

Background: Parkin is a ubiquitin ligase activated by a decrease in the mitochondrial membrane potential (ΔΨm). However, details regarding its mechanism remain limited.
Results: PINK1-dependent phosphorylation of Parkin at Ser-65 following dissipation of ΔΨm triggers ubiquitin-ester transfer by the RING2 domain of Parkin to Cys-431.
Conclusion: Parkin catalyzes trans- (ubiquitin-thioester)ification upon PINK1-dependent phosphorylation.
Significance: The molecular process of Parkin-catalyzed ubiquitylation has been determined.
Keywords: Mitochondria, Parkin, Pink1, Ubiquitin, Ubiquitin Ligase, RING Finger
Abstract
PINK1 and PARKIN are causal genes for autosomal recessive familial Parkinsonism. PINK1 is a mitochondrial Ser/Thr kinase, whereas Parkin functions as an E3 ubiquitin ligase. Under steady-state conditions, Parkin localizes to the cytoplasm where its E3 activity is repressed. A decrease in mitochondrial membrane potential triggers Parkin E3 activity and recruits it to depolarized mitochondria for ubiquitylation of mitochondrial substrates. The molecular basis for how the E3 activity of Parkin is re-established by mitochondrial damage has yet to be determined. Here we provide in vitro biochemical evidence for ubiquitin-thioester formation on Cys-431 of recombinant Parkin. We also report that Parkin forms a ubiquitin-ester following a decrease in mitochondrial membrane potential in cells, and that this event is essential for substrate ubiquitylation. Importantly, the Parkin RING2 domain acts as a transthiolation or acyl-transferring domain rather than an E2-recruiting domain. Furthermore, formation of the ubiquitin-ester depends on PINK1 phosphorylation of Parkin Ser-65. A phosphorylation-deficient mutation completely inhibited formation of the Parkin ubiquitin-ester intermediate, whereas phosphorylation mimics, such as Ser to Glu substitution, enabled partial formation of the intermediate irrespective of Ser-65 phosphorylation. We propose that PINK1-dependent phosphorylation of Parkin leads to the ubiquitin-ester transfer reaction of the RING2 domain, and that this is an essential step in Parkin activation.
Introduction
Parkinson disease is a neurodegenerative disorder that commonly arises sporadically. In some cases, however, the disease is familial and inherited. PINK1 and PARKIN have been identified as the causal genes responsible for hereditary recessive early-onset Parkinsonism (1, 2). PINK1 is a mitochondrial Ser/Thr kinase, whereas Parkin is a ubiquitin ligase (E3) that catalyzes ubiquitin transfer from the ubiquitin-activating enzyme (E1) and the ubiquitin-conjugating enzyme (E2) to specific substrates. Although the molecular mechanisms underlying sporadic Parkinson disease and familial Parkinsonism are complex, PINK1 and Parkin have been shown to cooperate in the identification, labeling, and clearance of damaged mitochondria (3, 4). Dysfunction of either likely causes an accumulation of low-quality depolarized mitochondria, which triggers familial Parkinsonism (3–5).
The molecular basis of how PINK1 and Parkin maintain mitochondrial integrity has eluded researchers for many years; however, relatively recent data have provided significant insights. After escaping mitochondrial membrane potential (ΔΨm)-dependent degradation (6–9), PINK1 selectively localizes on low-quality mitochondria and is subsequently activated by an autophosphorylation mechanism (10). PINK1 then recruits the latent form of Parkin from the cytosol to the same mitochondria. Once ΔΨm decreases, the E3 activity of Parkin is activated (6) and it ubiquitylates outer mitochondrial membrane substrates such as hexokinase I, MitoNEET/CISD1, mitofusin (Mfn),4 miro, and voltage-dependent anion channel 1 (see Refs. 4 and 11–16 and references therein). The ubiquitylated mitochondrial proteins are degraded via the proteasome. As a consequence, damaged mitochondria are quarantined by decreased mitochondrial fusion, separated from the destination by a pause in kinesin-dependent trafficking, and/or degraded via autophagy. The final destination of low-quality mitochondria remains controversial (11, 17–23). Furthermore, PINK1 may also possess alternative functions other than Parkin recruitment (24, 25). One of the most poorly understood events of PINK1/Parkin-mediated mitochondrial quality control is how the E3 activity of Parkin is re-established by damaged mitochondria.
Mechanistic insights into the ubiquitin-ligating reaction of Parkin have been developing since 2000 (26–32). Parkin possesses multiple RING finger motifs. In vitro reconstitution assays revealed that the most carboxyl-terminal RING finger motif (RING2) encompasses the E3 catalytic core (31, 33). The RING1 and RING2 finger motifs are spanned by an in-between RING (IBR) domain, thus this type of E3 is categorized as a RING-IBR-RING (RBR) E3 class. In addition to Parkin, a number of E3s, such as HOIP (HOIL-1L interacting protein) and human homologue of ariadne (HHARI), belong to this class of ligases (34, 35). Recently, HHARI and HOIL-1L interacting protein were shown to form a thioester adduct with ubiquitin on a consensus cysteine in the RING2 domain, similar to the ubiquitin-cascading reaction of HECT (homologous to E6-AP carboxyl terminus)-type E3s (36–38). These results suggest that Parkin may also form a ubiquitin-thioester intermediate, even though it was not observed in the aforementioned paper (36). In 2013, Lazarou et al. (39) showed that ubiquitin-oxyester formation of a Parkin C431S mutant depended on a decrease in the mitochondrial membrane potential, thereby partially solving the aforementioned contradiction. The impact of that article, however, was diminished by the lack of biochemical evidence demonstrating ubiquitin-thioester formation with recombinant Parkin and the absence of a mechanism for how PINK1 regulates a ubiquitin-thioester adduct on the catalytic cysteine of Parkin.
In this study, we found that Parkin forms the ubiquitin-thioester intermediate on Cys-431 both in vitro and in cells, and revealed that the function of the RING2 domain during ubiquitylation is not E2 recruitment as suggested but ubiquitin-thioester transfer. We further determined that PINK1-dependent phosphorylation of Ser-65 in Parkin leads to formation of the ubiquitin-ester intermediate. These results provide crucial insights into the mechanisms of Parkin activation.
EXPERIMENTAL PROCEDURES
Cells, Plasmids, and Reagents
PINK1−/− MEFs complemented by wild type (WT) or various mutants PINK1 were established by infecting PINK1−/− MEFs with recombinant retroviruses as follows. PINK1 mutants were subcloned into a pMXs-puro vector, transfected into PLAT-E retrovirus packaging cells (40), and cultured at 37 °C for 24 h. After changing the medium, PLAT-E cells were further incubated at 37 °C for 24 h and the viral supernatant was collected and used for infection. PINK1−/− MEFs (41) were plated on 35-mm dishes 24 h before infection, and the medium was replaced with the undiluted viral supernatant described above with 8 μg/ml of Polybrene (Sigma). Two days later, transformants were selected in medium containing 5 μg/ml of puromycin.
HeLa cells and MEFs were cultured at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM, Sigma) containing 10% fetal bovine serum, penicillin/streptomycin, 1× non-essential amino acids (Invitrogen), and 1× sodium pyruvate (Invitrogen). To depolarize the mitochondria, cells were treated with 10–30 μm CCCP (Sigma) for 60–90 min unless otherwise specified.
Plasmids for expressing WT or various PINK1 and Parkin mutants have been described previously (6, 10, 11, 42) or were newly constructed by conventional methods. Plasmid transfections were performed using the transfection reagent FuGENE6 (Roche Applied Science) for HeLa cells. For PINK1-complemented PINK1−/− MEFs, the transfection reagent polyethylenimine (Polyscience) and the electroporation device Neon (Life technologies) were used.
In Vitro Ubiquitylation Assay
To obtain maltose-binding protein (MBP)-fused Parkin and MBP-IBR-RING2, PARKIN, or IBR-RING2, the respective domains were subcloned into a pMAL vector (New England Biolabs) and transfected into a BL21(DE3) RIL codon plus Escherichia coli strain (Stratagene). Recombinant proteins were purified by conventional methods in elution buffer containing 20 mm Tris-HCl (pH 7.4), 200 mm NaCl, 1 mm dithiothreitol (DTT), 100 μm ZnSO4, and 10 mm maltose. In vitro ubiquitylation assays were performed essentially as described previously with a reaction volume of 40 μl in each experimental condition (31). Briefly, the purified MBP-Parkin protein (10 μg/ml) was incubated in reaction buffer (50 mm Tris-HCl (pH 8.5 unless otherwise specified), 5 mm MgCl2, 2.5 mm ATP, 2 mm DTT) with 300 μg/ml of ubiquitin (Sigma), 100 nm recombinant mouse E1, and 1/100 diluted E2 UbcH7 (BioMol) at 32 °C for 3 h, and then subjected to immunoblotting (IB) with an anti-Parkin antibody. For identification of ubiquitin in Parkin C431S mutants, recombinant MBP-Parkin C431S or MBP-IBR-RING2 C431S proteins were subjected to the in vitro ubiquitylation assay described above with 210 μg/ml of HA-ubiquitin (R&D Systems) instead of intact ubiquitin.
For labeling with a ubiquitin-vinyl sulfone probe (Ub-VS), recombinant MBP-Parkin or MBP-IBR-RING2 proteins (about 20 μg/ml) were incubated with saturating amounts (about 25 μg/ml) of Ub-VS (Boston Biochem) in 30 μl of reaction buffer (50 mm Tris-HCl (pH 8.5), 50 mm NaCl) at room temperature for 3 h. Preincubation with N-ethylmaleimide (NEM, Wako chemicals) was performed for 10 min at room temperature at a final concentration of 10 mm.
IB, immunoprecipitation, and Immunofluorescence
To detect ubiquitylation via IB, lysates of HeLa cells or MEFs were collected in TNE-N+ buffer (150 mm NaCl, 20 mm Tris-HCl (pH 8.0), 1 mm EDTA, and 1% Nonidet P-40) in the presence of 10 mm NEM to protect ubiquitylated proteins from deubiquitylase activity. For IB-based phosphorylation analyses, lysates from MEFs, HeLa, or HEK293T cells described above were collected in the presence of PhosSTOP (Roche Applied Science) to protect phosphorylated proteins from phosphatase activity. For immunoprecipitation experiments, lysates of HeLa cells transiently expressing HA-Parkin with or without Myc6-ubiquitin were extracted by TNE-N+ buffer and reacted with HA-agarose (Sigma) for 1 h at 4 °C. After washing completely, SDS-PAGE sample buffer was added to the precipitates and eluates were subjected to IB. The anti-Parkin antibody PRK8 (Sigma, 1:1,500 dilution), anti-Mfn2 antibody ab56889 (Abcam, 1:500 dilution), anti-LDH antibody ab2101 (Abcam, 1:1,000 dilution), anti-HA antibody TANA2 (MBL, 1:1,000 dilution), anti-Myc antibody 9E-10 (Santa Cruz, 1:500 dilution), and anti-PINK1 antibody BC100–494 (Novus, 1:1,000 dilution) were used for the IB experiments. For oxyester detection, HeLa cells expressing a Parkin C431S mutant treated with or without CCCP were incubated at 37 °C for 20 min with 0.1 n NaOH before being subjected to SDS-PAGE.
For immunofluorescence experiments, HeLa cells were fixed with 4% paraformaldehyde, permeabilized with 50 μg/ml of digitonin, and stained with a primary antibody (1:500 dilution of anti-HA antibody F7 or 1:3,000 dilution of anti-Tom20 antibody FL-145, Santa Cruz Biotechnology) and a 1:2,000 dilution of the secondary antibody (Alexa Fluor 488- or 568-conjugated anti-mouse or rabbit IgG antibody, Invitrogen). Cells were imaged using a laser-scanning microscope (LSM510; Carl Zeiss, Inc.) and image contrast and brightness were adjusted in Photoshop (Adobe).
Cell Fractionation
For fractionation experiments, HEK293 cells were treated with 10 μm CCCP for 90 min and subsequently treated with 1 mm dithiobis(succinimidyl propionate) (Pierce) in PBS ± CCCP for 90 min on ice, inactivated by 10 mm glycine in PBS three times, and suspended in chappell-perry buffer (0.15 m KCl, 20 mm HEPES-NaOH, pH 8.1, 5 mm MgCl2, protease and phosphatase inhibitor (Roche)). Cells were disrupted by passaging 30 times through a 25-gauge needle (1-ml syringe), debris was removed by centrifugation at 1,000 × g for 7 min, and the supernatant was subjected to 10,000 × g for 10 min to separate the mitochondria-rich fraction from the cytosol-rich fraction.
Phos-tag Assay and Protein Phosphatase Treatment
To detect phosphorylated proteins via SDS-PAGE, 7.5–8% polyacrylamide gels containing 50 μm Phos-tag acrylamide (Wako chemicals) and 100 μm MnCl2 were used. After electrophoresis, Phos-tag acrylamide gels were washed with gentle shaking in transfer buffer containing 0.01% SDS and 1 mm EDTA for 10 min and then incubated in transfer buffer containing 0.01% SDS without EDTA for 10 min according to the manufacturer's protocol. Proteins were transferred to PVDF membranes and analyzed by conventional IB as described above.
For protein phosphatase treatment, cell lysates of intact HEK293T cells ± CCCP treatment were incubated with λ-protein phosphatase (New England Biolabs) for 1 h at the indicated temperature in reaction buffer prepared according to the manufacturer's instructions, and then subjected to Phos-tag PAGE described above.
LC-MS/MS Analysis of GST-Parkin
GST-Parkin from CCCP-treated and untreated cells was subjected to SDS-PAGE and stained with Coomassie Brilliant Blue. GST-Parkin protein bands were excised, reduced, alkylated, and digested with trypsin (Promega) in 50 mm ammonium bicarbonate for 16 h at 37 °C. The resultant peptides were analyzed on a Q Exactive mass spectrometer (Thermo Scientific) with the raw data processed using Xcalibur (Thermo Scientific). The Mascot generic format files were searched against the NCBI non-redundant protein database restricted to Homo sapiens using the MS/MS ion search tool of the Mascot program (Matrix Science).
Cell-free Ubiquitylation Assays
HeLa cells expressing GFP-Parkin, HA-Parkin, or HA-Parkin with various mutations were homogenized in cell-free assay buffer (20 mm HEPES-KOH (pH 7.5), 220 mm sorbitol, 10 mm KAc, 70 mm sucrose) supplemented with protease inhibitor mixture minus EDTA (Roche). Cells were disrupted by passaging 30 times through a 25-gauge needle and cell homogenates were centrifuged at 800 × g for 10 min at 4 °C to obtain a postnuclear supernatant and then cytosolic fractions were collected by further centrifugation at 20,400 × g for 10 min at 4 °C. The final yields were 100 μl from 1 ml of confluent cell culture. For mitochondrial isolation, HeLa cells or MEFs expressing only endogenous or exogenous PINK1-FLAG were treated with 10 μm CCCP for 3 h followed by homogenization in the aforementioned cell-free assay buffer. Postnuclear supernatants were obtained by centrifugation as above and mitochondria were pelleted by further centrifugation at 10,000 × g for 20 min at 4 °C.
To initiate the cell-free ubiquitylation assay, HeLa cytosols with exogenous Parkin were supplemented with 2 mm DTT, 5 mm MgCl2, 5 mm ATP, and 1% glycerol. Mitochondria isolated from CCCP-treated confluent cells in 10 ml of medium were re-suspended in 100 μl of Mg/ATP-supplemented cytosol and incubated at 30 °C for 90 min.
RESULTS
A Parkin C431S Mutation Inhibits Substrate Ubiquitylation via Ubiquitin-Oxyester Adduct Formation
Pioneering work by Klevit's group (36) showed that the active cysteine (Cys-357) in the RING2 domain of RBR-type E3 HHARI forms a ubiquitin-thioester intermediate during the ubiquitin-ligating reaction. In the case of Parkin, Cys-431 is equivalent to HHARI Cys-357. Perplexingly, however, ester-linked ubiquitin of Parkin was not observed in that report even under thioester-stabilizing conditions (36). We previously demonstrated that the enzymatic function of Parkin in cells is activated upon dissipation of ΔΨm (6), and thus examined whether the ubiquitin-thioester formation on Cys-431 is specifically observed when cells were treated with the mitochondrial uncoupler CCCP. In this experiment, Parkin Cys-431 was mutated to Ser thereby converting an unstable ubiquitin-thioester bond to a stable ubiquitin-oxyester bond. When hemagglutinin (HA)-tagged Parkin (HA-Parkin) with the C431S mutation was expressed in HeLa cells, a higher molecular mass population compared with wild type Parkin (WT-Parkin) was observed following CCCP treatment (Fig. 1A, lane 6). The modification resulted in a 6–7 kDa increase in the molecular weight of Parkin, suggesting ubiquitin-oxyester formation at Ser-431. This modification disappeared with a C431A ester-deficient mutation (lane 4). Co-expression of Myc1-tagged ubiquitin retarded the mobility of this band (Fig. 1B). When Myc6-tagged ubiquitin was co-expressed with the HA-Parkin C431S mutant and Parkin was subjected to immunoprecipitation with an anti-HA antibody, the retarded band was specifically detected by the anti-Myc antibody. These results confirmed that the modification was derived from ubiquitin conjugation (Fig. 1C).
FIGURE 1.

A, a higher molecular mass population (indicated by the red asterisk) was specifically observed in the Parkin C431S mutant following CCCP treatment in HeLa cell lysates. B, immunoblotting of the Parkin C431S mutant was repeated in the absence or presence of Myc1-ubiquitin (Ub) co-expression. The slower migrating band resolved as a doublet because of the endogenous-ubiquitin adduct (indicated by a red arrow) and the Myc1-ubiquitin adduct (black arrow). C, HeLa cell lysates co-expressing HA-Parkin and Myc6-ubiquitin were immunoprecipitated with an anti-HA antibody, followed by immunoblotting with the indicated antibodies. The anti-Myc antibody specifically detected the modified Parkin(C431S) mutant. Red asterisk shows Parkin with the endogenous-ubiquitin adduct; blue asterisk shows Parkin with the exogenous Myc6-ubiquitin adduct; black asterisk indicates the cross-reacting band. D and E, Cys-431 of Parkin is important for substrate ubiquitylation. Both ubiquitylation of a pseudo-substrate (D) and a genuine substrate Mfn2 (E) were inhibited in the transfected cells by C431A and C431S mutations of Parkin. The red asterisks indicate the oxyester-linked ubiquitin and black asterisks indicate ordinary substrate ubiquitylation. F, HeLa cells expressing HA-Parkin mutants harboring the C431S mutation and one of the disease-relevant mutations (K211N, C352G, and T415N) were subjected to immunoblotting following CCCP treatment. The red asterisk indicates the ubiquitin-oxyester band. G, Parkin co-localization with mitochondria was analyzed in >100 cells per Cys-431 mutation. Example figures indicative of robust colocalization (counted as 1) and the absence of colocalization (counted as 0) are shown on the right (bars, 10 μm). Error bars represent the mean ± S.D. values of three experiments. Statistical significance was calculated using Welch's t test; NS, not significant. H, the ubiquitylated form of C431S Parkin (marked by a red asterisk) is sensitive to NaOH treatment, confirming the presence of the oxyester adduct.
We next checked whether Cys-431 is essential for substrate ubiquitylation. Amino-terminal fused lysine-rich proteins can function as Parkin pseudosubstrates (31). Consequently, lysine-rich GFP can be ubiquitylated in cells when fused in-frame with Parkin (6). The HA tag, in contrast, does not contain lysine residues and thus cannot function as a ubiquitylation pseudosubstrate. This is the reason why ubiquitylation of WT HA-Parkin was not observed in Fig. 1A. GFP-Parkin was ubiquitylated following CCCP treatment (Fig. 1D, lane 2), whereas the C431A mutation completely blocked ubiquitylation (lane 4). The GFP-Parkin C431S mutant was observed as a doublet with only a single additional band, which was putatively derived from ubiquitin-oxyester-Parkin (Fig. 1D, lane 6). Mitofusin 1/2 (Mfn1/2) is a genuine substrate of Parkin and is ubiquitylated upon dissipation of ΔΨm (17, 43–45). WT HA-Parkin ubiquitylated Mfn2 following CCCP treatment (Fig. 1E, lane 2), whereas no Mfn2 ubiquitylation was observed with either the C431A or the C431S mutation (lanes 4 and 6), confirming that Cys-431 is essential for substrate ubiquitylation. During preparation of this article, Lazarou et al. (39) independently published results showing that the Parkin C431S mutant forms a ubiquitin-oxyester upon CCCP treatment and is unable to ubiquitylate Mfn1. To examine whether pathogenic mutations of Parkin affect the thioester formation, we selected three mutants (K211N, C352G, and T415N) as representative defects for mitochondrial translocation, mitochondrial ubiquitylation, and E3 activity, respectively (6, 42). When HA-Parkin mutants harboring C431S and one of the disease-relevant mutations above (K211N, C352G, or T415N) were subjected to immunoblotting following CCCP treatment, no ubiquitin-oxyester adduct was observed (Fig. 1F), indicating that the three Parkin pathogenic mutations examined compromised formation of the ubiquitin-ester. The monoubiquitylated form of C431S Parkin was sensitive to NaOH treatment (Fig. 1H), confirming oxyester-linked ubiquitylation.
Direct Biochemical Evidence for a Ubiquitin-ester Adduct on Parkin Cys-431
Results shown in Fig. 1, D and E, imply that ubiquitin-ester formation on Cys-431 is essential for Parkin-catalyzed ubiquitylation. However, because the Parkin C431F and C431S mutants do not translocate to the mitochondria following CCCP treatment (39, 46), we are hesitant to overinterpret the previous results. We consequently further confirmed that both mutations strongly (albeit not completely) inhibited translocation of Parkin to depolarized mitochondria (Fig. 1G). Thus the defect in Mfn2 ubiquitylation shown in Fig. 1E could be attributable to mislocalization of the Parkin C431A/S mutants rather than enzymatic dysfunction. To separate the effect of subcellular mislocalization from biochemical function, we next measured the E3 activity of Parkin mutants.
Previously, we reconstituted the E3 activity of Parkin in vitro using recombinant Parkin or the IBR-RING2 domain (Fig. 2A) purified from E. coli, and found that the C431F pathogenic mutation completely inhibited E3 activity (31), indicating that Cys-431 is essential for E3 activity. We consequently performed an in vitro reconstitution assay using recombinant MBP-fused Parkin with the C431S mutation. Similar to the GFP-Parkin ubiquitylation observed in cells (Fig. 1D), MBP-Parkin in vitro exhibited multiple ubiquitylation bands, whereas the MBP-Parkin C431S resolved as only a doublet, which is equivalent to the singly ubiquitylated form (Fig. 2B, lanes 2 and 3). This band was not observed in the ester-deficient C431A mutant (lane 4). Furthermore, exclusion of ubiquitin from the reaction completely quenched modification of the Parkin C431S mutant (lane 5). Clearer results were obtained when the Parkin deletion mutant (MBP-IBR-RING2; Fig. 2A) was used (Fig. 2C). WT IBR-RING2 exhibited multiple ubiquitylation bands (lane 2), whereas the IBR-RING2 C431S mutant was observed as a singly ubiquitylated form (lane 4). When in vitro ubiquitylation was performed with recombinant HA-ubiquitin and followed by immunoblotting with anti-HA and anti-Parkin antibodies, the anti-HA antibody specifically detected the modified C431S mutants (Fig. 2D), confirming that in vitro modification was, as shown in cells, derived from ubiquitin conjugation. Next, we excluded each reaction component (ubiquitin, E1, and E2) from the assay because we previously showed that Parkin can catalyze E2-independent ubiquitylation at high pH in vitro (30). Ubiquitin-oxyester formation of the Parkin C431S mutant was completely inhibited by the exclusion of ubiquitin or E1 from the reaction (Fig. 2E, lanes 2, 3, 6, and 7). In contrast, even in the absence of E2, the ubiquitin-oxyester adduct was observed (lanes 4 and 8), suggesting that the IBR-RING2 domain catalyzes ubiquitin-oxyester formation not by E2 recruitment but by discharge and transfer of the ubiquitin-thioester moiety to itself (see “Discussion”). We checked the pH dependence of this reaction, and found that ubiquitin-oxyester formation was weak at pH 7.0 (Fig. 2F) but became evident when the reaction pH was increased to 8.5 (Fig. 2E). This is consistent with our previous results demonstrating that the E3 in vitro activity of MBP-Parkin is the highest under weak alkaline conditions (31). We speculate that the reactivity of the nucleophiles involved in the ubiquitin transfer reaction is affected by the relatively high pH.
FIGURE 2.

Reconstitution of ubiquitin-oxyester formation using recombinant MBP-Parkin protein in vitro. A, schematic depiction of the domain structure of Parkin and the deletion mutant (IBR-RING2). B, in vitro E3 activity of Parkin C431 mutants. MBP-Parkin with the C431A or C431S mutation was purified from E. coli and reconstituted with ATP, ubiquitin, E1, and E2. Ub indicates ubiquitin, the red asterisk indicates the oxyester-linked ubiquitin and the black asterisk indicates conventional isopeptide-linked ubiquitylation, unless otherwise specified. C, E3 activity of the Parkin IBR-RING2 domain ± C431S mutation. D, observed variances in the MBP-Parkin C431S and MBP-IBR-RING2 C431S mutants are the result of ubiquitylation. In vitro ubiquitylation was performed with recombinant HA-ubiquitin and followed by immunoblotting with the indicated antibodies. The anti-HA antibody specifically detected the modified Parkin(C431S) mutants. E, ubiquitin-oxyester formation of MBP-Parkin and MBP-IBR-RING2 with the C431S mutation in the absence of ubiquitin, E1 or E2, or in the presence of all three components. F, ubiquitin-oxyester formation of MBP-IBR-RING2 (C431S) was repeated as E, except at neutral pH conditions (pH 7.0).
We further examined whether ubiquitin is attached to the WT Parkin catalytic cysteine. Ubiquitin- or ubiquitin-like protein-derived probes with electrophilic C-terminal ends, such as Ub-VS and ubiquitin-vinylmethyl ester (Ub-VME) react specifically with the cognate-conjugating and -deconjugating enzymes (47, 48). For example, Ub-VME attached to E1, E2, and HECT-type E3s (47). Importantly, these adducts are formed through a Michael addition reaction of the active-site cysteine thiol of the conjugating or deconjugating enzyme with the C-terminal electrophilic (e.g. vinyl sulfone) moiety (48) (Fig. 3A).
FIGURE 3.

A, reaction scheme for in vitro labeling experiments performed in B to E using the active site-directed probe Ub-VS. B, Ub-VS conjugates to Parkin and IBR-RING2 proteins. C and D, the Ub-VS adduct was inhibited by preincubation of IBR-RING2 with NEM (C) or the C431S mutation (D). E, C431S is the lone free cysteine mutation to specifically inhibit Ub-VS conjugation. F, E3 activity of Parkin lacking the Ubl and RING1 domains in cells. HeLa cells expressing GFP-Parkin or GFP-IBR-RING2 with the C431A or T415N mutation were treated with CCCP and subjected to immunoblotting. G, GFP-IBR-RING2 catalyzes autoubiquitylation in cells irrespective of a decrease in ΔΨm. H, cytosolic localization of GFP-IBR-RING2 following CCCP treatment. The mitochondrial localization of GFP-Parkin following CCCP treatment is shown as a control.
When Parkin was incubated with Ub-VS, it led to the appearance of a faint modification with a molecular weight consistent with a Parkin-Ub-VS adduct (Fig. 3B, lane 2). A clearer signal was obtained when MBP-IBR-RING2 was used (lane 4). Incubation of Parkin with WT ubiquitin lacking the C-terminal vinyl sulfone moiety did not lead to this modification (lanes 1 and 3), suggesting that covalent modification of Parkin depends on the C-terminal electrophilic substituent of ubiquitin. This modification by Ub-VS was blocked by pretreatment with the cysteine-directed sulfhydryl alkylating agent NEM, confirming the cysteine-dependence of adduct formation (Fig. 3C, lanes 2 and 4). Moreover, this reaction was not observed in the Parkin C431S mutant (Fig. 3D, lane 4). The IBR-RING2 domain has 17 cysteine residues, 14 of which coordinate with zinc ions (49, 50). Consequently only 3 cysteine residues are free in IBR-RING2 domain: Cys-323, Cys-431, and Cys-451. We serially substituted these 3 Cys residues with Ser, and found that Ub-VS still conjugated to C323S and C451S mutants equivalent to WT (Fig. 3E), suggesting that Cys-431 is the only ligatable cysteine within the IBR-RING2 domain. Taken together, we conclude that Cys-431 is an active-site cysteine in Parkin crucial for ubiquitin ligation and is consequently labeled by Ub-VS.
Collectively, the results shown in Figs. 2 and 3 reveal that Parkin forms a ubiquitin-thioester on Cys-431, and suggest that impaired substrate ubiquitylation by the Parkin C431S mutant (Fig. 1, D and E) is attributable to both aberrant subcellular localization and the trapping of ubiquitin in this dead-end pseudo-intermediate on Ser-431. We observed no difference in the mitochondrial localization between the C431A (ubiquitin-ester deficient) and C431S (ubiquitin-oxyester stabilized) mutants (Fig. 1G), suggesting that the ubiquitin-ester itself does not promote the translocation of Parkin to depolarized mitochondria.
We also examined the ubiquitylation activity of the IBR-RING2 domain of Parkin toward a pseudosubstrate (GFP) in cells, and found that GFP-IBR-RING2 catalyzed ubiquitylation, which was blocked by a T415N or C431A mutation (Fig. 3F). The RING1 domain of HHARI functions as the “ubiquitin-conjugated E2” recruiting domain and is essential for ubiquitin ligation (36), thus a lone IBR-RING2 domain without the Parkin RING1 domain catalyzing pseudosubstrate ubiquitylation both in vitro (Fig. 2C) and in cells (Fig. 3F) is unexpected. We consequently examined the CCCP dependence and subcellular localization of GFP-IBR-RING2, and found that GFP-IBR-RING2 undergoes autoubiquitylation irrespective of CCCP treatment and mitochondrial localization (Fig. 3, G and H). These results suggest that IBR-RING2 becomes a constitutively active form because the autoinhibitory effect is prevented. The Parkin structure (50) is consistent with this result as RING0 occludes Cys-431 of RING2 via RING0-RING2 interactions. Although IBR-RING2 can catalyze ubiquitylation, the results shown in Figs. 2 and 3 do not indicate that RING1 and the interaction with E2 are physiologically dispensable because there are many pathogenic missense mutations in the Parkin RING1 domain (3). Rather Figs. 2 and 3 imply that the underlying mechanism during the ubiquitin ligating reaction is different between the RING1 and RING2 domains, even though both domains contribute cooperatively to ubiquitin ligation (see “Discussion”).
PINK1 Is Essential for Formation of the Ester-linked Parkin-Ubiquitin Intermediate
We next checked the effect of PINK1 on the ubiquitin-ester formation of Parkin. In MEFs prepared from PINK1 knock-out (PINK1−/−) mice (41), the formation of the ubiquitin-oxyester in the Parkin C431S mutant was completely impeded even following CCCP treatment (Fig. 4A, lane 1). Subsequent transfection of WT PINK1 complemented the defect (lane 2), revealing that PINK1 is essential for Parkin ubiquitin-oxyester formation. To investigate the role of mitochondrial localization, kinase activity, and the effect of various pathogenic mutations of PINK1 on Parkin ubiquitin-oxyester formation, we co-expressed the various PINK1 mutants with the Parkin C431S mutant in PINK1−/− MEFs. A PINK1 N-terminal deletion mutant lacking the terminal 155 amino acids, which are critical for mitochondrial localization of PINK1 (51), and kinase-dead (KD) mutations (K219A, D362A, and D384A) that abolish PINK1 kinase activity (52), completely blocked complementation of ubiquitin-oxyester formation in the Parkin C431S mutant (Fig. 4A, lanes 3 and 4). Among the various pathogenic PINK1 mutations that cause early-onset familial Parkinson disease, the C92F mutant supported Parkin ubiquitin-oxyester formation equivalent to WT PINK1 (Fig. 4A, lane 5). In contrast, the other PINK1 point mutations (i.e. A168P, E240K, H271Q, G309D, L347P, G386A, G409V, E417G, and 534insQ) severely hindered ubiquitin-oxyester formation (lanes 6–14). The C92F mutation was identified from a sporadic case carrying the compound heterozygote missense mutations (C92F and R464H) but not identified in the lineage (53), suggesting that C92F may represent a natural rare variant and is not a true disease-causing mutation.
FIGURE 4.
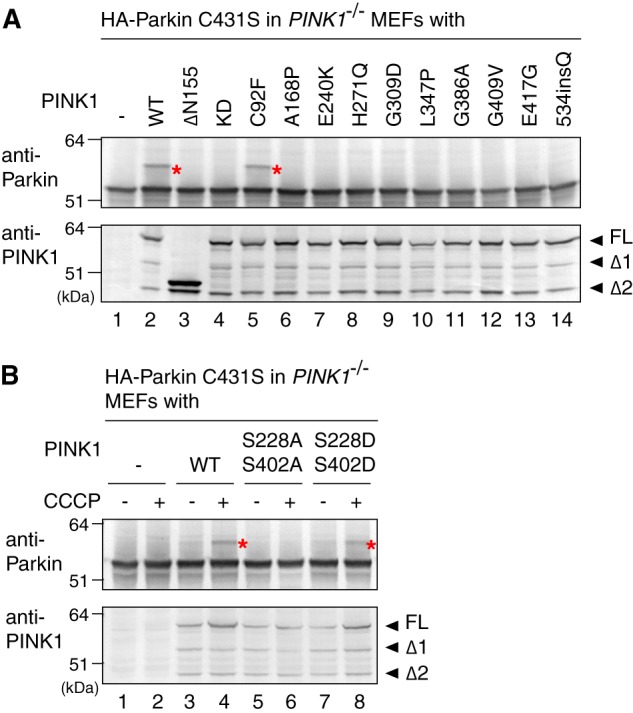
A and B, PINK1−/− MEFs co-expressing C431S Parkin mutant and various pathogenic (A) or autophosphorylation-relevant (B) PINK1 mutants were subjected to immunoblotting using an anti-Parkin antibody for detection of ubiquitin-oxyester formation (upper panel) or an anti-PINK1 antibody to confirm expression of the PINK1 mutants (lower panel). The red asterisks indicate the ubiquitin-oxyester band. FL, full-length PINK1; Δ1 and Δ2, the amino-terminal processed forms as reported in Ref. 6.
We next examined the effect of PINK1 autophosphorylation on formation of the Parkin-ubiquitin intermediate. We recently showed that dissipation of ΔΨm triggers PINK1 autophosphorylation of Ser-228 and Ser-402. This autophosphorylation is critical for Parkin recruitment to the same mitochondria (10). When WT PINK1, PINK1 with a S228A/S402A double mutation (autophosphorylation-deficient form), or PINK1 with a S228D/S402D double mutation (autophosphorylation mimic form) were expressed in PINK1−/− MEFs at the appropriate expression level, we found that the S228D/S402D mutant promoted ubiquitin-oxyester formation of the Parkin C431S mutant similar to WT PINK1 (Fig. 4B, lane 8). The S228A/S402A mutant, in contrast, failed to support formation of the ubiquitin adduct on Parkin (lane 6). Taken together, the results shown in Fig. 4 suggest that mitochondrial localization, kinase activity, and autophosphorylation of PINK1 are essential for formation of the ubiquitin-thioester intermediate on Parkin Cys-431.
Parkin Phosphorylation Is Dependent on Both ΔΨm Dissipation and PINK1
Because PINK1 is a Ser/Thr kinase, the simplest model is that PINK1 phosphorylation of Parkin accelerates formation of the ubiquitin-thioester intermediate. However, PINK1 phosphorylation of Parkin has, until recently, been controversial (7, 54–56). To detect a potentially phosphorylated form of Parkin with high sensitivity, we performed electrophoresis using polyacrylamide gels conjugated with a 1,3-bis[bis(pyridin-2-yl-methyl)amino]propan-2-olato diMn(II) complex (referred to hereafter as Phos-tag). Because Phos-tag can capture phosphomonoester dianions (ROPO32−), the phosphorylated Parkin can be easily distinguished from the non-phosphorylated form as a slower migrating band (57). Because mobility in Phos-tag PAGE does not reflect the molecular weight (58), the necessity of molecular weight markers is not critical.
When Parkin overexpressed in HeLa cells was subjected to Phos-tag PAGE, a clear mobility shift was observed following CCCP treatment, implying that Parkin underwent phosphorylation in response to mitochondrial damage (Fig. 5A, lanes 1 and 2; note that the ester-ubiquitin derived band was only observed with the C431S mutation and was thus undetectable in this experiment). Unlike HeLa cells, HEK293T cells express the PARKIN gene (59, 60). We consequently used this cell line to determine whether endogenous Parkin is also phosphorylated. Although the extended exposure necessary to detect endogenous Parkin resulted in an intense cross-reacting band (Fig. 5A, arrow), we confirmed that endogenous Parkin is also phosphorylated (lanes 3 and 4). λ-Phosphatase treatment abolished the high-molecular weight shift of this endogenous Parkin (Fig. 5B), confirming that Parkin is indeed phosphorylated. We used PINK1−/− MEFs to further examine the role of PINK1 on Parkin phosphorylation. In PINK1+/+ MEFs, Parkin underwent phosphorylation upon CCCP treatment (Fig. 5C, lane 4). In contrast, no detectable phosphorylation was observed in PINK1−/− MEFs (lane 5). However, the introduction of PINK1 in PINK1−/− MEFs complemented the phosphorylation of Parkin (lane 6), confirming that the defect was caused by the lack of endogenous PINK1. Co-expression of pathogenic PINK1 mutations with HA-Parkin in PINK1−/− MEFs severely compromised Parkin phosphorylation (Fig. 5D). The lone exception was the C92F mutation. This effect of the PINK1 disease-relevant mutations on Parkin phosphorylation is consistent with the observed effect on Parkin ubiquitin-oxyester formation (compare Fig. 4A with Fig. 5D), implying an underlying link between these two events. In fractionation experiments using HEK293 cells, endogenous Parkin is recovered in the mitochondria-rich fraction following CCCP treatment (6). Phos-tag PAGE confirmed that Parkin recovered in the mitochondrial fraction is the phosphorylated form (Fig. 5E, lane 6), suggesting that Parkin phosphorylation is important for mitochondrial localization.
FIGURE 5.

A, exogenous or endogenous Parkin underwent phosphorylation following CCCP treatment. The cell lysate of exogenous Parkin-expressing HeLa cells or intact HEK293T cells ± CCCP treatment were subjected to Phos-tag PAGE. Note that mobility does not reflect the molecular weight of proteins in Phos-tag PAGE (58) and thus molecular weight markers are not shown. The blue asterisks indicate phosphorylated Parkin, whereas the arrows indicate the cross-reacting band unless otherwise specified. B, protein phosphatase treatment in cell lysates caused the high-molecular shift of endogenous Parkin to disappear. C, phosphorylation of Parkin following CCCP treatment in cells depends on PINK1. Parkin was not phosphorylated in PINK1−/− MEFs following CCCP treatment, whereas exogenous PINK1 complemented the aforementioned defect. D, phosphorylation of Parkin was severely compromised by various pathogenic mutations of PINK1. Parkin C431S mutant-expressing PINK1−/− MEFs complemented by disease-relevant PINK1 were treated with CCCP and subjected to immunoblotting. FL, full-length PINK1; Δ1 and Δ2, the amino-terminal processed form as described in the legend to Fig. 4A. E, intact HEK293T cells ± CCCP treatment were subjected to fractionation experiments. Cyt and Mt indicate the cytosol-rich supernatant and the mitochondria-rich membrane pellet, respectively. Arrowheads indicate endogenous Parkin.
We next sought to identify the Parkin phosphorylation site. Although numerous papers have reported Parkin phosphorylation, the phosphorylation site(s) remains debatable. To date, Ser-65 (56, 61), Ser-101 (62, 63), Ser-108 (64), Ser-127 (63), Ser-131 (56, 62, 63, 65), Ser-136 (62, 65), Thr-175 (66), Thr-217 (66), Ser-296 (62), and Ser-378 (62, 63) have been reported as phosphorylation sites. We consequently serially substituted these Ser/Thr residues with Asp. When these Parkin mutants were subjected to Phos-tag PAGE following CCCP treatment, almost all of the mutations, excluding S65D (described later), had a clear phosphorylation-derived signal following CCCP treatment (Fig. 6A). We thus suggest that these amino acids are not the Parkin phosphorylation sites associated with mitochondrial damage. In sharp contrast, mutating Ser-65 in the amino-terminal ubiquitin-like (Ubl) domain of Parkin (Fig. 2A) to Ala, Asp, or Glu prevented phosphorylation following CCCP treatment (Fig. 6B, lanes 4, 6, and 8). This is consistent with the claim by Muqit and Hattori (56, 61) and co-workers that Ser-65 is the genuine Parkin phosphorylation site. To determine the phosphorylation site directly, we performed mass spectrometric analysis of phosphorylated Parkin. Because Parkin autoubiquitylation impedes the detection of phosphorylation, a T415N Parkin mutant was used. Glutathione S-transferase (GST)-fused Parkin (T415N) was integrated into the genome of HeLa cells by retrovirus-mediated transformation. GST-Parkin(T415N) was then purified from this stable cell line following ±CCCP treatment, and subjected to LC-MS/MS analysis after trypsin digestion. A peptide equivalent to amino acids 52–75 (NDWTVQNCDLDQQSIVHIVQRPWR) was identified as a putative phosphorylated peptide. Although the unphosphorylated peptide was detected from both CCCP-treated and untreated cells, the phosphorylated peptide was only detected in CCCP-treated cells (Fig. 6C). MS/MS data further suggested that Ser-65 was phosphorylated (Fig. 6D). Taken together, Fig. 6 indicates that Ser-65 is the Parkin phosphorylation site.
FIGURE 6.

Parkin is phosphorylated at Ser-65 following CCCP treatment. A, Ser to Asp substitution at various putative phosphorylation sites did not alter the phosphorylation pattern of Parkin in HeLa cells upon dissipation of ΔΨm. Blue asterisks indicate phosphorylated Parkin unless otherwise specified. Note that mobility does not reflect the molecular weight in Phos-tag PAGE (58) and thus molecular weight markers are not shown. B, the S65A, S65D, and S65E mutations dramatically decreased the phosphorylation of Parkin in cells. C, mass spectrometric analysis of the in cell phosphorylation site of Parkin. GST-Parkin purified from cells ± CCCP treatment was subjected to LC-MS/MS analysis. A phosphorylated peptide equivalent to amino acids 52–75 was detected only in CCCP-treated cells. D, the MS/MS data suggest that phosphorylation occurs at Ser-65.
Phosphorylation of Ser-65 Is Essential for Ubiquitin-Ester Formation of Parkin
We examined the role of this phosphorylation on Parkin ubiquitin-oxyester formation. Phos-tag PAGE analysis of a C431S Parkin mutant harboring the S65A, S65D, or S65E mutations revealed that the phosphorylation-derived band was absent in all cases (Fig. 7A) as well as the individual S65A, S65D, or S65E mutations (Fig. 6B). We subsequently examined formation of the ubiquitin-oxyester in these mutants. The S65A/C431S mutant was unable to form the ubiquitin-oxyester band on HA-Parkin (Fig. 7B, lanes 7–9), whereas the S65D/C431S and S65E/C431S mutants partially complemented ubiquitin-oxyester formation (Fig. 7B, lanes 12 and 15), although neither underwent phosphorylation (Fig. 7A). When these HA-Parkin mutants were co-expressed with Myc6-tagged ubiquitin and subjected to immunoprecipitation with an anti-HA antibody, the retarded band was specifically detected by the anti-Myc antibody, confirming that the modification was derived from ubiquitin conjugation (Fig. 7C). We also generated a Parkin S65T mutant that can potentially act as a substrate for the Ser/Thr kinase PINK1. The Parkin S65T mutant underwent phosphorylation equivalent to WT Parkin as expected (Fig. 7D), and formed a clear ubiquitin-oxyester band (Fig. 7E). The Ser-65 mutation affected mitochondrial localization of Parkin(C431S) following CCCP treatment (Fig. 7F). The S65A mutation in particular had a pronounced negative effect on the translocation of Parkin to damaged mitochondria. Thus the S65A mutation could inhibit ubiquitin-oxyester formation via subcellular mislocalization.
FIGURE 7.

A, the S65A/S65D/S65E mutations decreased the phosphorylation of ubiquitin ester-stabilized Parkin C431S mutants in cells. Blue asterisks indicate phosphorylated Parkin, unless otherwise specified. B, straight immunoblotting of HeLa cell lysates expressing Parkin harboring double mutations (i.e. C431S with the S65A, S65D, or S65E mutation). The red asterisks indicate ubiquitin-oxyester formation unless otherwise specified. The S65A/C431S Parkin mutant disturbed the ubiquitin-ester formation completely, whereas the S65D/C431S and S65E/C431S mutants partially complemented ubiquitin-ester formation. C, immunoprecipitation with an anti-HA antibody followed by immunoblotting with the indicated antibodies confirmed that Myc6-ubiquitin is conjugated to the Parkin C431S, S65D/C431S, and S65E/C431S mutants. Red asterisks show Parkin with the Myc6-ubiquitin adduct. D and E, the Parkin S65T or S65T/C431S mutants underwent phosphorylation (D) and exerted ubiquitin-oxyester formation (E) equivalent to WT or C431S mutant in cells. F, the number of HeLa cells with Parkin localized to the mitochondria per C431S/S65X mutation following CCCP treatment for 1 h was counted in >100 cells as Fig. 1G. Error bars represent the mean ± S.D. values and statistical significance was calculated using Welch's t test.
To demonstrate the importance of Ser-65 phosphorylation for ubiquitin-ester formation more convincingly, we established the in vitro assay referred to by Lazarou et al. (39). When cytosol-containing GFP-Parkin derived from mitochondria-intact HeLa cells was incubated in vitro with mitochondria isolated from CCCP-treated or non-treated cells, ubiquitylation of GFP-Parkin and Mfn2 were specifically observed in the reaction containing CCCP-pretreated mitochondria (Fig. 8A). Addition of recombinant ubiquitin, E1, and E2 accelerates the ubiquitylation (lane 6). Ubiquitylation of GFP-Parkin and Mfn2 was not observed in the reaction containing mitochondria isolated from CCCP-treated PINK1−/− MEFs, whereas exogenous PINK1 complement the ubiquitylation (Fig. 8B), revealing that PINK1 is essential. A Parkin C431S mutant specifically forms an ubiquitin-oxyester in this assay in the presence of CCCP-pretreated mitochondria (Fig. 8C). We then examined the effect of Ser-65 mutations on ubiquitin-oxyester formation at Ser-431. Even under in vitro experimental conditions, the phosphorylation-deficient S65A mutation completely inhibited ubiquitin-oxyester conjugation of HA-Parkin (Fig. 8D, lane 6), whereas the S65E/C431S mutation weakly complemented ubiquitin-oxyester formation (Fig. 8D, lane 8). These results suggest that phosphorylation of Parkin Ser-65 is important for ubiquitin-oxyester formation.
FIGURE 8.

A, mitochondria collected from cells ± CCCP pretreatment were incubated at 30 °C with cytosol expressing GFP-Parkin collected from cells with intact mitochondria. In this cell-free ubiquitylation assay, CCCP-treated mitochondria stimulate autoubiquitylation of GFP-Parkin and substrate ubiquitylation toward Mfn2. B, activation of GFP-Parkin by CCCP-pretreated mitochondria depends on PINK1. Mitochondria collected from PINK1−/− MEFs following CCCP treatment do not activate GFP-Parkin in the cell-free ubiquitylation assay, whereas exogenous PINK1 complements the aforementioned defect. C, formation of an apparent ubiquitin-oxyester adduct on the Parkin C431S mutant is dependent on the presence of CCCP-pretreated mitochondria in the cell-free assay. The red asterisks indicate ubiquitin-oxyester formation unless otherwise specified. D, ubiquitin-oxyester formation of Parkin harboring C431S, C431S/S65A, or C431S/S65E mutations in the cell-free ubiquitylation experiments. The S65A mutation disrupted ubiquitin-ester formation completely, whereas the C431S/S65E mutant has weak but detectable ubiquitin-oxyester formation. E, a model for Parkin activation on damaged mitochondria. See text for details.
DISCUSSION
Biochemical Evidence for Ubiquitin-Ester Conjugation of Parkin Cys-431
Ubiquitin ligases (E3) can be classified into three groups, namely HECT, U-box, and RING-type E3s. In HECT-type E3s, ubiquitin is transferred from the ubiquitin-conjugation enzyme (E2) to a HECT domain as a ubiquitin-thioester relay, and is finally passed to the substrate. On the other hand, the most basic role of RING-type E3 is that it unites both E2 and the substrate facilitating ubiquitin transfer from E2 to the substrate by placing them in close physical proximity. During this process, the RING finger motif functions as an E2-binding domain (67). Since the reports of E3 activity in 2000 (26–28), Parkin has been considered a RING-type E3. This activity was subsequently shown to reside in the RING2 domain (31, 33).
In 2011, based on analysis of the HHARI E3, Wenzel et al. (36) proposed that RING-IBR-RING type E3s such as Parkin function as a “RING-HECT hybrid” because HHARI forms an ubiquitin-thioester on a conserved cysteine residue in the rear RING finger motif similar to the HECT domain. Although thought provoking, this hypothesis was inconsistent with results showing that the Parkin C431S mutant was unable to stabilize ubiquitin-oxyester linkage. Using intact cell lysate and mitochondria collected from CCCP-treated cells, Lazarou et al. (39) reported formation of ubiquitin-oxyester on a Parkin C431S mutant dependent on PINK1 and a decrease in ΔΨm, thereby partially solving the aforementioned contradiction. However, even in that paper, reconstitution of ubiquitin-ester formation using recombinant Parkin protein was missing and the mechanism of how PINK1 regulates the ubiquitin-ester formation of Parkin was vague. To address this issue, we performed in vitro biochemical analyses using recombinant Parkin, and confirmed ubiquitin-oxyester formation of recombinant Parkin C431S mutant (Fig. 2) and an active site-directed ubiquitin probe (Ub-VS) targets Cys-431 of WT Parkin (Fig. 3).
Molecular Mechanism Underlying Parkin Catalysis of Ubiquitylation
We found in the current study that the IBR-RING2 domain of Parkin catalyzes in vitro ubiquitin-oxyester formation with ATP, ubiquitin, and E1, even in the absence of E2 (Fig. 2E). The ubiquitin-oxyester formation of IBR-RING2 was dependent on E1, suggesting that IBR-RING2 is unable to form the ubiquitin-ester bond de novo but is able to discharge the ubiquitin-thioester from E1 and then transfer it to Parkin Cys-431. In the recently proposed model, the RING1 domain functions as an “ubiquitin-charged E2” binding domain. Because the IBR-RING2 domain functions in conjunction with the neighboring RING0 and RING1 domains in cells, experiments incorporating only IBR-RING2 might be artificial. Nevertheless, the aforementioned results clearly showed that the IBR-RING2 domain catalyzes transthiolation and/or acyl transfer rather than E2-recuitment. Another interesting result is that the IBR-RING2 domain catalyzes ubiquitylation irrespective of dissipation of ΔΨm and mitochondrial localization in cells (Fig. 3, F–H), suggesting that IBR-RING2 is converted to a constitutively active form.
While this manuscript was in preparation, the Parkin structure was resolved (50). Interestingly, structural analysis revealed that RING2 domain topology is distinct from other typical RING fingers, consistent with our anticipation that the IBR-RING2 is not a conventional RING finger E2-recuitment domain (67). Moreover, the RING0 domain occludes the Cys-431 ubiquitin acceptor site in RING2, suggesting that deletion of the RING0 domain de-repressed the ester-transfer (transthioesterification) activity of the RING2 domain, and thus GFP-IBR-RING2 exhibited constitutive activity (Fig. 3, G, and H).
PINK1-dependent Phosphorylation of Parkin Ser-65 Is Important for Formation of the Ubiquitin-Thioester
Genetic analyses of PINK1 using Drosophila melanogaster mutants have shown that it acts as an upstream factor of Parkin (68–70). We and other groups have since reported that PINK1 is essential for translocation of Parkin to damaged mitochondria, revealing that PINK1 regulates the subcellular localization of Parkin (6, 7, 43, 55, 71). In addition, we have demonstrated that the E3 activity of Parkin is also up-regulated by a decrease in ΔΨm and PINK1 (6), although the molecular details remain obscure.
We confirmed in this study that Parkin is phosphorylated at Ser-65 following a decrease in ΔΨm as reported (56, 61), and showed that ubiquitin-thioester formation of Parkin is regulated by this PINK1-dependent phosphorylation event. Although the lack of a detectable ubiquitin adduct in the absence of CCCP (Fig. 7B, lanes 10 and 13) suggests that the putative phosphomimic mutations (S65D/S65E) do not bypass the decrease in ΔΨm requirement for ubiquitin-oxyester formation, we speculate that Parkin is not a unique PINK1 substrate and that phosphorylation of other PINK1 substrate(s) is important for full Parkin activation. Alternatively, Glu-65 and Asp-65 might be incomplete mimics of phosphor-Ser-65. We showed that the ubiquitin-oxyester adduct was not formed on unphosphorylated Parkin (Fig. 4A) and that the phosphorylation-deficient S65A mutation inhibited ubiquitin-oxyester formation of Parkin (Figs. 7B and 8D). To our knowledge, this is the first report that connects PINK1-dependent phosphorylation and ubiquitin-ester formation of Parkin.
Conclusion
In this study, we have shown that PINK1-dependent phosphorylation of Parkin Ser-65 plays an important role in ubiquitin-ester formation on Cys-431 following a decrease in ΔΨm. Moreover, biochemical analyses indicate that RING2 is not an E2-recruiting domain but is rather an ubiquitin-ester-transferring domain. Although other mode(s) of activation (e.g. Ser-65 phosphorylation strengthens the interaction of Parkin with mitochondria) cannot be ruled out completely, our model for Parkin activation is that phosphorylation of Ser-65 in the Parkin Ubl domain by PINK1 de-represses its autoinhibitory activity. This allows the RING2 domain to transfer the ubiquitin-thioester to Cys-431 and thus catalyze substrate ubiquitylation (depicted in Fig. 8E). The results presented in this work provide insights into the molecular details of Parkin activation, and are potentially beneficial for disease treatment by ameliorating the E3 activity of Parkin.
Acknowledgments
We thank Dr. J. Shen for providing PINK1−/− MEFs, Dr. M. Shindo for evaluation of MS data, and Dr. T. Kitamura for the PLAT-E cells. We also thank Drs. N. Tani and H. Taniguchi for help with the LC-MS/MS analysis.
Note Added in Proof
While our manuscript was under review, four groups independently published results that are related to those described herein (72–75).
This work was supported in part by Japan Society for the Promotion of Science (JSPS)/Ministry of Education, Culture, Sports, Science and Technology (MEXT) KAKENHI Grants 23-6061 (to K. O.), 23791001 (to Y. K.), 21000012 (to K. T.), and 23687018, 24111557, and 25112522 (to N. M.); the Tomizawa Jun-ichi and Keiko Fund for Young Scientist (to N. M.); and the Takeda Science Foundation (to H. K., N. M., and K. T.).
- Mfn
- mitofusin
- HHARI
- human homologue of ariadne
- MEF
- mouse embryonic fibroblast
- CCCP
- carbonyl cyanide p-chlorophenylhydrazone
- IB
- immunoblotting
- Ub-VS
- ubiquitin-vinyl sulfone
- NEM
- N-ethylmaleimide.
REFERENCES
- 1. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 2. Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 3. Corti O., Lesage S., Brice A. (2011) What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol. Rev. 91, 1161–1218 [DOI] [PubMed] [Google Scholar]
- 4. Exner N., Lutz A. K., Haass C., Winklhofer K. F. (2012) Mitochondrial dysfunction in Parkinson's disease. Molecular mechanisms and pathophysiological consequences. EMBO J. 31, 3038–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Imaizumi Y., Okada Y., Akamatsu W., Koike M., Kuzumaki N., Hayakawa H., Nihira T., Kobayashi T., Ohyama M., Sato S., Takanashi M., Funayama M., Hirayama A., Soga T., Hishiki T., Suematsu M., Yagi T., Ito D., Kosakai A., Hayashi K., Shouji M., Nakanishi A., Suzuki N., Mizuno Y., Mizushima N., Amagai M., Uchiyama Y., Mochizuki H., Hattori N., Okano H. (2012) Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y. S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greene A. W., Grenier K., Aguileta M. A., Muise S., Farazifard R., Haque M. E., McBride H. M., Park D. S., Fon E. A. (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Okatsu K., Oka T., Iguchi M., Imamura K., Kosako H., Tani N., Kimura M., Go E., Koyano F., Funayama M., Shiba-Fukushima K., Sato S., Shimizu H., Fukunaga Y., Taniguchi H., Komatsu M., Hattori N., Mihara K., Tanaka K., Matsuda N. (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Okatsu K., Iemura S., Koyano F., Go E., Kimura M., Natsume T., Tanaka K., Matsuda N. (2012) Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem. Biophys. Res. Commun. 428, 197–202 [DOI] [PubMed] [Google Scholar]
- 12. Narendra D., Walker J. E., Youle R. (2012) Mitochondrial quality control mediated by PINK1 and Parkin. Links to parkinsonism. Cold Spring Harb. Perspect. Biol. 4, a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koh H., Chung J. (2010) PINK1 and Parkin to control mitochondria remodeling. Anat. Cell Biol. 43, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vives-Bauza C., Przedborski S. (2011) Mitophagy. The latest problem for Parkinson's disease. Trends Mol. Med. 17, 158–165 [DOI] [PubMed] [Google Scholar]
- 15. Springer W., Kahle P. J. (2011) Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 7, 266–278 [DOI] [PubMed] [Google Scholar]
- 16. Pogson J. H., Ivatt R. M., Whitworth A. J. (2011) Molecular mechanisms of PINK1-related neurodegeneration. Curr. Neurol. Neurosci. Rep. 11, 283–290 [DOI] [PubMed] [Google Scholar]
- 17. Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D. F., Karbowski M., Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L., Hess S., Chan D. C. (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshii S. R., Kishi C., Ishihara N., Mizushima N. (2011) Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y. L., Selkoe D., Rice S., Steen J., LaVoie M. J., Schwarz T. L. (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu S., Sawada T., Lee S., Yu W., Silverio G., Alapatt P., Millan I., Shen A., Saxton W., Kanao T., Takahashi R., Hattori N., Imai Y., Lu B. (2012) Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8, e1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rakovic A., Shurkewitsch K., Seibler P., Grünewald A., Zanon A., Hagenah J., Krainc D., Klein C. (2013) Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy. Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 288, 2223–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vilain S., Esposito G., Haddad D., Schaap O., Dobreva M. P., Vos M., Van Meensel S., Morais V. A., De Strooper B., Verstreken P. (2012) The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 8, e1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yacobi-Sharon K., Namdar Y., Arama E. (2013) Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev. Cell 25, 29–42 [DOI] [PubMed] [Google Scholar]
- 26. Shimura H., Hattori N., Kubo Si., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., Suzuki T. (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y., Gao J., Chung K. K., Huang H., Dawson V. L., Dawson T. M. (2000) Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. U. S. A. 97, 13354–13359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Imai Y., Soda M., Takahashi R. (2000) Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275, 35661–35664 [DOI] [PubMed] [Google Scholar]
- 29. Chaugule V. K., Burchell L., Barber K. R., Sidhu A., Leslie S. J., Shaw G. S., Walden H. (2011) Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 30, 2853–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chew K. C., Matsuda N., Saisho K., Lim G. G., Chai C., Tan H. M., Tanaka K., Lim K. L. (2011) Parkin mediates apparent E2-independent monoubiquitination in vitro and contains an intrinsic activity that catalyzes polyubiquitination. PloS One 6, e19720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsuda N., Kitami T., Suzuki T., Mizuno Y., Hattori N., Tanaka K. (2006) Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 281, 3204–3209 [DOI] [PubMed] [Google Scholar]
- 32. Trempe J. F., Fon E. A. (2013) Structure and Function of Parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front. Neurol. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hampe C., Ardila-Osorio H., Fournier M., Brice A., Corti O. (2006) Biochemical analysis of Parkinson's disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 15, 2059–2075 [DOI] [PubMed] [Google Scholar]
- 34. Kirisako T., Kamei K., Murata S., Kato M., Fukumoto H., Kanie M., Sano S., Tokunaga F., Tanaka K., Iwai K. (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 25, 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moynihan T. P., Ardley H. C., Nuber U., Rose S. A., Jones P. F., Markham A. F., Scheffner M., Robinson P. A. (1999) The ubiquitin-conjugating enzymes UbcH7 and UbcH8 interact with RING finger/IBR motif-containing domains of HHARI and H7-AP1. J. Biol. Chem. 274, 30963–30968 [DOI] [PubMed] [Google Scholar]
- 36. Wenzel D. M., Lissounov A., Brzovic P. S., Klevit R. E. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smit J. J., Monteferrario D., Noordermeer S. M., van Dijk W. J., van der Reijden B. A., Sixma T. K. (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 31, 3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stieglitz B., Morris-Davies A. C., Koliopoulos M. G., Christodoulou E., Rittinger K. (2012) LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 13, 840–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lazarou M., Narendra D. P., Jin S. M., Tekle E., Banerjee S., Youle R. J. (2013) PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J. Cell Biol. 200, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning. Powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 41. Gautier C. A., Kitada T., Shen J. (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 105, 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y. S., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., Matsuda N. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cell 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ziviani E., Tao R. N., Whitworth A. J. (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U. S. A. 107, 5018–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gegg M. E., Cooper J. M., Chau K. Y., Rojo M., Schapira A. H., Taanman J. W. (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glauser L., Sonnay S., Stafa K., Moore D. J. (2011) Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 118, 636–645 [DOI] [PubMed] [Google Scholar]
- 46. Joselin A. P., Hewitt S. J., Callaghan S. M., Kim R. H., Chung Y. H., Mak T. W., Shen J., Slack R. S., Park D. S. (2012) ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 21, 4888–4903 [DOI] [PubMed] [Google Scholar]
- 47. Love K. R., Pandya R. K., Spooner E., Ploegh H. L. (2009) Ubiquitin C-terminal electrophiles are activity-based probes for identification and mechanistic study of ubiquitin conjugating machinery. ACS Chem. Biol. 4, 275–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hemelaar J., Borodovsky A., Kessler B. M., Reverter D., Cook J., Kolli N., Gan-Erdene T., Wilkinson K. D., Gill G., Lima C. D., Ploegh H. L., Ovaa H. (2004) Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins. Mol. Cell. Biol. 24, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rankin C. A., Roy A., Zhang Y., Richter M. (2011) Parkin, a top level manager in the cell's sanitation department. Open Biochem. J. 5, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trempé J. F., Sauvé V., Grenier K., Seirafi M., Tang M. Y., Ménade M., Al-Abdul-Wahid S., Krett J., Wong K., Kozlov G., Nagar B., Fon E. A., Gehring K. (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455 [DOI] [PubMed] [Google Scholar]
- 51. Zhou C., Huang Y., Shao Y., May J., Prou D., Perier C., Dauer W., Schon E. A., Przedborski S. (2008) The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. U. S. A. 105, 12022–12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beilina A., Van Der Brug M., Ahmad R., Kesavapany S., Miller D. W., Petsko G. A., Cookson M. R. (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. U. S. A. 102, 5703–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Valente E. M., Salvi S., Ialongo T., Marongiu R., Elia A. E., Caputo V., Romito L., Albanese A., Dallapiccola B., Bentivoglio A. R. (2004) PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 56, 336–341 [DOI] [PubMed] [Google Scholar]
- 54. Sha D., Chin L. S., Li L. (2010) Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-κB signaling. Hum. Mol. Genet. 19, 352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrané J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U. S. A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H. I., Campbell D. G., Gourlay R., Burchell L., Walden H., Macartney T. J., Deak M., Knebel A., Alessi D. R., Muqit M. M. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2, 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 58. Kinoshita E., Kinoshita-Kikuta E., Koike T. (2012) Phos-tag SDS-PAGE systems for phosphorylation profiling of proteins with a wide range of molecular masses under neutral pH conditions. Proteomics 12, 192–202 [DOI] [PubMed] [Google Scholar]
- 59. Denison S. R., Wang F., Becker N. A., Schüle B., Kock N., Phillips L. A., Klein C., Smith D. I. (2003) Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 22, 8370–8378 [DOI] [PubMed] [Google Scholar]
- 60. Pawlyk A. C., Giasson B. I., Sampathu D. M., Perez F. A., Lim K. L., Dawson V. L., Dawson T. M., Palmiter R. D., Trojanowski J. Q., Lee V. M. (2003) Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J. Biol. Chem. 278, 48120–48128 [DOI] [PubMed] [Google Scholar]
- 61. Shiba-Fukushima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., Hattori N. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2, 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamamoto A., Friedlein A., Imai Y., Takahashi R., Kahle P. J., Haass C. (2005) Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J. Biol. Chem. 280, 3390–3399 [DOI] [PubMed] [Google Scholar]
- 63. Rubio de la Torre E., Luzón-Toro B., Forte-Lago I., Minguez-Castellanos A., Ferrer I., Hilfiker S. (2009) Combined kinase inhibition modulates parkin inactivation. Hum. Mol. Genet. 18, 809–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Avraham E., Rott R., Liani E., Szargel R., Engelender S. (2007) Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J. Biol. Chem. 282, 12842–12850 [DOI] [PubMed] [Google Scholar]
- 66. Kim Y., Park J., Kim S., Song S., Kwon S. K., Lee S. H., Kitada T., Kim J. M., Chung J. (2008) PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 377, 975–980 [DOI] [PubMed] [Google Scholar]
- 67. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 68. Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., Chung J. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161 [DOI] [PubMed] [Google Scholar]
- 69. Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 70. Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J. W., Yang L., Beal M. F., Vogel H., Lu B. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U. S. A. 103, 10793–10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on voltage-dependent anion channel 1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 72. Riley B. E., Lougheed J. C., Callaway K., Velasquez M., Brecht E., Nguyen L., Shaler T., Walker D., Yang Y., Regnstrom K., Diep L., Zhang Z., Chiou S., Bova M., Artis D. R., Yao N., Baker J., Yednock T., Johnston J. A. (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 4, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spratt D. E., Julio Martinez-Torres R., Noh Y. J., Mercier P., Manczyk N., Barber K. R., Aguirre J. D., Burchell L., Purkiss A., Walden H., Shaw G. S. (2013) A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nat. Commun. 4, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wauer T., Komander D. (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 10.1038/emboj.2013.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zheng X., Hunter T. (2013) Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res. 23, 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]