] [Google Scholar]
- 12. Ahn E.-Y., Pan G., Oh J. H., Tytler E. M., McDonald J. M. (2003) The combination of calmodulin antagonists and interferon-γ induces apoptosis through caspase-dependent and -independent pathways in cholangiocarcinoma cells. Am. J. Pathol. 163, 2053–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahn E. Y., Lim S. T., Cook W. J., McDonald J. M. (2004) Calmodulin binding to the Fas death domain. Regulation by Fas activation. J. Biol. Chem. 279, 5661–5666 [DOI] [PubMed] [Google Scholar]
- 14. Pawar P., Ma L., Byon C. H., Liu H., Ahn E. Y., Jhala N., Arnoletti J. P., McDonald J. M., Chen Y. (2009) Molecular mechanisms of tamoxifen therapy for cholangiocarcinoma. Role of calmodulin. Clin. Cancer res. 15, 1288–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pawar P. S., Micoli K. J., Ding H., Cook W. J., Kappes J. C., Chen Y., McDonald J. M. (2008) Calmodulin binding to cellular FLICE-like inhibitory protein modulates Fas-induced signalling. Biochem. J. 412, 459–468 [DOI] [PubMed] [Google Scholar]
- 16. Que F. G., Phan V. A., Phan V. H., Celli A., Batts K., LaRusso N. F., Gores G. J. (1999) Cholangiocarcinomas express Fas ligand and disable the Fas receptor. Hepatology 30, 1398–1404 [DOI] [PubMed] [Google Scholar]
- 17. Pan G., Vickers S. M., Pickens A., Phillips J. O., Ying W., Thompson J. A., Siegal G. P., McDonald J. M. (1999) Apoptosis and tumorigenesis in human cholangiocarcinoma cells. Involvement of Fas/APO-1 (CD95) and calmodulin. Am. J. Pathol. 155, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yuan K., Jing G., Chen J., Liu H., Zhang K., Li Y., Wu H., McDonald J. M., Chen Y. (2011) Calmodulin mediates Fas-induced FADD-independent survival signaling in pancreatic cancer cells via activation of Src-extracellular signal-regulated kinase (ERK). J. Biol. Chem. 286, 24776–24784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wei J. W., Morris H. P., Hickie R. A. (1982) Positive correlation between calmodulin content and hepatoma growth rates. Cancer Res. 42, 2571–2574 [PubMed] [Google Scholar]
- 20. Chin D., Means A. R. (2000) Calmodulin. A prototypical calcium sensor. Trends Cell Biol. 10, 322–328 [DOI] [PubMed] [Google Scholar]
- 21. Hoeflich K. P., Ikura M. (2002) Calmodulin in action. Diversity in target recognition and activation mechanisms. Cell 108, 739–742 [DOI] [PubMed] [Google Scholar]
- 22. Ishida H., Vogel H. J. (2006) Protein-peptide interaction studies demonstrate the versatility of calmodulin target protein binding. Protein Pept. Lett. 13, 455–465 [DOI] [PubMed] [Google Scholar]
- 23. Osawa M., Tokumitsu H., Swindells M. B., Kurihara H., Orita M., Shibanuma T., Furuya T., Ikura M. (1999) A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase kinase. Nat. Struct. Biol. 6, 819–824 [DOI] [PubMed] [Google Scholar]
- 24. Vetter S. W., Leclerc E. (2003) Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem. 270, 404–414 [DOI] [PubMed] [Google Scholar]
- 25. Yamniuk A. P., Vogel H. J. (2004) Calmodulin's flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 27, 33–57 [DOI] [PubMed] [Google Scholar]
- 26. Kretsinger R. H. (1996) EF-hands reach out. Nat. Struct. Biol. 3, 12–15 [DOI] [PubMed] [Google Scholar]
- 27. Moorthy A. K., Murthy M. (2001) Conformation and structural transitions in the EF-hands of calmodulin. J. Biomol. Struct. Dyn. 19, 47–57 [DOI] [PubMed] [Google Scholar]
- 28. Yap K. L., Ames J. B., Swindells M. B., Ikura M. (1999) Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins 37, 499–507 [DOI] [PubMed] [Google Scholar]
- 29. Huang B., Eberstadt M., Olejniczak E. T., Meadows R. P., Fesik S. W. (1996) NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384, 638–641 [DOI] [PubMed] [Google Scholar]
- 30. Park W. S., Oh R. R., Kim Y. S., Park J. Y., Lee S. H., Shin M. S., Kim S. Y., Kim P. J., Lee H. K., Yoo N. Y., Lee J. Y. (2001) Somatic mutations in the death domain of the Fas (apo-1/CD95) gene in gastric cancer. J. Pathol. 193, 162–168 [DOI] [PubMed] [Google Scholar]
- 31. Takakuwa T., Dong Z., Takayama H., Matsuzuka F., Nagata S., Aozasa K. (2001) Frequent mutations of Fas gene in thyroid lymphoma. Cancer Res. 61, 1382–1385 [PubMed] [Google Scholar]
- 32. Suever J. D., Chen Y., McDonald J. M., Song Y. (2008) Conformation and free energy analyses of the complex of calcium-bound calmodulin and the Fas death domain. Biophys. J. 95, 5913–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watanabe-Fukunaga R., Brannan C. I., Copeland N. G., Jenkins N. A., Nagata S. (1992) Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356, 314–317 [DOI] [PubMed] [Google Scholar]
- 34. Vickers S. M., Jhala N. C., Ahn E. Y., McDonald J. M., Pan G., Bland K. I. (2002) Tamoxifen (TMX)/Fas induced growth inhibition of human cholangiocarcinoma (HCC) by γ interferon (IFN-γ). Ann. Surg. 235, 872–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dewhurst L. O., Gee J. W., Rennie I. G., MacNeil S. (1997) Tamoxifen, 17β-oestradiol and the calmodulin antagonist J8 inhibit human melanoma cell invasion through fibronectin. Br. J. Cancer 75, 860–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ito H., Wang J. Z., Shimura K. (1991) Inhibition of lung metastasis by a calmodulin antagonist, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W-7), in mice bearing Lewis lung carcinoma. Anticancer Res. 11, 249–252 [PubMed] [Google Scholar]
- 37. Samal A. B., Ghanam R. H., Fernandez T. F., Monroe E. B., Saad J. S. (2011) NMR, biophysical, and biochemical studies reveal the minimal calmodulin-binding domain of the HIV-1 matrix protein. J. Biol. Chem. 286, 33533–33543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lebowitz J., Lewis M. S., Schuck P. (2002) Modern analytical ultracentrifugation in protein science. A tutorial review. Protein. Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schuck P. (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 41. Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation. Strategies and application to model systems. Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 43. Johnson B. A., Blevins R. A. (1994) NMRview. A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 44. Ikura M., Kay L. E., Bax A. (1990) A novel approach for sequential assignment of 1H, 13C, and 15N spectra of larger proteins. Heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 29, 4659–4667 [DOI] [PubMed] [Google Scholar]
- 45. Ghanam R. H., Fernandez T. F., Fledderman E. L., Saad J. S. (2010) Binding of calmodulin to the HIV-1 matrix protein triggers myristate exposure. J. Biol. Chem. 285, 41911–41920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, New York [Google Scholar]
- 47. Kay L. E., Clore G. M., Bax A., Gronenborn A. M. (1990) Four-dimensional heteronuclear triple-resonance NMR spectroscopy of interleukin-1β in solution. Science 249, 411–414 [DOI] [PubMed] [Google Scholar]
- 48. Esposito D., Sankar A., Morgner N., Robinson C. V., Rittinger K., Driscoll P. C. (2010) Solution NMR investigation of the CD95/FADD homotypic death domain complex suggests lack of engagement of the CD95 C terminus. Structure 18, 1378–1390 [DOI] [PubMed] [Google Scholar]
- 49. Ferguson B. J., Esposito D., Jovanović J., Sankar A., Driscoll P. C., Mehmet H. (2007) Biophysical and cell-based evidence for differential interactions between the death domains of CD95/Fas and FADD. Cell Death Differ. 14, 1717–1719 [DOI] [PubMed] [Google Scholar]
- 50. Majava V., Kursula P. (2009) Domain swapping and different oligomeric states for the complex between calmodulin and the calmodulin-binding domain of calcineurin A. PLoS ONE 4, e5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ikura M., Clore G. M., Gronenborn A. M., Zhu G., Klee C. B., Bax A. (1992) Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 256, 632–638 [DOI] [PubMed] [Google Scholar]
- 52. Meador W. E., Means A. R., Quiocho F. A. (1992) Target enzyme recognition by calmodulin. 2.4 Å structure of a calmodulin-peptide complex. Science 257, 1251–1255 [DOI] [PubMed] [Google Scholar]
- 53. Osawa M., Swindells M. B., Tanikawa J., Tanaka T., Mase T., Furuya T., Ikura M. (1998) Solution structure of calmodulin-W-7 complex. The basis of diversity in molecular recognition. J. Mol. Biol. 276, 165–176 [DOI] [PubMed] [Google Scholar]
- 54. Cook W. J., Walter L. J., Walter M. R. (1994) Drug binding by calmodulin. Crystal structure of a calmodulin-trifluoperazine complex. Biochemistry 33, 15259–15265 [DOI] [PubMed] [Google Scholar]
- 55. Vertessy B. G., Harmat V., Böcskei Z., Náray-Szabó G., Orosz F., Ovádi J. (1998) Simultaneous binding of drugs with different chemical structures to Ca2+-calmodulin. Crystallographic and spectroscopic studies. Biochemistry 37, 15300–15310 [DOI] [PubMed] [Google Scholar]
- 56. Vandonselaar M., Hickie R. A., Quail J. W., Delbaere L. T. (1994) Trifluoperazine-induced conformational change in Ca2+-calmodulin. Nat. Struct. Biol. 1, 795–801 [DOI] [PubMed] [Google Scholar]
- 57. Feldkamp M. D., O'Donnell S. E., Yu L., Shea M. A. (2010) Allosteric effects of the antipsychotic drug trifluoperazine on the energetics of calcium binding by calmodulin. Proteins 78, 2265–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu Z., Johnson K. W., Choi Y., Ciardelli T. L. (1995) Ligand binding analysis of soluble interleukin-2 receptor complexes by surface plasmon resonance. J. Biol. Chem. 270, 16045–16051 [DOI] [PubMed] [Google Scholar]
- 59. Yap K. L., Kim J., Truong K., Sherman M., Yuan T., Ikura M. (2000) Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14 [DOI] [PubMed] [Google Scholar]
- 60. Chen Y., Xu J., Jhala N., Pawar P., Zhu Z. B., Ma L., Byon C. H., McDonald J. M. (2006) Fas-mediated apoptosis in cholangiocarcinoma cells is enhanced by 3,3′-diindolylmethane through inhibition of AKT signaling and FLICE-like inhibitory protein. Am. J. Pathol. 169, 1833–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Larsson G., Schleucher J., Onions J., Hermann S., Grundström T., Wijmenga S. S. (2005) Backbone dynamics of a symmetric calmodulin dimer in complex with the calmodulin-binding domain of the basic-helix-loop-helix transcription factor SEF2–1/E2–2. A highly dynamic complex. Biophys. J. 89, 1214–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yuan T., Vogel H. J. (1998) Calcium-calmodulin-induced dimerization of the carboxyl-terminal domain from petunia glutamate decarboxylase. A novel calmodulin-peptide interaction motif. J. Biol. Chem. 273, 30328–30335 [DOI] [PubMed] [Google Scholar]
- 63. Lafitte D., Heck A. J., Hill T. J., Jumel K., Harding S. E., Derrick P. J. (1999) Evidence of noncovalent dimerization of calmodulin. Eur. J. Biochem. 261, 337–344 [DOI] [PubMed] [Google Scholar]
- 64. Chow J. Y., Jeffries C. M., Kwan A. H., Guss J. M., Trewhella J. (2010) Calmodulin disrupts the structure of the HIV-1 MA protein. J. Mol. Biol. 400, 702–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gietzen K., Sadorf I., Bader H. (1982) A model for the regulation of the calmodulin-dependent enzymes erythrocyte Ca2+-transport ATPase and brain phosphodiesterase by activators and inhibitors. Biochem. J 207, 541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stec B. (2012) Hinge sequences as signaling agents? FEBS Lett. 586, 1675–1677 [DOI] [PubMed] [Google Scholar]
- 67. Rasmussen C. D., Lu K. P., Means R. L., Means A. R. (1992) Calmodulin and cell cycle control. J. Physiol. Paris 86, 83–88 [DOI] [PubMed] [Google Scholar]
- 68. Jing G., Yuan K., Liang Q., Sun Y., Mao X., McDonald J. M., Chen Y. (2012) Reduced CaM/FLIP binding by a single point mutation in c-FLIP(L) modulates Fas-mediated apoptosis and decreases tumorigenesis. Lab. Invest. 92, 82–90 [DOI] [PubMed] [Google Scholar]