

Background: Pin1 regulates several signaling proteins by isomerizing the cis/trans conformation of the Ser(P)-Pro peptide bond.
Results: Pin1 is up-regulated in dopaminergic neurons in cell culture, animal models, and human PD brains. Pin1 inhibition protects dopaminergic neurons in PD models.
Conclusion: Pin1 up-regulation plays a proapoptotic function in PD.
Significance: Pin1 inhibition may be a viable translational strategy in PD.
Keywords: Dopamine, Neurobiology, Neurodegenerative Diseases, Neuroprotection, Parkinson Disease, MPTP, Pin1, Caspase-3, Juglone
Abstract
Parkinson disease (PD) is a chronic neurodegenerative disease characterized by a slow and progressive degeneration of dopaminergic neurons in substantia nigra. The pathophysiological mechanisms underlying PD remain unclear. Pin1, a major peptidyl-prolyl isomerase, has recently been associated with certain diseases. Notably, Ryo et al. (Ryo, A., Togo, T., Nakai, T., Hirai, A., Nishi, M., Yamaguchi, A., Suzuki, K., Hirayasu, Y., Kobayashi, H., Perrem, K., Liou, Y. C., and Aoki, I. (2006) J. Biol. Chem. 281, 4117–4125) implicated Pin1 in PD pathology. Therefore, we sought to systematically characterize the role of Pin1 in PD using cell culture and animal models. To our surprise we observed a dramatic up-regulation of Pin1 mRNA and protein levels in dopaminergic MN9D neuronal cells treated with the parkinsonian toxicant 1-methyl-4-phenylpyridinium (MPP+) as well as in the substantia nigra of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model. Notably, a marked expression of Pin1 was also observed in the substantia nigra of human PD brains along with a high co-localization of Pin1 within dopaminergic neurons. In functional studies, siRNA-mediated knockdown of Pin1 almost completely prevented MPP+-induced caspase-3 activation and DNA fragmentation, indicating that Pin1 plays a proapoptotic role. Interestingly, multiple pharmacological Pin1 inhibitors, including juglone, attenuated MPP+-induced Pin1 up-regulation, α-synuclein aggregation, caspase-3 activation, and cell death. Furthermore, juglone treatment in the MPTP mouse model of PD suppressed Pin1 levels and improved locomotor deficits, dopamine depletion, and nigral dopaminergic neuronal loss. Collectively, our findings demonstrate for the first time that Pin1 is up-regulated in PD and has a pathophysiological role in the nigrostriatal dopaminergic system and suggest that modulation of Pin1 levels may be a useful translational therapeutic strategy in PD.
Introduction
Parkinson disease (PD)4 is a chronic and progressive neurodegenerative movement disorder affecting >1% of the population over 65 years of age (1). Pathologically, the disease is characterized by gliosis, progressive loss of dopaminergic neurons and their terminals in the nigrostriatal axis, and appearance of cytoplasmic inclusions known as Lewy bodies in the surviving neurons of substantia nigra (SN) (2–4). In recent years the occurrence of non-dopaminergic neuronal loss and non-motor symptoms has been recognized in PD pathology (5, 6).
Reversible phosphorylation on Ser/Thr-Pro motifs regulated by proline-directed protein kinases and phosphatases is an important molecular switch in controlling various cellular processes (7, 8). Proline-directed kinases and some of the Ser/Thr phosphatases phosphorylate or dephosphorylate such motifs to regulate cellular signaling (9). Pin1 is unique among the peptidyl-prolyl isomerases because it specifically recognizes phosphorylated serine or threonine residues immediately preceding proline (Ser(P)/Thr-Pro) in a subset of proteins and isomerizes the cis/trans conformation of the peptide bond (10, 11). Several studies have shown that Pin1-mediated conformational regulation can have a profound impact on the regulation of cell growth, stress responses, immune function, germ cell development, neuronal differentiation, and survival (12, 13). Dysregulation of Pin1 signaling is implicated in some pathological conditions such as Alzheimer disease (14, 15), asthma (16, 17), corticobasal degeneration (18, 19), and cancer (20). Significant expression of Pin1 in terminally differentiated and post-mitotic neurons suggests that it may play an important role in the nervous system (21, 22). Pin1 interacts with mitochondrial BH3-only protein BIMEL and activates c-Jun to regulate the apoptotic machinery (23). Interestingly, Pin1 has been shown to be present in Lewy bodies in PD patients and is known to facilitate the formation of α-synuclein inclusions in a cellular model of α-synuclein aggregation (24). Recently, we reported that mixed-lineage kinase 3 (MLK3) phosphorylates Pin1 to regulate its nuclear translocation and function (25). Because the role of Pin1 has not been explored in Parkinson disease, herein we systematically characterized the role of Pin 1 in PD using cell culture, animal models, and postmortem human PD brains.
Surprisingly, we found that Pin1 is highly up-regulated in cell culture and animal models of PD as well as in human PD brains. Consistent with these data, Pin1 functions as a proapoptotic factor in degeneration of dopaminergic neurons because knockdown of Pin1 attenuates apoptotic events in cell culture models of PD. Inhibition of Pin1 function with the pharmacological inhibitors juglone, PiB, or cyclic peptide inhibitor F also abolished MPP+-induced Pin1 expression in a cellular model of PD. Notably, juglone treatment attenuated Pin1 expression and protected the nigrostriatal axis in a preclinical mouse model of PD.
EXPERIMENTAL PROCEDURES
Chemicals and Biological Reagents
1-Methyl-4-phenyl tetrahydropteridine (MPP+ iodide), Pin1 inhibitor PiB, and MPTP-HCl were purchased from Sigma. Pin1 inhibitor juglone was purchased from Calbiochem. Caspase substrate (Ac-DEVD-aminofluoromethylcoumarin) was obtained from Bachem Biosciences (King of Prussia, PA). Bradford protein assay reagent was purchased from Bio-Rad. Neurobasal medium, RPMI 1640 medium, hygromycin B, B27 supplement, fetal bovine serum, l-glutamine, penicillin, and streptomycin were purchased from Invitrogen. The Pin1-cyclic peptide inhibitor (peptide inhibitor F, sequence cyclo(d-Arg-d-Arg-d-Thr(P)-Pip-Nal-Arg-Gln), where Pip is l-piperidine-2-carboxylic acid and Nal is l-2-naphthylalanine) was kindly provided by Dr. Pei Dehua (Ohio State University) and generated as described previously (26).
Cell Culture
The MN9D dopaminergic cell line originates from fusion of rostral mesencephalic neurons from embryonic C57BL/BJ (embryonic day 14 mice) with N18TG2 neuroblastoma cells (27). MN9D cells were grown in a high glucose (4500 mg/liter) Dulbecco's modified Eagle's medium (Sigma) containing 10% Tet-approved fetal bovine serum (Invitrogen), 3.7 g liter−1 NaHCO3 and 4 mm l-glutamine in a 5% CO2 atmosphere at 37 °C. The human wild-type α-synuclein or empty vector stably transfected N27 rat dopaminergic neuronal cells were cultured in 200 μg/ml hygromycin added to N27 growth media, as described previously (28).
Treatment Paradigm
MPP+ (300 μm) was added to the cells in the presence or absence of juglone (1 μm), PiB (5 μm), or peptide inhibitor F (5, 10, and 15 μm) for the duration of the experiment. The cells were detached from the flask using a rubber policeman and centrifuged at 200 × g for 5 min, washed with ice-cold PBS twice, and homogenized with radioimmunoprecipitation assay buffer. Cell lysates, collected by spinning down the cell fragments at 20,000 × g for 45 min at 4 °C, were used to determine changes to Pin1 expression.
siRNA Transfection
MN9D cells were plated in 6-well tissue culture plates at 70–75% confluency. The Pin1-specific siRNA (SI01378923) was purchased from Qiagen (Valencia, CA), and scrambled negative control siRNA (4611) was obtained from Ambion (Austin, TX). Transfections were performed using LipofectamineTM 2000 (Invitrogen) according to the manufacturer's instructions. Four to six hours after transfection media were replaced with fresh growth media. The following day cells were treated with MPP+ or vehicle and processed for further experiments.
Quantitative Real-time-PCR
After treatment total RNA was extracted from the MN9D cells or the mouse brains using an RNA Miniprep Kit (Agilent Technologies, Santa Clara, CA) following the manufacturer's protocol. Total RNA was treated with DNase I to remove DNA contamination and then reversibly transcribed into first-strand cDNA using the SuperScript III first-strand synthesis system (Invitrogen) as described in the kit instructions. SYBR Green quantitative PCR was performed with validated primers of Pin1 and control β-actin primers (Qiagen) using either FastStart Universal SYBR Green master (Rox) (Roche Applied Science) for the MN9D cells or RT2 SYBR® Green qPCR Master Mix (SABiosciences, Frederick, MD) for the mouse tissue.
Sytox Cell Death Assay
The MN9D cells were incubated with 300 μm MPP+ for up to 24 h in the presence or absence of 1 μm juglone, and cell death was determined using cell impermeable Sytox green (Molecular Probes, Eugene, OR). Sytox green intercalates with the DNA in the membrane-compromised cells to produce a green fluorescence that is quantifiable at excitation 485 nm and emission 538 nm using the fluorescence microplate system (Synergy 4, Biotek, Winooski, VT). The fluorescence can also be viewed under a fluorescence microscope. Fluorescence and phase contrast were taken after exposure to MPP+ with a NIKON TE2000 microscope, and pictures were captured with a SPOT digital camera.
Caspase-3 Activity Assay
Caspase-3 activity was determined as previously described (29). After the MPP+ treatment, cells were lysed and centrifuged, and the resulting supernatants were incubated with 50 μm Ac-DEVD-amidomethylcoumarin (a caspase-3 substrate) at 37 °C for 1 h, and caspase activity was measured by fluorescence plate reader (Synergy 4, Biotek) with excitation at 380 nm and emission at 460 nm.
DNA Fragmentation Assay
DNA fragmentation was measured using a Cell Death Detection ELISA Plus Assay kit as described previously (30). After MPP+ treatment, cells were centrifuged and washed with PBS once. DNA fragmentation was measured in the cell lysates according to the manufacturer's protocol. The absorbance of the ELISA reaction was measured at 490 and 405 nm using a microplate reader (SpectraMAX 190, Molecular Devices Corp., Sunnyvale, CA).
High Affinity [3H]Dopamine Uptake Assay
Cells in each well were washed twice with 1 ml of Krebs-Ringer buffer (16 mm NaH2PO4, 16 mm Na2HPO4, 119 mm NaCl, 4.7 mm KCl, 1.8 mm CaCl2, 1.2 mm MgSO4, 1.3 mm EDTA, and 5.6 mm glucose, pH 7.4). The cells were then incubated with 25 nm [3H]dopamine in Krebs-Ringer buffer (0.4 ml/well) for 20 min at 37 °C. Nonspecific uptake of dopamine was determined in parallel wells incubated with tritiated dopamine and 10 μm mazindol, an inhibitor of neuronal dopamine uptake. Briefly, the cells were washed 3 times with ice-cold Krebs-Ringer buffer (1 ml/well) and lysed with 1 n NaOH (0.5 ml/well). After washing, the lysates were mixed with 5 ml of scintillation fluid (Scintiverse BD), and radioactivity was determined with a liquid scintillation counter (Tri-Carb 1600 TR; Packard, Meriden, CT). The specific dopaminergic uptake was calculated by subtracting the amount of radioactivity observed in the presence of mazindol from that observed in the absence of mazindol.
Mesencephalic Primary Neuron Cultures and Treatment
Primary mesencephalic neuronal culture was prepared from the ventral mesencephalon of gestational 14–15-day-old mice embryos as described previously (31). Briefly, mesencephalic and striatal tissues from E14 to E15 mouse embryos were dissected and maintained in ice-cold calcium-free Hanks' balanced salt solution and then dissociated in Hanks' balanced salt solution containing trypsin, 0.25% EDTA for 20 min at 37 °C. The dissociated cells were then plated at equal density of 0.6 million cells per well on 12-mm coverslips precoated with 0.1 mg/ml poly-d-lysine. Cultures were maintained in neurobasal medium fortified with B-27 supplement, 500 mm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). The cells were maintained in a humidified CO2 incubator (5% CO2 and 37 °C) for 24 h. Half of the culture medium was replaced every 2 days. Approximately 6–7-day-old cultures were used for experiments. Primary mesencephalic and striatal dopaminergic neuronal cells were exposed to 10 μm MPP+ in the presence or absence of juglone (3 μm) for 24 h.
Immunocytochemistry
The primary mesencephalic neurons were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min and processed for immunocytochemical staining. First, nonspecific sites were blocked with 2% bovine serum albumin, 0.5% Triton X-100, and 0.05% Tween 20 in PBS for 45 min at room temperature. Cells were then incubated with different primary antibodies such as anti-TH (1:1600, mouse monoclonal; Millipore, Billerica, MA) and Pin1 (1:400, goat polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA) at 4 °C overnight. Appropriate secondary antibodies (Alexa Fluor 488 or 594 or 555; Invitrogen) were used followed by incubation with 10 μg/ml Hoechst 33342 (Invitrogen) for 5 min at room temperature to stain the nucleus. The coverslip-containing stained cells were washed twice with PBS and mounted on poly-D lysine-coated slides (Sigma). Cells were viewed under a NIKON inverted fluorescence microscope (model TE-2000U; NIKON, Tokyo, Japan), and images were captured with a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI).
For immunostaining of α-synuclein aggregates in α-synuclein stably expressing and vector control N27 cells, cells grown on coverslips in media containing 0.2% FBS were pretreated with the Pin1 inhibitor PiB (3 μm) for 12 h after which the medium was removed, and cells were exposed to MPP+ (300 μm) along with PiB (3 μm) for 24 h. The cells were then washed with PBS and fixed in 4% paraformaldehyde for 25 min. After washing, the cells were permeabilized and blocked with 0.1% Triton X-100, 0.1% sodium azide, and 10% donkey serum in PBS for 1 h. Cells were then incubated with the antibody against human α-synuclein (1:100; Invitrogen, clone LB509) for 1 h. FITC-conjugated secondary antibody (1:500) was used to visualize the protein. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/ml). Finally, images were viewed using a 100× oil immersion objective on an inverted fluorescence microscope (model Eclipse TI; Nikon) and captured with a Photometrics K4 CCD camera.
Animals and MPTP Treatment
8–10-week-old male C57BL/6 mice weighing 24–28 g were housed in standard conditions: constant temperature (22 ± 1 °C), humidity (relative, 30%), and a 12-h light/dark cycle. Mice were allowed free access to food and water. Use of the animals and protocol procedures were approved and supervised by the Institutional Animal Care and Use Committee at Iowa State University (Ames, IA). Mice received either a single high dose of MPTP-HCl (30 mg/kg free base) or four injections of MPTP-HCl (15 mg/kg of free base; Sigma) in 1× PBS at 2-h intervals intraperitoneally. The control mice received only 1× PBS.
Juglone Treatment in Mice
Juglone was dissolved in 10% ethanol and administered via intraperitoneal injection. Mice received the first dose of juglone (3 mg/kg) 24 h before the first MPTP injection, the second dose of juglone 3 h before the first MPTP injection, and the third dose of juglone 3 h after the last MPTP injection. For neuroprotection studies mice were treated with a single dose of juglone each day for an additional 6 days.
Immunoblotting
Cells or brain tissues were collected and resuspended in modified radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitor mixture. Cell suspensions were sonicated after resuspension, whereas tissues were homogenized, sonicated, and then centrifuged at 14,000 × g for 1 h at 4 °C. Lysates were separated on 8–15% SDS-polyacrylamide electrophoresis gels. After the separation, proteins were transferred to a nitrocellulose membrane, and nonspecific binding sites were blocked by treating with Odyssey® blocking buffer (LiCor, Lincoln, NE). Anti-Pin1 (1:2,000; mouse monoclonal; R&D Systems, Minneapolis, MN), anti-TH (1:1,500, mouse monoclonal; Millipore), and anti-β-actin (1:10,000; mouse monoclonal; Sigma) were used to blot the membranes. Secondary IR-680-conjugated anti-mouse (1:10,000, donkey anti-mouse; Molecular Probes, Carlsbad, CA) was used for antibody detection with the Odyssey IR imaging system (LiCor).
Quantification of Tyrosine Hydroxylase (TH)-positive Cell Counts and Neuronal Processes
Tyrosine hydroxylase-positive dopaminergic neurons and neuronal processes in primary neuronal cultures from each coverslip were measured using MetaMorph software, Version 5.0 (Molecular Devices) as described previously (31). For counting TH cells, pictures were first taken at 20× magnification, then thresholded. After that, neurons were counted using the Integrated Morphometry Analysis function. For measurement of neuronal processes, pictures were taken using a 60× oil immersion lens, and the lengths of the processes were marked by applying the region and length measurement function in the Integrated Morphometry Analysis. TH-positive neurons and their processes were counted in at least six individual cultures for each treatment.
Diaminobenzidine Immunostaining, Stereological Counting of TH-positive Neurons, and Striatal Optical Density
Tyrosine hydroxylase and Pin1 diaminobenzidine (DAB) immunostaining were performed in striatal and substantia nigral sections as described previously (4, 32). Briefly, 7 days after MPTP injection, mice were sacrificed and perfused with 4% paraformaldehyde in 1× PBS and post-fixed with paraformaldehyde and 30% sucrose. The fixed brains were subsequently cut into 30-μm coronal sections using a cryostat in 30% sucrose-ethylene glycol solutions at −20 °C. On the day of staining, sections were first washed with PBS and incubated with the anti-TH antibody (1:1600, rabbit anti-mouse; Calbiochem) or with the anti-Pin1 antibody (1:400, goat anti-mouse; Santa Cruz Biotechnology) overnight at 4 °C. Biotinylated anti-rabbit or anti-goat secondary antibodies were used for 2 h at room temperature followed by incubation with avidin peroxidase (Vectastain ABC Elite kit, Burlingame, CA). Immunolabeling was observed using DAB, which yielded a brown-colored stain. Total numbers of TH-stained neurons in SN were counted stereologically with Stereo Investigator software (MicroBrightField, Inc., Williston, VT) using an optical fractionator (32). Quantification of striatal TH fibers was performed using Metamorph software, Version 5.0 (Molecular Devices). Briefly, striatal pictures were first taken at 2× magnification, then thresholded. After that, the optical density of fibers was counted using the Integrated Morphometry Analysis function.
HPLC Analysis of Striatal Dopamine and Its Metabolite Levels
The striatal dopamine, dihydrophenylacetic acid (DOPAC), and homovanillic acid (HVA) levels were quantified using high performance liquid chromatography (HPLC) with electrochemical detection. Samples were prepared and quantified as described previously (33). Briefly, 7 days after MPTP treatment, mice were sacrificed, striata were collected, and neurotransmitters from striatal tissues were extracted in 0.1 m perchloric acid solutions containing 0.05% Na2EDTA and 0.1% Na2S2O5 and isoproterenol (internal standard). The extracts were filtered through 0.22-μm spin tubes, and 20 μl of the sample was loaded for analysis. Dopamine and its metabolites DOPAC and HVA were separated isocratically in a C-18 reversed-phase column using a flow rate of 0.6 ml/min. An HPLC system (ESA Inc., Bedford, MA) with an automatic sampler equipped with a refrigerated temperature control (model 542; ESA Inc.) was used in these experiments. The electrochemical detection system consisted of a Coulochem-II model 5100A with an analytical cell (model 5014A) and a guard cell (model 5020) (ESA Inc.). The data acquisition and analysis were performed using EZStart HPLC Software (ESA Inc.).
Immunohistochemistry
Twenty-four hours after MPTP treatment, mice were perfused with 4% paraformaldehyde and post-fixed with paraformaldehyde and 30% sucrose. Next, 30-μm coronal SN free-floating sections were blocked with 2% bovine serum albumin, 0.5% Triton X-100, and 0.05% Tween 20 in PBS for 1 h at room temperature. Sections were incubated with different primary antibodies such as anti-Pin1 antibody (1:500; goat polyclonal; Santa Cruz Biotechnology), anti-NeuN (1:500; mouse monoclonal; Chemicon, Temecula, CA), and anti-TH (1:1600; mouse monoclonal; Chemicon) overnight at 4 °C. After washing with PBS, sections were incubated in appropriate secondary antibodies (Alexa Fluor 488 or 594 or 555 from Invitrogen) for 2 h followed by incubation with 10 μg/ml Hoechst 33342 for 5 min at room temperature to stain the nucleus. Sections were viewed under a NIKON inverted fluorescence microscope (model TE-2000U; NIKON, Tokyo, Japan); images were captured with a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI) using MetaMorph software, Version 5.0 (Molecular Devices).
Behavioral Measurements
We performed the open-field experiment for testing the locomotor activities (33, 34) and the rotarod experiment (33) to test foot movement of mice after MPTP and juglone treatments. An automated device (AccuScan, model RXYZCM-16, Columbus, OH) was used to measure the spontaneous activity of mice. The activity chamber was 40 × 40 × 30.5 cm made of clear Plexiglas and covered with a Plexiglas lid with holes for ventilation. The infrared monitoring sensors were located every 2.54 cm along the perimeter (16 infrared beams along each side) and 2.5 cm above the floor. Two additional sets of 16 sensors were located 8.0 cm above the floor on opposite sides. Data were collected and analyzed by a VersaMax Analyzer (AccuScan, model CDA-8, Columbus, OH). Before any treatment, mice were placed inside the infrared monitor for 10 min daily for three consecutive days to train them. Five days after the last MPTP injection, both open-field and rotarod experiments were conducted. In the open-field experiment, mice were monitored for horizontal activity, vertical activity, total distance traveled (cm), total movement time (s), total rest time (s), and rearing activity over a 10-min test session. Using Versaplot and Versadat software we analyzed the data among the three groups. In the rotarod, foot movements of mice were observed using a constant 20-rpm speed. Mice were given a 5–7 min rest interval to eliminate stress and fatigue.
Statistical Analysis
Data were analyzed with Prism 3.0 software (GraphPad Software, San Diego, CA). Bonferroni and Dunnett multiple comparison testing were used. Differences with p < 0.05 were considered significant.
RESULTS
Pin1 Expression in the Cell Culture and Animal Model of PD
To determine the role of Pin1 in PD, we used both cell culture and animal models and then compared the results in human PD brains. For the cell culture model, we adopted widely used MN9D mouse dopaminergic neuronal cells because of the amenability of the cells for biochemical studies relevant to dopaminergic neurodegeneration (35, 36). First, we sought to understand changes to the Pin1 message and protein levels upon treatment with the parkinsonian toxicant MPP+. We treated MN9D dopaminergic cells with 300 μm MPP+ and determined Pin1 message levels by quantitative real-time PCR. As evident from Fig. 1, A and B, MPP+ induced Pin1 message expression in MN9D dopaminergic cells in a time-dependent manner, with a maximal expression at 3 h (***, p < 0.001, compared with control). Next we examined whether Pin1 mRNA induction translated to changes in Pin1 protein levels by immunoblotting. Fig. 1, C and D, illustrates that the parkinsonian toxicant MPP+ induced Pin1 protein expression in MN9D mouse dopaminergic cells starting from 3 h with maximal expression at 24 h (***, p < 0.001, compared with control).
FIGURE 1.

MPP+ and MPTP induce Pin1 expression in cell culture and animal model of PD. A, MN9D cells were treated with 300 μm MPP+ for different time points and assayed for Pin1 mRNA using quantitative real-time PCR. B, data represent results from six individual measurements and are expressed as the mean ± S.E. MN9D cells were treated with 300 μm MPP+ for different time points, and proteins were separated by 15% SDS-PAGE and probed with Pin1 antibody to observe for Pin1 bands. C, shown is a Pin1 immunoblot after MPP+ treatment. D, the graph represents densitometric analysis of Pin1 protein levels normalized to β-actin (loading control). Mice were administered a single dose of MPTP (30 mg/kg) intraperitoneally and sacrificed at different time points (ranging from 1.5 to 24 h). Expression of Pin1 in mice SN (E) and striatum (F) at different time points after MPTP administration is shown. G, the graph represents densitometric analysis of Pin1 protein levels in SN normalized to β-actin (loading control). H, the graph represents densitometric analysis of Pin1 protein levels in striatum normalized to β-actin (loading control). Results are the mean ± S.E. either of at least three independent experiments or six mice per group. *, p < 0.05 versus control; **, p < 0.01 versus control, ***, p < 0.001 versus control.
Next we wanted to examine whether Pin1 is up-regulated in the MPTP-treated mouse brain. The expression of Pin1 in the nigrostriatal tissues was determined at different time points after a single dose of MPTP (30 mg/kg). We observed the highest expression of Pin1 in the SN (**, p < 0.01 compared with control, Fig. 1, E and G) and the striatum (***, p < 0.001 compared with control, Fig. 1, F and H) 6 h after MPTP injection. Time course analysis revealed that MPTP-induced Pin1 expression returned to basal levels in both striatum and SN 24 h after the MPTP treatment.
Pin1 Expression in Human Postmortem PD Tissues
Following the identification of Pin1 up-regulation in cell culture and animal models of PD, we aimed to compare the Pin1 levels in PD human brains with that of age-matched control brains. To determine the Pin1 level in nigral dopaminergic neurons, we performed a double labeling of TH and Pin1 in human nigral tissues. As depicted in Fig. 2, A–D, we found increased expression of Pin1 in SN of PD brains, and it also co-localized in the cytoplasm of TH-positive dopaminergic neurons (Fig. 2A). Very low levels of Pin1 expression were detected in the control human SN brain sections (Fig. 2, A and B). Human midbrain contains both pigmented (with neuromelanin) and non-pigmented (without neuromelanin) dopaminergic neurons (37), and in the diseased condition, the neuromelanin-containing pigmented dopaminergic neurons degenerate more than the non-pigmented (38). In our study we observed increased expression of Pin1 in the pigmented dopaminergic neurons of PD brain as characterized by dark staining (Fig. 2C), whereas less expression of Pin1 was observed in non-pigmented dopaminergic neurons (Fig. 2D). To further confirm our findings, we checked Pin1 protein levels in the SN of human midbrains by Western blotting. We detected robust increase in Pin1 expression in SN of PD patients (***, p < 0.001 compared with control; Fig. 2, E and F). Consistent with immunohistochemical data, Western blot analysis also showed a very low expression of Pin1 protein levels in control SN regions. Collectively, these results clearly demonstrate that Pin1 level is highly elevated in the nigral dopaminergic neurons in PD.
FIGURE 2.

Increased expression of Pin1 in human PD midbrain. A, human midbrain SN sections of PD patients and age-matched controls were immunostained for TH and Pin1. B, an enlarged image of control section shows decreased co-localization of TH and Pin1 in substantia nigra. C, expression of Pin1 in neuromelanin-containing pigmented TH-positive neurons. The inset shows co-localization of TH and Pin1 in a pigmented dopaminergic neuron. D, shown is expression of Pin1 in non-pigmented TH-positive neurons. The inset shows reduced co-localization of TH and Pin1 in a non-pigmented dopaminergic neuron. E, shown is a representative Western blot illustrating the expression of Pin1 in human midbrain lysates. Con, control. F, the graph represents densitometric analysis of Pin1 protein levels normalized to β-actin (loading control). Results are the mean ± S.E. from 10 different human midbrain lysates per group. ***, p < 0.001 versus control.
Pin1 Plays a Proapoptotic Function in Dopaminergic Neuronal Death
To understand the role of Pin1 in dopaminergic neuronal cell death, we carried out a series of functional studies. First, we knocked down Pin1 expression with a Pin1-specific siRNA and then examined the extent of MPP+ induced apoptotic cell death by caspase-3 activation and DNA fragmentation. MN9D dopaminergic cells were transfected with Pin1-specific siRNA, which effectively suppressed Pin1 levels as compared with nonspecific siRNA-treated control cells (Fig. 3A). Importantly, knockdown of Pin1 by siRNA effectively attenuated MPP+-induced caspase-3 activation (###, p < 0.001 compared with nonspecific siRNA treated with MPP+; Fig. 3B) and DNA fragmentation (###, p < 0.001 compared with nonspecific MPP+; Fig. 3C). Together, these results revealed that Pin1 plays a pro-apoptotic function in dopaminergic neuronal cells.
FIGURE 3.

siRNA-mediated knockdown of Pin1 attenuates MPP+-induced apoptotic events. MN9D dopaminergic neuronal cells were cultured, transfected with Pin1-specific siRNA, and exposed to 300 μm MPP+ for 24 h. A, shown is immunoblotting for Pin1 from siRNA-transfected MN9D cell lysates. B, shown is caspase-3 activation in MN9D cells after MPP+ treatment. C, shown is DNA fragmentation in MN9D cells after MPP+ treatment. Results are the mean ± S.E. of at least three independent experiments. ***, p < 0.001 versus control (nonspecific siRNA); ###, p < 0.001 versus MPP+ (nonspecific siRNA).
To further confirm the proapoptotic function of Pin1 in the dopaminergic neurodegenerative process, we tested the efficacy of a pharmacological inhibitor of Pin1, juglone, against MPP+ apoptotic cell death in MN9D mouse dopaminergic cells. The cells were co-treated with juglone and MPP+, and then cytotoxicity was measured at various time points by Sytox fluorescent dye assay. As shown in Fig. 4, A and B, 1 μm juglone treatment almost completely prevented 300 μm MPP+-induced cell death for the entire treatment period of 24 h. Furthermore, we also examined the effect of juglone on MPP+-induced apoptosis by measuring caspase-3 activation in the MN9D dopaminergic cells. Juglone significantly attenuated MPP+-induced caspase-3 activation (###, p < 0.001 compared with MPP+; Fig. 4C), confirming the protective effect of the Pin1 inhibitor. We further determined whether juglone has any effect on Pin1 levels in MN9D dopaminergic neuronal cells. To our surprise we found that MPP+-induced Pin1 expression was significantly attenuated in the presence of juglone at 12 h (a, p < 0.001 compared with MPP+ 12 h) and 24 h (b, p < 0.001 compared with MPP+ 24 h) (Fig. 4, D and E). To further rule out any nonspecific effect of juglone, we used two additional classes of Pin1 inhibitors and examined their efficacy in attenuating MPP+-induced Pin1 up-regulation in MN9D cells. PiB, a small chemically synthesized compound, was originally recognized as a potent Pin1 inhibitor through a library screen (39). The cyclic peptide inhibitor F (sequence, cyclo(d-Arg-d-Arg-d-Thr(P)-Pip-Nal-Arg-Gln)) belongs to a novel family of cyclic peptidyl Pin1 inhibitors showing levels of affinity and specificity higher than small molecular Pin1 inhibitors (26). The cyclic peptide inhibitor F inhibits Pin1 both in vitro and in intact cells at low micromolar concentrations (26). MN9D cells were treated with 300 μm MPP+ for 24 h in the presence or absence of PiB (5 μm), juglone (1 μm), or peptide inhibitor (5, 10, and 15 μm), and Pin1 expression was determined by Western blot. As shown in Fig. 4, F and G, PiB and peptide inhibitor F both abrogated MPP+-induced Pin1 up-regulation, similar to that achieved with juglone, suggesting that the observed effect of juglone on MPP+-induced Pin1 up-regulation is attributable to its inhibitory action on Pin1 activity.
FIGURE 4.

Effects of Pin1 inhibitors on MPP+-induced Pin1 expression, cytotoxicity, and caspase-3 activation in MN9D dopaminergic neuronal cells. A–E, MN9D cells were treated with 300 μm MPP+ for different time points in the presence or absence of 1 μm juglone. A, shown is cytotoxicity of MN9D cells during 300 μm MPP+ treatment in the presence or absence of 1 μm juglone. Cytotoxicity was determined by Sytox green nucleic acid stain by measuring the fluorescence at regular intervals as described “under Experimental Procedures.” B, the two panels demonstrate phase contrast images (right) and fluorescence micrographs (left) to show the extent of Sytox green staining of cells in the field of view. C, shown is a caspase-3 assay in the presence or absence of 1 μm juglone. D, shown is immunoblotting for Pin1. E, a graph represents densitometric analysis of Pin1 protein levels normalized to β-actin (loading control). F, MN9D cells were treated with 300 μm MPP+ for 24 h in the presence or absence of juglone (1 μm), PiB (5 μm), or peptide inhibitor F (5, 10, 15 μm), and cell lysates were prepared for immunoblotting with anti-Pin1 antibody. G, the graph represents densitometric analysis of Pin1 levels in panel F. Results are the mean ± S.E. of at least three independent experiments. M, MPP+. a, p < 0.001 versus MPP+ 12-h treatment; b, p < 0.001 versus MPP+ 24-h treatment; c, p < 0.01 versus MPP+ 24-h treatment; d, p < 0.001 versus MPP+ 24-h treatment; ***, p < 0.001 versus control; ###, p < 0.001 versus MPP+.
Because the Pin1 inhibitor juglone was very effective at blocking cell death and Pin1 expression in the MN9D dopaminergic cells, we further verified that the effect can be replicated in primary dopaminergic neurons from mesencephalic cultures. Primary mesencephalic neuronal cultures were isolated from E15 mouse embryos and grown on laminin-coated coverslips. The neuronal cultures were treated with 10 μm MPP+ in the presence or absence of 3 μm juglone for 24 h, and double immunocytochemistry was performed for Pin1 and TH. As evident from Fig. 5A, MPP+ induced increased expression of Pin1 in TH+ve dopaminergic neurons in primary mesencephalic nigral cultures. Notably, juglone attenuated MPP+-induced expression of Pin1 in dopaminergic neurons (Fig. 5A). Next, we quantified whether juglone treatment protected dopaminergic neurons from MPP+-induced neurodegeneration. We measured the lengths of processes of dopaminergic neurons from mesencephalic nigral culture (Fig. 5B) and found that MPP+ and juglone co-treated dopaminergic neuronal processes were significantly longer than the processes of neurons treated only with MPP+ (**, p < 0.01 compared with MPP+; Fig. 5B). Integrity of dopaminergic neurons can also be assessed by dopamine reuptake capacity, which serves as a functional indicator of the healthiness of the neurites (31). Thus, we also measured dopaminergic neuron function by dopamine uptake assay (Fig. 5C). As expected, treatment with MPP+ resulted in a significant loss of dopamine uptake activity of TH+ve neurons (***, p < 0.001 compared with control; Fig. 5C). However, juglone ameliorated MPP+-induced loss of dopamine uptake activity (###, p < 0.001 compared with MPP+; Fig. 5C). Taken together, these results demonstrate that the Pin1 inhibitor juglone has a neuroprotective effect in cell culture models of dopaminergic neurodegeneration.
FIGURE 5.

Juglone inhibits MPP+-induced expression of Pin1 in primary neuronal culture. Mesencephalic tissues from E15 mouse embryos were cultured and grown on laminin-coated coverslips. The neuronal cultures were treated with 10 μm MPP+ for 24 h in the presence or absence of 3 μm juglone. After culture, primary neurons were fixed with 4% paraformaldehyde and incubated with anti-TH and anti-Pin1 antibodies and viewed under a NIKON TE2000 fluorescence microscope. A, double labeling of TH and Pin1 in primary mesencephalic culture from substantia nigra is shown. M, MPP+. B, mesencephalic neuronal process lengths were quantified using MetaMorph image analysis software as mentioned “under Experimental Procedures.” C, shown is dopamine uptake assay from primary striatal culture. Results are the mean ± S.E. of at least three independent experiments. ***p < 0.001 versus control; ###, p < 0.001 versus MPP+; **, p < 0.01 versus MPP+.
Pin1 Mediates the Formation of α-Synuclein Aggregates in Response to MPP+ in N27 Dopaminergic Cells
Because Ryo et al. previously reported that overexpression of Pin1 in a kidney 293T cell line led to the formation of α-synuclein inclusions (24), we further examined whether MPP+-induced Pin1 up-regulation promotes the formation of α-synuclein aggregates. For this purpose we analyzed the status of α-synuclein aggregates in a stable rat dopaminergic N27 neuronal cell line that constitutively expresses human wild-type α-synuclein. These cells displayed significant α-synuclein protein aggregates upon treatment with neurotoxic compounds (28), representing an excellent dopaminergic neuronal model for studying α-synuclein aggregation. N27 dopaminergic cells grown in media supplemented with 0.2% FBS were pretreated with 3 μm PiB for 12 h followed by exposure to MPP+ (300 μm) along with PiB (3 μm) for 24 h, and then α-synuclein aggregates were determined by immunostaining. As shown in Fig. 6A, exposure of α-synuclein-expressing cells to MPP+ induced a significant increase in α-synuclein aggregates compared with control. Notably, inhibition of Pin1 function by PiB dramatically suppressed α-synuclein aggregates. PiB alone did not show any effect on the formation of α-synuclein aggregates. To further confirm that MPP+-induced up-regulation of Pin1 expression also occurs in human α-synuclein-expressing N27 cells, we treated the cells with MPP+ and measured Pin1 expression by Western blot. Consistent with the findings of MN9D cells, Pin1 expression in N27 cells was induced by MPP+ (Fig. 6, B and C). Together, these results suggest that Pin1 up-regulation in dopaminergic neuronal cells during neurotoxic stress may play a role in α-synuclein aggregation.
FIGURE 6.
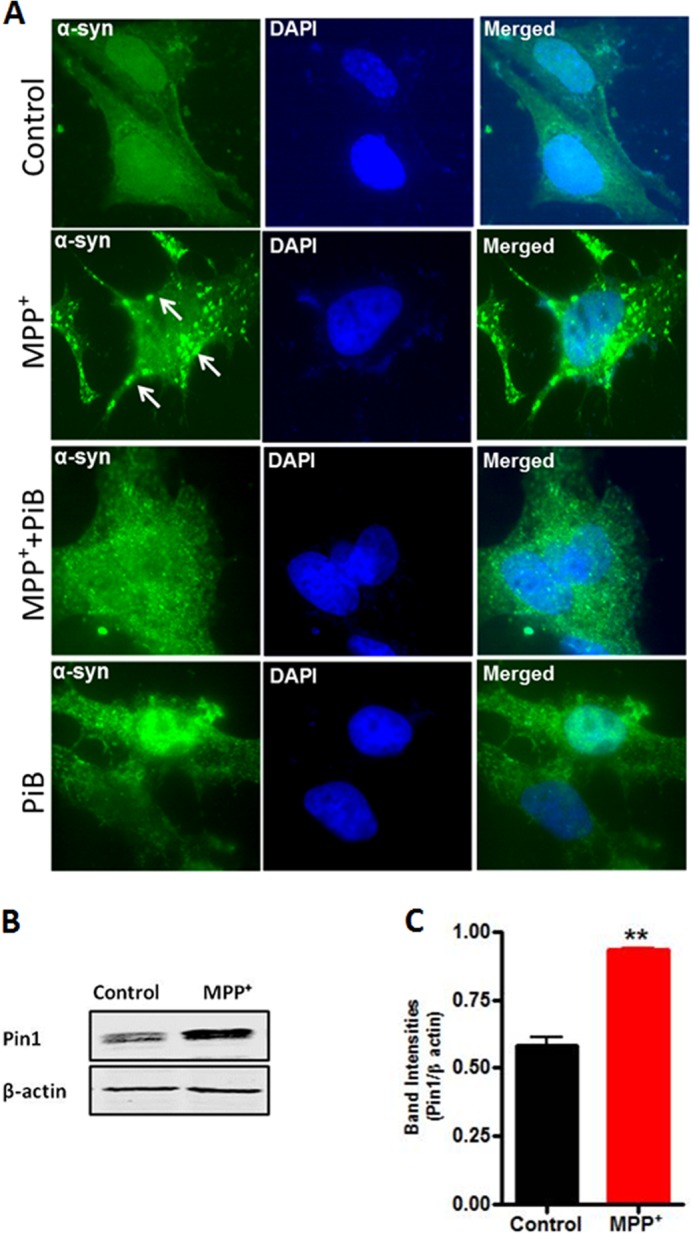
Pin1 mediates the formation of α-synuclein aggregates in response to MPP+ in N27 dopaminergic cells. A, α-synuclein stably expressing N27 cells were pretreated with the Pin1 inhibitor PiB (3 μm) for 12 h and then exposed to MPP+ (300 μm) along with PiB (3 μm) for 24 h. Cells were fixed and immunostained for α-synuclein. The nuclei were counterstained by DAPI. Images were obtained using a Nikon Eclipse T1 fluorescence microscope. Green, α-synuclein; blue, nucleus. The white arrows point to α-synuclein-positive inclusions. Magnification, 100×. Representative immunofluorescence images are shown. B, MPP+ induced Pin1 up-regulation in α-synuclein stably expressing N27 cells. Cells were treated with MPP+ for 24 h, and Pin1 expression was measured by Western blot. C, the graph represents densitometric analysis of Pin1 levels in panel B. Results are the mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control.
Juglone Inhibits Pin1 Expression in Vivo in the Nigrostriatum of MPTP-treated Mice
To expand the neuroprotective effect of Pin1 to animal models, we adopted the four-dose MPTP mouse model of PD. First, we characterized the time course of Pin1 expression in mice treated with 4 doses of MPTP on a single day at 2-h intervals. As evident from Fig. 7, A and B, Western blotting for Pin1 showed a significant increase in Pin1 expression in SN at 12 and 24 h time points with maximum expression at 24 h after MPTP injections (***, p < 0.001 compared with control). However, in striatum (Fig. 7, C and D), we detected a significant increase in Pin1 expression at 6 and 12 h (***, p < 0.001 compared with control) and 24 and 72 h (**, p < 0.01 compared with control) time points. After the characterization of Pin1 expression in the MPTP model, we examined if juglone was capable of suppressing the expression of Pin1 in vivo in the nigrostriatum of MPTP mouse model. First, we sought to determine the effect of juglone on Pin1 mRNA levels in the SN by measurement with quantitative real-time PCR. It is evident from Fig. 7E that MPTP treatment markedly induced the expression of Pin1 mRNA levels in the SN at the 24-h time point (**, p < 0.01 compared with control). Importantly, mice that were treated with juglone (3 mg/kg/dose, intraperitoneal) from 24 h before the MPTP treatment exhibited greatly decreased expression of Pin1 (a, p < 0.05 compared with MPTP; Fig. 7E), suggesting that juglone is capable of inhibiting the message level of Pin1 in the animal model of PD. Next, we also examined whether juglone could block the Pin1 protein expression in the nigrostriatal regions in MPTP-treated mice. As shown by Western blotting (Fig. 7, F–I), MPTP administration led to markedly increased Pin1 protein levels both in the SN and striatum, and juglone strongly attenuated MPTP-induced expression of Pin1 in the SN (a, p < 0.01 compared with MPTP; Fig. 7, F and H) and the striatum (a, p < 0.05 compared with MPTP; Fig. 7, G and I).
FIGURE 7.

Juglone attenuates MPTP-induced Pin1 message and protein expression in mouse SN and striatum. Mice were injected with four doses (intraperitoneal) of MPTP (18 mg/kg) on a single day and sacrificed at different time points. A, expression of Pin1 in mice substantia nigra at different time points after MPTP administration is shown. B, the graph represents densitometric analysis of Pin1 protein levels in SN normalized to β-actin (loading control). C, shown is expression of Pin1 in mice striatum at different time points after MPTP administration. D, the graph represents densitometric analysis of Pin1 protein levels in striatum normalized to β-actin (loading control). Another group of MPTP-treated mice also received juglone intraperitoneally at a dose of 3 mg/kg. Twenty-four hours after MPTP injection, mice were sacrificed and checked for Pin1 expression in SN by quantitative real-time PCR (E) and Western blot (F) and in striatum by Western blot (G). H, the graph represents densitometric analysis of Pin1 protein levels in SN normalized to β-actin (loading control). I, the graph represents densitometric analysis of Pin1 protein levels in striatum normalized to β-actin (loading control). Results are the mean ± S.E. from six mice per group. *, p < 0.05 versus control; **, p < 0.01 versus control; ***, p < 0.001 versus control; a, p < 0.05 versus MPTP; b, p < 0.01 versus MPTP.
To further determine the effect of juglone on nigral dopaminergic neurons, we performed immunohistochemical analysis. DAB immunostaining analysis for Pin1 in ventral midbrain sections showed that MPTP treatment led to a marked increase in Pin1 expression in the nigral regions and that juglone suppressed MPTP-induced expression of Pin1 (Fig. 8A). Furthermore, we performed double-labeling immunohistochemistry for Pin1 and NeuN (common neuronal marker) in the nigral regions to determine the cellular types that show increased Pin1 expression. We observed that several NeuN-positive neurons, but not all, displayed Pin1 immunoreactivity in MPTP-treated mice, whereas little or no co-localization of Pin1 and NeuN was observed in saline-treated control sections (data not shown). To determine specific neuronal type, we did double-labeling immunohistochemistry of TH (marker for dopaminergic neuron) and Pin1 in the SN. It was clearly evident from Fig. 8B that Pin1 predominantly co-localized with TH+ve dopaminergic neurons in MPTP-treated mice. Enlarged insets clearly show colocalization of TH and Pin1 in cytosol (Fig. 8B). Notably, juglone treatment attenuated MPTP-induced expression of Pin1 in TH+ve dopaminergic neurons (Fig. 8B). Taken together, these results clearly suggest that juglone inhibits MPTP induced Pin1 expression in the nigrostriatal dopaminergic neurons in the commonly used MPTP mouse model of PD. To further evaluate whether the up-regulation of Pin1 is specific to the nigrostriatum region of the brain, we examined Pin1 expression levels in cortex and hippocampus 24 h after MPTP treatment. We found no change in protein expression levels of Pin1 in MPTP-treated mice when compared with saline-treated control mice (data not shown). Together, these results demonstrate that Pin1 is up-regulated in the nigrostriatal dopaminergic system in experimental models of PD.
FIGURE 8.
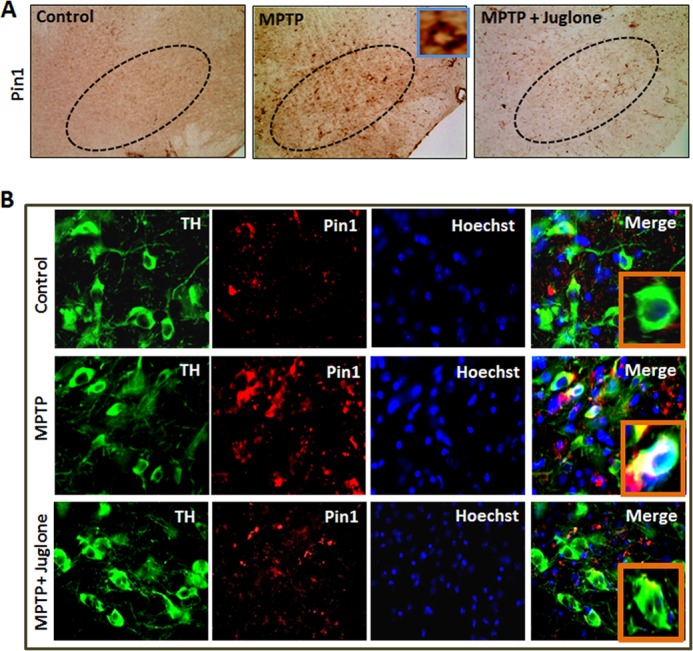
Juglone attenuates MPTP-induced Pin1 expression in mouse SN. Mice were treated with juglone (3 mg/kg) 24 h before MPTP treatment. Twenty-four hours after the last dose of MPTP, mice were sacrificed, and SN sections were immunolabeled with different antibodies. A, DAB immunostaining of Pin1 in SN is shown. The inset in MPTP group shows a single enlarged Pin1-positive cell. B, mice SN tissue sections were immunolabeled for TH (marker of dopaminergic neurons) and Pin1. Insets in Merge panel of each group show higher magnified double-labeled pictures of TH and Pin1.
Juglone Improved Locomotor Functions in MPTP-treated Mice
Following the effect of juglone on MPTP-induced Pin1 expression in the MPTP model, we examined whether juglone protects against neurobehavioral deficits caused by MPTP. Five days after MPTP and juglone treatments, mice were tested for locomotor activities by using locomotor activity monitor and rotarod. Representative maps using Versaplot software (Accuscan, OH) depicts the locomotor activity pattern of mice over a 10-min period (Fig. 9A). We observed a marked decrease in horizontal activities (Fig. 9B), vertical activities (Fig. 9C), total distance traveled (Fig. 9D), total movement time (Fig. 9E), and rearing activities (Fig. 9F) in the MPTP-treated mice. However, juglone significantly improved MPTP-induced hypolocomotion (Fig. 9, A–F). Likewise, in the rotarod with a 20-rpm speed, MPTP-treated mice showed (75%) a decrease in time spent on the rod (***, p < 0.001 compared with control; Fig. 9G). Juglone treatment significantly restored (50%) rotarod activities in MPTP-treated mice (*, p < 0.05 compared with control; Fig. 9G). Collectively, these results demonstrate that juglone restored the motor function impairments in the MPTP mouse model of PD.
FIGURE 9.

Juglone improves motor function and loss of neurotransmitters in MPTP-treated mice. Mice received juglone (3 mg/kg) intraperitoneally 1 day before MPTP treatment, and treatment was continued for another 7 days. Five days after MPTP treatment mice were tested for motor functions. A, shown is the moving track of mice using Versaplot software. B, shown is horizontal activity. C, shown is vertical activity. D, shown is total distance traveled (cm). E, shown is total movement time (seconds). F, shown is the number of rearing activities. G, shown is time spent on rotarod. Seven days after the last dose of MPTP treatment mice were sacrificed and dopamine (H), DOPAC (I), and HVA (J) were measured from striatum by HPLC as mentioned under “Experimental Procedures.” Results are the mean ± S.E. of 10 mice per group. M, MPTP. ***p < 0.001 versus control; **, p < 0.01 versus control; *, p < 0.05 versus MPTP; #, p < 0.01 versus MPTP.
Juglone Increased Striatal Dopamine and Its Metabolite Levels in MPTP-treated Mice
After establishing the role of juglone in improving motor functions, we next determined whether juglone also protects against MPTP-induced neurochemical depletion. Mice were sacrificed following the behavioral measurements described above, and striatum tissue samples were processed for neurotransmitter analysis by HPLC (as mentioned under “Experimental Procedures”). We observed an 80% decrease in dopamine (***, p < 0.001 compared with control; Fig. 9H), 85% decrease in DOPAC (***, p < 0.001 compared with control; Fig. 9I), and 90% decrease in HVA (**, p < 0.01 compared with control; Fig. 9J) in MPTP-treated mice compared with the striata of saline-injected control mice. In contrast, juglone-treated MPTP mice showed only a 50% decrease in striatal dopamine (#, p < 0.01 compared with MPTP, Fig. 9H). Additionally, juglone also restored DOPAC and HVA levels by ≈33% (#, p < 0.01 compared with MPTP; Fig. 9I) and ≈18% (Fig. 9J, not significant), respectively, in MPTP-treated mice. Taken together, these results suggest that the Pin1 inhibitor juglone can improve neurochemical and behavioral deficits in the MPTP mouse model of PD.
Juglone Protects Nigrostriatal Dopaminergic Neurons from MPTP-induced Neurotoxicity
Because juglone significantly restored the behavioral and striatal neurotransmitter deficits in MPTP-treated mice, we wanted to determine whether juglone treatment could protect against MPTP-induced dopaminergic neurodegeneration in the nigrostriatal axis. Mice were treated with juglone (3 mg/kg/day) 24 h before MPTP injections and were continued for another 6 days after MPTP injections. Seven days after MPTP treatment, mice were sacrificed, and TH DAB immunostaining was performed in the SN and striatum regions of ventral midbrain (Fig. 10, A and B). MPTP treatment led to an ∼67% loss of nigral TH-positive neurons (***, p < 0.001 compared with control; Fig. 10, B and D) and 60% reduction of striatal TH optical density (***, p < 0.001 compared with control; Fig. 10, A and C). Higher magnified (10×) pictures clearly showed MPTP-induced loss of TH-positive dopaminergic neurons, mainly in the substantia nigra lateralis (SNl), substantia nigra pars compacta (SNc), and substantia nigra ventricularis (SNr) regions (Fig. 10B, lower panel). Notably, juglone-treated MPTP-injected mice showed less reduction in the nigral TH-positive neurons (*, p < 0.05 compared with MPTP; Fig. 10, B and D) and the striatal TH optical density (a, p < 0.01 compared with MPTP; Fig. 10, A and C), demonstrating clear protection against dopaminergic neuronal degeneration. In addition to immunohistochemical studies, we also confirmed the neuroprotective effect of juglone by determining the TH protein level by Western blot analysis. We observed attenuation of TH expression in the striatum and the SN of MPTP-treated mice (Fig. 10, E–H), whereas juglone treatment effectively attenuated the loss of TH levels in the striatum (**, p < 0.01 compared with MPTP; Fig. 10, E and G) and in the SN (**, p < 0.01 compared with MPTP; Fig. 10, F and H) of MPTP-treated mice. Collectively, these data demonstrate the neuroprotective effect of juglone against dopaminergic neuronal degeneration in the MPTP mouse model.
FIGURE 10.

Effects of juglone on nigrostriatum in MPTP-treated mice. Mice were administered juglone (3 mg/kg intraperitoneal) 1 day before MPTP injection, and treatment was continued for 7 days (1 day co-treatment with MPTP and 6 days post-treatment after MPTP). Mice were sacrificed 7 days after MPTP treatment, and TH-DAB immunostaining was performed in striatum (2× magnification) (A) and SN (upper panel, 2X magnification; lower panel, 10× magnification) (B). SNl, substantia nigra lateralis; SNc, substantia nigra pars compacta; SNr, substantia nigra ventricularis. C, quantification of TH-positive fibers in the striatum is shown. D, stereological counting of TH-positive neurons in the SN is shown. Representative Western blots illustrate the expression of TH in the striatum (E) and SN (F). The graph represents densitometric analysis of Pin1 protein levels normalized to β-actin (loading control) in the striatum (G) and SN (H). Results are the mean ± S.E. of eight mice per group. ***p < 0.001 versus control; **, p < 0.01 versus MPTP; *, p < 0.05 versus MPTP; a, p < 0.001 versus MPTP.
DISCUSSION
Several lines of evidence presented in our study clearly show up-regulation of Pin1 expression in dopaminergic cell culture models in an animal model of PD and in human PD midbrain. We also demonstrate that a pharmacological inhibitor of Pin1 capable of reducing the induction of Pin1 expression protects dopaminergic neurons from neurotoxic insults. Our conclusions are based on the following observations. First, we show that Pin1 is highly up-regulated in MN9D and N27 dopaminergic cells and in primary nigrostriatal cultures after parkinsonian neurotoxicant MPP+ treatment. Second, we also observed increased expression of Pin1 in the nigral dopaminergic neurons of postmortem PD brains. Third, by employing various pharmacological and genetic tools in cultured neuronal cells, we establish that Pin1 has a proapoptotic role in the degeneration of dopaminergic neurons and promotes α-synuclein protein aggregation. Fourth, the Pin1 inhibitor juglone ameliorated functional motor deficits, neurochemical depletion, and nigral degeneration in the MPTP mouse model of PD. To our knowledge, Pin1 transcriptional regulation in the degeneration of nigral dopaminergic neurons has never been explored, and our results reveal that up-regulation of Pin1 mRNA and protein may be a critical neurotoxic event in the pathogenesis of PD. Also, our pharmacological inhibitor studies in the MPTP animal model of PD provide preclinical evidence for targeting Pin1 for therapeutic intervention of dopaminergic neuronal degeneration in PD.
Several cellular regulatory processes such as proliferation, differentiation, growth, and apoptosis are controlled by Pin1, which has been linked to the disease processes of cancer (20), asthma (16), and Alzheimer disease (14). Proline-directed phosphorylation is important for the regulation of post-mitotic development of neurons (40), but the role of Pin1 in neurodegenerative disorders is a subject of contention. Down-regulation of Pin1 during oxidative stress has been linked with tautopathy in Alzheimer disease (41, 42). Also, inhibition of Pin1 led to attenuation of neurofilament-H phosphorylation and its perikaryal accumulation in Alzheimer disease and amyotrophic lateral sclerosis (ALS) models (43, 44). Recently, Ryo et al. (24) showed the presence of Pin1 in the LBs of human PD brain tissues and described an intriguing function of Pin1 as facilitating the formation of α-synuclein inclusions via binding to synphilin-1 using the non-neuronal 293T and COS-1 cell models. Their studies, however, do not provide direct evidence regarding up-regulation of Pin1 during neurotoxic stress or its functional relevance to dopaminergic neuronal degeneration using a cell culture and animal models of Parkinson disease. Our experimental data provide several novel insights into the role of Pin1 in the pathogenesis of PD. We show for the first time that neurotoxic stress induces Pin1 up-regulation in dopaminergic neurons in both cell culture and animal models of PD. Our cell culture findings reveal Pin1 is up-regulated at both protein and message levels during MPP+-induced neurotoxic stress in MN9D and N27 dopaminergic cells as well as in primary mesencephalic neurons (Figs. 1, 5, and 6). Additionally, we identified up-regulation of Pin1 expression in nigrostriatum of C57 black mice after MPTP insult, corroborating our in vitro findings (Figs. 1, 7, and 8). Another interesting finding is that Pin1 inhibitor significantly suppressed MPP+-induced α-synuclein protein aggregation in the N27 dopaminergic cell model (Fig. 6A), suggesting that up-regulation of Pin during neurotoxic stress may promote α-synuclein protein misfolding and aggregation. Although the cellular mechanism of Pin1 up-regulation in α-synuclein protein aggregation is not currently well understood, Pin1 may contribute to protein aggregation via modulating kinase and phosphatase activity and thereby influence the phosphorylation status of α-synuclein. For example, evidence exists that α-synuclein phosphorylation can be influenced by Polo-like-kinases, casein kinases, G protein-coupled receptor kinases, and phosphatase 2A (45–52). Also, a recent study by Ryo et al. (24) showed that Pin1 overexpression in 293T cells prolongs the half-life and insolubility of α-synuclein. Therefore, it is possible that the Pin1-dependent α-synuclein aggregation observed in our study may be due to the prolonged half-life and insolubility of α-synuclein.
We also extended our key finding of Pin1 up-regulation to postmortem human PD brains. We first identified that Pin1 is highly expressed in the pigmented dopaminergic neurons of PD patients in comparison to age-matched controls (Fig. 2). Human PD cases are mainly associated with loss of pigmented dopaminergic neurons in SN, and therefore, our findings of elevated Pin1 expression only in pigmented dopaminergic neurons suggest a possible role of Pin1 in pathogenesis of PD. Precedence for up-regulation of Pin1 expression in diseased states has been reported in lung cancer patients (53) and in oral squamous cell carcinoma (54). However, changes in Pin1 expression have never been reported in the nigral dopaminergic system. Together, the data provided in our manuscript provide compelling new evidence that neurotoxic stress induces dramatic up-regulation of Pin1 in both in vitro and in vivo models of PD as well as that postmortem PD brains have increased levels of Pin1.
The role of Pin1 in cell death and survival has recently been recognized. Although inhibition of Pin1 led to apoptosis in vascular smooth muscle cells (55), Pin1 was required for pro-survival signaling in eosinophils (56). In the nervous system, Pin1 has been shown to regulate oligodendrocyte apoptosis after spinal injury by binding and stabilizing Mcl-1 after JNK3 activation (57). Also, Pin1 dissociates from the neuron-specific JNK scaffold protein JIP3 to associate with phosphorylated BH3-only BIMEL and activate the mitochondrial apoptotic machinery (58). Recently, we reported that mixed-lineage kinase 3 (MLK3) regulates nuclear translocation and function of Pin1 in breast cancer cells (25). Using pharmacological inhibitors and siRNA knockdown (Figs. 3 and 4), we demonstrate that Pin1 has proapoptotic function in nigral dopaminergic neurons.
We employed a known inhibitor of Pin1, juglone, to determine the neuroprotective efficacy of the compound in an established MPTP model of PD. Juglone (5-hydroxy-1,4-napthalenedione (C10H6O3)), an aromatic organic napthoquinone naturally found in the leaves, roots, and bark of black walnut plants, has been widely used as a Pin1 inhibitor (59, 60). Juglone irreversibly inactivates enzymatic activity of human Pin1 by modification of thiol groups of cysteine residues (61). Juglone at a dose of 5.7 μm in vitro blocked Pin1 isomerase activity completely without any effect on other peptidyl prolyl cis-trans isomerases (61). In our study we found that micromolar concentrations (1 μm) of juglone attenuated MPP+-induced Pin1 expression in MN9D cells (Fig. 4, D and E). In addition to juglone, we also demonstrated that two structurally different Pin1 inhibitors, PiB and peptide inhibitor F, significantly inhibited MPP+-induced Pin1 up-regulation (Fig. 4, F and G). These results further provide direct evidence for specific involvement of Pin1 in mediating MPP+-induced neurotoxicity. Juglone also significantly restored dopamine reuptake in primary mesencephalic neurons and also protected mesencephalic and striatal primary neurons and their neurites from MPP+ toxicity (Fig. 5). Inhibition of Pin1 by juglone has shown anticancer effects in experimental models. Esnault et al. (59) showed that juglone prevented acute and chronic rejection of MHC mismatched, orthotopic rat lung transplants by reducing the expression of IFN-γ and CXCL-10 mRNA stability, accumulation, and protein expression after cell activation. Another report showed that juglone at a dose of 5 mg/kg can attenuate rheumatoid arthritis development and COX-2 expression in human primary cultured RA chondrocytes and in type II collagen (CII)-treated DBA/1J mice (60). In our study we injected juglone (3 mg/kg) intraperitoneally to C57BL/6 mice, which attenuated MPTP-induced expression of Pin1 in nigrostriatal axis (Figs. 7 and 8). Because juglone suppresses Pin1 expression and apoptotic events in cell culture models, we decided to investigate the efficacy of juglone at protecting dopaminergic neurons in the acute MPTP model of PD. The data presented in this manuscript clearly show that juglone can protect dopaminergic neurons from parkinsonian toxicity. We show that juglone restored the behavioral activities as well as dopamine and its metabolite levels in striatum of MPTP-treated mice (Fig. 9). Additionally, juglone also protected against MPTP-induced loss of TH-positive neurons and terminals in the nigrostriatum (Fig. 10). Taken together, the observed protective effect of the Pin1 inhibitor juglone in an MPTP model of PD strongly suggests efficacy of juglone as a neuroprotective agent at a reasonable dose level. Importantly, the 3 mg/kg dose of juglone used in our study was nontoxic. However, we noted some adverse effects after administration of larger repeated doses of juglone in mice, indicating that further optimization of juglone-related compounds may provide better neuroprotective effects against dopaminergic neuronal degeneration in vivo.
The mechanism of Pin1 suppression by juglone, PiB, and Pin1 peptide inhibitor F is not entirely clear. Because juglone inhibited Pin1 expression both in vitro and in vivo, juglone may act through another mechanism besides direct inhibition of Pin1 activity. Possibly Pin1 autoregulates the transcription of Pin1 gene expression; however, no clear evidence exists in the literature to support this notion. Although very little is known about the transcriptional regulation of the Pin1 gene, Ryo et al. (62) showed that Pin1 transcription was initiated by oncogenic Neu/Ras signaling via E2F activation. Similarly, PI3K/p38 kinase-dependent E2F activation has been shown to induce Pin1 (63). In addition, Pin1 binds to the phosphorylated T254P residue of NF-κB-p65 and through this interaction stabilizes p65 in the nucleus (64). Pin1 also promotes tumor cell death by dissociating tumor suppressor p53 from apoptosis inhibitor iASPP (65). Furthermore, Pin1 controls p53 stability and p21 transactivation during DNA damage (66). However, it is not known whether Pin1 interacts with other transcriptional regulators such as NF-κB or p53 in dopaminergic neurons during neurotoxic insult. Further research in this area could elucidate new mechanistic insights into the role of Pin1 in the pathogenesis of PD.
In conclusion, we demonstrate for the first time to our knowledge that Pin1 is up-regulated in cell culture and animal models of PD. Importantly, we also show that pigmented dopaminergic neurons in PD brains express high levels of Pin1. By employing pharmacological and genetic tools to inhibit Pin1, we report that Pin1 has a proapoptotic role in PD pathogenesis and that the Pin1 inhibitor juglone effectively attenuates Pin1 expression during dopaminergic neuronal degeneration in the nigrostriatal system. Additionally, Pin1 inhibitor improved behavioral deficits and neurochemical depletion in an animal model of PD. Overall, these results clearly suggest that Pin1 has a proapoptotic role in PD pathogenesis and that the development of interventional strategies targeting Pin1 signaling may offer neuroprotection against dopaminergic neurodegeneration.
Acknowledgment
We thank Gary Zenitsky for assistance in the preparation of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health RO1 Grants NS074443 (to A. G. K.), NS65167 (to A. K.), and GM55835 (to A. R.).
- PD
- Parkinson disease
- SN
- substantia nigra
- DAB
- diaminobenzidine
- DOPAC
- dihydrophenylacetic acid
- HVA
- homovanillic acid
- TH
- tyrosine hydroxylase
- MPP+
- 1-methyl-4-phenylpyridinium
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
REFERENCES
- 1. Lees A. J., Hardy J., Revesz T. (2009) Parkinson disease. Lancet 373, 2055–2066 [DOI] [PubMed] [Google Scholar]
- 2. Dauer W., Przedborski S. (2003) Parkinson's disease. Mechanisms and models. Neuron 39, 889–909 [DOI] [PubMed] [Google Scholar]
- 3. Dunnett S. B., Björklund A. (1999) Prospects for new restorative and neuroprotective treatments in Parkinson's disease. Nature 399, A32–A39 [DOI] [PubMed] [Google Scholar]
- 4. Ghosh A., Roy A., Matras J., Brahmachari S., Gendelman H. E., Pahan K. (2009) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson's disease. J. Neurosci. 29, 13543–13556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chaudhuri K. R., Schapira A. H. (2009) Non-motor symptoms of Parkinson's disease. Dopaminergic pathophysiology and treatment. Lancet Neurol. 8, 464–474 [DOI] [PubMed] [Google Scholar]
- 6. Martinez-Martin P., Rodriguez-Blazquez C., Abe K., Bhattacharyya K. B., Bloem B. R., Carod-Artal F. J., Prakash R., Esselink R. A., Falup-Pecurariu C., Gallardo M., Mir P., Naidu Y., Nicoletti A., Sethi K., Tsuboi Y., van Hilten J. J., Visser M., Zappia M., Chaudhuri K. R. (2009) International study on the psychometric attributes of the non-motor symptoms scale in Parkinson's disease. Neurology 73, 1584–1591 [DOI] [PubMed] [Google Scholar]
- 7. Göthel S. F., Marahiel M. A. (1999) Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol. Life Sci. 55, 423–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu K. P., Finn G., Lee T. H., Nicholson L. K. (2007) Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3, 619–629 [DOI] [PubMed] [Google Scholar]
- 9. Shaw P. E. (2007) Peptidyl-prolyl cis/trans isomerases and transcription. Is there a twist in the tail? EMBO Rep. 8, 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joseph J. D., Yeh E. S., Swenson K. I., Means A. R., Winkler (2003) The peptidyl-prolyl isomerase Pin1. Prog. Cell Cycle Res. 5, 477–487 [PubMed] [Google Scholar]
- 11. Lu K. P. (2003) Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell 4, 175–180 [DOI] [PubMed] [Google Scholar]
- 12. Atchison F. W., Means A. R. (2004) A role for Pin1 in mammalian germ cell development and spermatogenesis. Front. Biosci. 9, 3248–3256 [DOI] [PubMed] [Google Scholar]
- 13. Braithwaite A. W., Del Sal G., Lu X. (2006) Some p53-binding proteins that can function as arbiters of life and death. Cell Death Differ. 13, 984–993 [DOI] [PubMed] [Google Scholar]
- 14. Akiyama H., Shin R. W., Uchida C., Kitamoto T., Uchida T. (2005) Pin1 promotes production of Alzheimer's amyloid β from β-cleaved amyloid precursor protein. Biochem. Biophys. Res. Commun. 336, 521–529 [DOI] [PubMed] [Google Scholar]
- 15. Maruszak A., Safranow K., Gustaw K., Kijanowska-Haładyna B., Jakubowska K., Olszewska M., Styczyńska M., Berdyński M., Tysarowski A., Chlubek D., Siedlecki J., Barcikowska M., Zekanowski C. (2009) PIN1 gene variants in Alzheimer's disease. BMC Med. Genet. 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Esnault S., Shen Z. J., Malter J. S. (2008) Pinning down signaling in the immune system. The role of the peptidyl-prolyl isomerase Pin1 in immune cell function. Crit. Rev. Immunol. 28, 45–60 [DOI] [PubMed] [Google Scholar]
- 17. Shen Z. J., Esnault S., Rosenthal L. A., Szakaly R. J., Sorkness R. L., Westmark P. R., Sandor M., Malter J. S. (2008) Pin1 regulates TGF-β1 production by activated human and murine eosinophils and contributes to allergic lung fibrosis. J. Clin. Invest. 118, 479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutton M., Lewis J., Dickson D., Yen S. H., McGowan E. (2001) Analysis of tauopathies with transgenic mice. Trends Mol. Med. 7, 467–470 [DOI] [PubMed] [Google Scholar]
- 19. Lee V. M., Goedert M., Trojanowski J. Q. (2001) Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 20. Li H., Wang S., Zhu T., Zhou J., Xu Q., Lu Y., Ma D. (2006) Pin1 contributes to cervical tumorigenesis by regulating cyclin D1 expression. Oncol. Rep. 16, 491–496 [PubMed] [Google Scholar]
- 21. Becker E. B., Bonni A. (2006) Pin1 mediates neural-specific activation of the mitochondrial apoptotic machinery. Neuron 49, 655–662 [DOI] [PubMed] [Google Scholar]
- 22. Lu P. J., Wulf G., Zhou X. Z., Davies P., Lu K. P. (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788 [DOI] [PubMed] [Google Scholar]
- 23. Becker E. B., Bonni A. (2007) Pin1 in neuronal apoptosis. Cell Cycle 6, 1332–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryo A., Togo T., Nakai T., Hirai A., Nishi M., Yamaguchi A., Suzuki K., Hirayasu Y., Kobayashi H., Perrem K., Liou Y. C., Aoki I. (2006) Prolyl-isomerase Pin1 accumulates in lewy bodies of Parkinson disease and facilitates formation of α-synuclein inclusions. J. Biol. Chem. 281, 4117–4125 [DOI] [PubMed] [Google Scholar]
- 25. Rangasamy V., Mishra R., Sondarva G., Das S., Lee T. H., Bakowska J. C., Tzivion G., Malter J. S., Rana B., Lu K. P., Kanthasamy A., Rana A. (2012) Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. U.S.A. 109, 8149–8154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu T., Liu Y., Kao H. Y., Pei D. (2010) Membrane permeable cyclic peptidyl inhibitors against human peptidylprolyl isomerase Pin1. J. Med. Chem. 53, 2494–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi H. K., Won L. A., Kontur P. J., Hammond D. N., Fox A. P., Wainer B. H., Hoffmann P. C., Heller A. (1991) Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 552, 67–76 [DOI] [PubMed] [Google Scholar]
- 28. Sun F., Anantharam V., Latchoumycandane C., Kanthasamy A., Kanthasamy A. G. (2005) Dieldrin induces ubiquitin-proteasome dysfunction in α-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. J. Pharmacol. Exp. Ther. 315, 69–79 [DOI] [PubMed] [Google Scholar]
- 29. Anantharam V., Kaul S., Song C., Kanthasamy A., Kanthasamy A. G. (2007) Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. Neurotoxicology 28, 988–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song C., Kanthasamy A., Anantharam V., Sun F., Kanthasamy A. G. (2010) Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells. Relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 77, 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang D., Anantharam V., Kanthasamy A., Kanthasamy A. G. (2007) Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson's disease. J. Pharmacol. Exp. Ther. 322, 913–922 [DOI] [PubMed] [Google Scholar]
- 32. Ghosh A., Kanthasamy A., Joseph J., Anantharam V., Srivastava P., Dranka B. P., Kalyanaraman B., Kanthasamy A. G. (2012) Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson's disease. J. Neuroinflammation 9, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghosh A., Chandran K., Kalivendi S. V., Joseph J., Antholine W. E., Hillard C. J., Kanthasamy A., Kanthasamy A., Kalyanaraman B. (2010) Neuroprotection by a mitochondria-targeted drug in a Parkinson's disease model. Free Radic. Biol. Med. 49, 1674–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy A., Ghosh A., Jana A., Liu X., Brahmachari S., Gendelman H. E., Pahan K. (2012) Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson's disease. PloS ONE 7, e38113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu X., Weng Z., Chu C. T., Zhang L., Cao G., Gao Y., Signore A., Zhu J., Hastings T., Greenamyre J. T., Chen J. (2011) Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J. Neurosci. 31, 247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castro D. S., Hermanson E., Joseph B., Wallén A., Aarnisalo P., Heller A., Perlmann T. (2001) Induction of cell cycle arrest and morphological differentiation by Nurr1 and retinoids in dopamine MN9D cells. J. Biol. Chem. 276, 43277–43284 [DOI] [PubMed] [Google Scholar]
- 37. Hirsch E., Graybiel A. M., Agid Y. A. (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature 334, 345–348 [DOI] [PubMed] [Google Scholar]
- 38. Kastner A., Hirsch E. C., Lejeune O., Javoy-Agid F., Rascol O., Agid Y. (1992) Is the vulnerability of neurons in the substantia nigra of patients with Parkinson's disease related to their neuromelanin content? J. Neurochem. 59, 1080–1089 [DOI] [PubMed] [Google Scholar]
- 39. Uchida T., Takamiya M., Takahashi M., Miyashita H., Ikeda H., Terada T., Matsuo Y., Shirouzu M., Yokoyama S., Fujimori F., Hunter T. (2003) Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem. Biol. 10, 15–24 [DOI] [PubMed] [Google Scholar]
- 40. Rudrabhatla P., Pant H. C. (2010) Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by Pin1. Implications for therapeutics in neurodegeneration. J. Alzheimers Dis. 19, 389–403 [DOI] [PubMed] [Google Scholar]
- 41. Butterfield D. A., Abdul H. M., Opii W., Newman S. F., Joshi G., Ansari M. A., Sultana R. (2006) Pin1 in Alzheimer's disease. J. Neurochem. 98, 1697–1706 [DOI] [PubMed] [Google Scholar]
- 42. Sultana R., Boyd-Kimball D., Poon H. F., Cai J., Pierce W. M., Klein J. B., Markesbery W. R., Zhou X. Z., Lu K. P., Butterfield D. A. (2006) Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus. A redox proteomics analysis. Neurobiol. Aging 27, 918–925 [DOI] [PubMed] [Google Scholar]
- 43. Kesavapany S., Patel V., Zheng Y. L., Pareek T. K., Bjelogrlic M., Albers W., Amin N., Jaffe H., Gutkind J. S., Strong M. J., Grant P., Pant H. C. (2007) Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol. Biol. Cell 18, 3645–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rudrabhatla P., Albers W., Pant H. C. (2009) Peptidyl-prolyl isomerase 1 regulates protein phosphatase 2A-mediated topographic phosphorylation of neurofilament proteins. J. Neurosci. 29, 14869–14880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lou H., Montoya S. E., Alerte T. N., Wang J., Wu J., Peng X., Hong C. S., Friedrich E. E., Mader S. A., Pedersen C. J., Marcus B. S., McCormack A. L., Di Monte D. A., Daubner S. C., Perez R. G. (2010) Serine 129 phosphorylation reduces the ability of α-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 285, 17648–17661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mbefo M. K., Paleologou K. E., Boucharaba A., Oueslati A., Schell H., Fournier M., Olschewski D., Yin G., Zweckstetter M., Masliah E., Kahle P. J., Hirling H., Lashuel H. A. (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Machiya Y., Hara S., Arawaka S., Fukushima S., Sato H., Sakamoto M., Koyama S., Kato T. (2010) Phosphorylated α-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J. Biol. Chem. 285, 40732–40744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pronin A. N., Morris A. J., Surguchov A., Benovic J. L. (2000) Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522 [DOI] [PubMed] [Google Scholar]
- 49. Sakamoto M., Arawaka S., Hara S., Sato H., Cui C., Machiya Y., Koyama S., Wada M., Kawanami T., Kurita K., Kato T. (2009) Contribution of endogenous G-protein-coupled receptor kinases to Ser-129 phosphorylation of α-synuclein in HEK293 cells. Biochem. Biophys. Res. Commun. 384, 378–382 [DOI] [PubMed] [Google Scholar]
- 50. Okochi M., Walter J., Koyama A., Nakajo S., Baba M., Iwatsubo T., Meijer L., Kahle P. J., Haass C. (2000) Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J. Biol. Chem. 275, 390–397 [DOI] [PubMed] [Google Scholar]
- 51. Inglis K. J., Chereau D., Brigham E. F., Chiou S. S., Schöbel S., Frigon N. L., Yu M., Caccavello R. J., Nelson S., Motter R., Wright S., Chian D., Santiago P., Soriano F., Ramos C., Powell K., Goldstein J. M., Babcock M., Yednock T., Bard F., Basi G. S., Sham H., Chilcote T. J., McConlogue L., Griswold-Prenner I., Anderson J. P. (2009) Polo-like kinase 2 (PLK2) phosphorylates α-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee K. W., Chen W., Junn E., Im J. Y., Grosso H., Sonsalla P. K., Feng X., Ray N., Fernandez J. R., Chao Y., Masliah E., Voronkov M., Braithwaite S. P., Stock J. B., Mouradian M. M. (2011) Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J. Neurosci. 31, 6963–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ai Z., Zhang W., Luo W., Huang L., Pan Y. (2009) Expression of Pin1 mRNA in non-small-cell lung cancer patients. Asian Cardiovasc. Thorac. Ann. 17, 157–161 [DOI] [PubMed] [Google Scholar]
- 54. Miyashita H., Mori S., Motegi K., Fukumoto M., Uchida T. (2003) Pin1 is overexpressed in oral squamous cell carcinoma and its levels correlate with cyclin D1 overexpression. Oncol. Rep. 10, 455–461 [PubMed] [Google Scholar]
- 55. Lv L., Zhou Z., Huang X., Zhao Y., Zhang L., Shi Y., Sun M., Zhang J. (2010) Inhibition of peptidyl-prolyl cis/trans isomerase Pin1 induces cell cycle arrest and apoptosis in vascular smooth muscle cells. Apoptosis 15, 41–54 [DOI] [PubMed] [Google Scholar]
- 56. Shen Z. J., Esnault S., Schinzel A., Borner C., Malter J. S. (2009) The peptidyl-prolyl isomerase Pin1 facilitates cytokine-induced survival of eosinophils by suppressing Bax activation. Nat. Immunol. 10, 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li Q. M., Tep C., Yune T. Y., Zhou X. Z., Uchida T., Lu K. P., Yoon S. O. (2007) Opposite regulation of oligodendrocyte apoptosis by JNK3 and Pin1 after spinal cord injury. J. Neurosci. 27, 8395–8404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barone M. C., Desouza L. A., Freeman R. S. (2008) Pin1 promotes cell death in NGF-dependent neurons through a mechanism requiring c-Jun activity. J. Neurochem. 106, 734–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Esnault S., Rosenthal L. A., Shen Z. J., Sedgwick J. B., Szakaly R. J., Sorkness R. L., Malter J. S. (2007) A critical role for Pin1 in allergic pulmonary eosinophilia in rats. J. Allergy Clin. Immunol. 120, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 60. Jeong H. G., Pokharel Y. R., Lim S. C., Hwang Y. P., Han E. H., Yoon J. H., Ahn S. G., Lee K. Y., Kang K. W. (2009) Novel role of Pin1 induction in type II collagen-mediated rheumatoid arthritis. J. Immunol. 183, 6689–6697 [DOI] [PubMed] [Google Scholar]
- 61. Hennig L., Christner C., Kipping M., Schelbert B., Rücknagel K. P., Grabley S., Küllertz G., Fischer G. (1998) Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37, 5953–5960 [DOI] [PubMed] [Google Scholar]
- 62. Ryo A., Liou Y. C., Wulf G., Nakamura M., Lee S. W., Lu K. P. (2002) PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 22, 5281–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee K. Y., Lee J. W., Nam H. J., Shim J. H., Song Y., Kang K. W. (2011) PI3-kinase/p38 kinase-dependent E2F1 activation is critical for Pin1 induction in tamoxifen-resistant breast cancer cells. Mol. Cells 32, 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y. C., Wulf G., Rottapel R., Yamaoka S., Lu K. P. (2003) Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12, 1413–1426 [DOI] [PubMed] [Google Scholar]
- 65. Mantovani F., Tocco F., Girardini J., Smith P., Gasco M., Lu X., Crook T., Del Sal G. (2007) The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat. Struct. Mol. Biol. 14, 912–920 [DOI] [PubMed] [Google Scholar]
- 66. Wulf G. M., Liou Y. C., Ryo A., Lee S. W., Lu K. P. (2002) Role of Pin1 in the regulation of p53 stability and p21 transactivation and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 277, 47976–47979 [DOI] [PubMed] [Google Scholar]