

Background: tBid is a Bcl-2 family protein that promotes apoptosis at the mitochondria.
Results: tBid undergoes a reversible conformational change at membranes before activation that is accelerated by Mtch2.
Conclusion: The Mtch2 accelerated conformational change in membrane-bound tBid enables it to activate Bax.
Significance: The conformational change in tBid is a novel potential site of apoptosis regulation.
Keywords: Apoptosis, Bax, Bcl-2 Proteins, Fluorescence Correlation Spectroscopy, Fluorescence Resonance Energy Transfer (FRET), Protein Conformation, Bid, Mtch2, Fluorescence Spectroscopy, tBid
Abstract
Bid is a Bcl-2 family protein that promotes apoptosis by activating Bax and eliciting mitochondrial outer membrane permeabilization (MOMP). Full-length Bid is cleaved in response to apoptotic stimuli into two fragments, p7 and tBid (p15), that are held together by strong hydrophobic interactions until the complex binds to membranes. The detailed mechanism(s) of fragment separation including tBid binding to membranes and release of the p7 fragment to the cytoplasm remain unclear. Using liposomes or isolated mitochondria with fluorescently labeled proteins at physiological concentrations as in vitro models, we report that the two components of the complex quickly separate upon interaction with a membrane. Once tBid binds to the membrane, it undergoes slow structural rearrangements that result in an equilibrium between two major tBid conformations on the membrane. The conformational change of tBid is a prerequisite for interaction with Bax and is, therefore, a novel step that can be modulated to promote or inhibit MOMP. Using automated high-throughput image analysis in cells, we show that down-regulation of Mtch2 causes a significant delay between tBid and Bax relocalization in cells. We propose that by promoting insertion of tBid via a conformational change at the mitochondrial outer membrane, Mtch2 accelerates tBid-mediated Bax activation and MOMP. Thus the interaction of Mtch2 and tBid is a potential target for therapeutic control of Bid initiated cell death.
Introduction
Loss of integrity of the mitochondrial outer membrane (MOM)4 is an event that commits a eukaryotic cell to undergo apoptosis (1, 2). The intrinsic and extrinsic pathways of apoptosis converge at the MOM where the Bcl-2 family of proteins plays a pivotal role in the regulation of apoptosis through protein-protein and protein-membrane interactions (3, 4). The Bcl-2 family includes more than 20 members with either pro-apoptotic or anti-apoptotic functions. It is divided into three groups based on function and the presence of conserved Bcl-2 homology (BH) regions. The pro-apoptotic proteins comprise two groups; those with all four homology regions, such as Bax and Bak, and those that contain only the BH3 region (5, 6). The multiregion family members Bax and Bak undergo complex conformational changes that mediate oligomerization and permeabilization of the MOM, thereby releasing apoptogenic factors such as cytochrome c and Smac (7, 8). These conformational changes are initiated by binding BH3 proteins such as tBid, Bim, and Puma, termed activators because they activate the latent pro-apoptotic activity of Bax and Bak (9, 10). The third group includes the anti-apoptotic proteins such as Bcl-2 and Bcl-XL that contain all four homology regions and function by binding either activated Bax/Bak or activator BH3 proteins. Bad and Noxa are examples of sensitizer BH3 proteins that promote apoptosis indirectly by competing for binding to Bcl-XL, thus displacing tBid and Bim to allow Bax/Bak activation (11). A summary of the properties of the Bcl-2 family proteins most relevant to this work is given in Table 1.
TABLE 1.
Properties of Bcl-2 family proteins
| Protein | Properties and function | Size |
|---|---|---|
| kDa | ||
| Bid | Full-length Bid; α-helical; cannot activate Bax | 22 |
| cBid | Bid cleaved by caspase 8; N-terminal and C-terminal fragments remain bound due to hydrophobic interactions; activator to Bax | 22 |
| tBid (p15) | Truncated C-terminal fragment; activator to Bax | 15 |
| p7 | Truncated N-terminal fragment; inhibitor to tBid | 7 |
| Bax | α-Helical, forms pores in the MOM | 22 |
| Bcl-XL | α-Helical, inhibits MOMP by sequestering tBid and Bax | 27 |
Binding to membranes has been shown to trigger structural rearrangements in both anti- and pro-apoptotic multi BH region members of the Bcl-2 family (8, 12–15). These conformational changes alter their affinity for different binding partners and consequently their role in apoptosis. On the other hand, the role of membrane binding in modifying the dynamic structure and function of BH3 proteins has not yet been studied (16).
Bid is a BH3 protein that is required for the transmission of many death signals originating outside the cell to the MOM (17). Bid is crucial for death receptor induced apoptosis in hepatocytes and pancreatic β cells. Binding of ligands to the cell surface death receptors or exposure to the contents of vesicles secreted from cytotoxic lymphocytes results in a single proteolytic cleavage of Bid by caspase 8, calpain, or granzyme B (18–20). Although the exact sites of cleavage are different, they are sufficiently close that the two fragments generated are generically referred to as p7 (7 kDa) and tBid (15 kDa, also called p15). In solution, the two cleaved fragments remain bound to one another in a complex termed cBid (cleaved Bid) due to tight non-covalent interactions (see Fig. 1) (21–24). In cells, the MOM protein Mtch2 facilitates the recruitment of tBid to the MOM by an unknown mechanism (25, 26), where tBid interacts with Bax and Bcl-XL to regulate MOM integrity. Mtch2 is a part of the mitochondrial carrier family proteins and has been recently identified as a novel binding partner of tBid on the MOM. Mtch2 anchors in the MOM by six transmembrane helices and is a structural homologue to the mitochondrial inner membrane protein ANT (26).
FIGURE 1.

There is a pronounced lag phase in membrane permeabilization by Bax when cBid is used as the activator. A, shown is a schematic outlining the preparation of different forms of Bid. Full-length Bid was first subjected to cleavage by caspase 8 to obtain cBid (cleaved Bid). The cleavage site is indicated with a dotted line. Although cleaved, the two fragments of Bid remain in a complex due to strong hydrophobic interactions. To isolate the active, membrane binding tBid fragment (truncated Bid), cBid is treated with octylglucoside to remove the p7 fragment. The BH3 region (black), BH4 region (light gray), and the membrane binding helices (gray) are shown. The boxes indicate the individual α-helices (numbered α symbols above). B, shown is cytochrome c release from mitochondria isolated from bak−/− mice. Mitochondria (1 mg/ml) were incubated with 1 nm tBid (gray, open circles) or cBid (black, closed circles) and with 50 nm Bax. Cytochrome c release was quantified by immunoblotting the bound protein in the pellet and the free protein in the supernatant at the indicated times. The data (circles) are fit with a delayed single exponential function (line), (mean ± S.E., n = 3). Dashed lines indicate the value of the lag time obtained from the fit. C, permeabilization of liposomes encapsulating ANTS and DPX by incubation with 20 nm tBid (gray, open circles) or cBid (black, closed circles) and 100 nm Bax is shown. The increase in the fluorescence intensity of ANTS over time is due to membrane permeabilization of the liposomes. One representative series of measurements from three independent experiments is shown. The data (circles) are fit with a function accounting for a lag phase followed by a single exponential function (line). Dashed lines indicate the value of the lag time obtained from the fit.
Unlike other well studied BH3 proteins that are unstructured, Bid shares a similar three-dimensional structural arrangement of its constituent α-helices with the multiregion family members Bcl-XL and Bax (27–29). Thus Bid contains eight α-helices, of which α6 and α7 are central hydrophobic helices necessary for binding to the membrane that are surrounded by the remaining amphipathic helices (21, 30–32). The BH3 region located within α3 is critical for binding to other Bcl-2 family members to regulate apoptosis (19, 33). In addition, a BH4 region is present close to the N terminus of Bid (6, 24); this feature as well as the specific arrangement of the core hydrophobic helices differentiates Bid from other BH3 proteins (34, 35). Additional clues from phylogenomic analysis and other aspects of the mechanism of action strongly suggest that Bid is not a bona fide BH3 protein and may function as a “pseudo-Bax” that is unable to oligomerize efficiently, and permeabilize the MOM itself but that functions to recruit Bax to the membrane and to activate it (for review, see Ref. 35).
Despite the critical role of Bid in apoptosis, the detailed mechanism of the events that follow the cleavage of Bid until it assumes its fully active form at the MOM, including how membrane binding may participate in this process, remains to be understood. To address these questions, we used fluorescence spectroscopy to investigate the mechanism of activation of Bid using a cell-free system with either isolated mitochondria or biomimetic liposomes. We used physiological nanomolar concentrations of recombinant proteins to accurately reflect cellular conditions (36). By labeling Bid at selected positions, the individual steps in this process were identified and studied separately. Our results indicate that the activation of cBid begins when binding to membranes triggers the separation of the two cleaved fragments. Then an elaborate conformational change results in the insertion of anchoring helices in the membrane to achieve the fully active form. Furthermore, we show that the conformational change in tBid after binding to the membrane is the specific step that is accelerated by Mtch2 on the MOM.
EXPERIMENTAL PROCEDURES
Protein Purification and Labeling
Different variants of recombinant His6-tagged full-length murine Bid were obtained by site-directed mutagenesis and purified as described (15). All of the Bid proteins used in our studies contain an N-terminal His tag, except tBid, in which the His-tagged p7 fragment was removed. For clarity in the nomenclature, we have omitted the His prefix from the names of the proteins. Wild-type (WT) Bid has two endogenous cysteine residues at positions 30 and 126. Plasmids encoding single cysteine versions of Bid with a cysteine at one of the endogenous positions and a no-cysteine variant were generated through QuikChange mutagenesis (Stratagene). The endogenous cysteine residues in Bid were mutated to serine residues for the no-cysteine variant. Subsequently, Bid plasmids containing a single cysteine at various locations within the protein were generated from the no-cysteine Bid plasmid by using QuikChange mutagenesis. By convention, the Bid variants are identified by the location of the single cysteine residue used for labeling; thus, tBid 126C refers to the tBid fragment of Bid with a single cysteine at position 126. A plasmid was also generated in which all six lysine residues in Bid 126C were replaced with arginine residues (Bid ΔK 126C). All the lysine residues in murine Bid are located in close proximity; therefore, silent mutations were created to excise the coding region for Bid from amino acid Ala-130 to Val-175, and the sequence was replaced with an annealed pair of oligonucleotides containing the desired mutations.
All of the Bid proteins contained an N-terminal His tag and were purified using nickel-affinity chromatography (Qiagen) as described (23). At this stage, purified Bid mutants intended for labeling were dialyzed in storage buffer (10 mm Hepes, 7.3, 100 mm NaCl, 0.5 mm EDTA, 10% glycerol). The protein was labeled with the indicated dyes in the storage buffer in the presence of 0.5% CHAPS for 2–3 h at room temperature with rotation. For labeling, a 10–15-fold molar excess of dye dissolved in DMSO was added to the protein slowly while ensuring that the final DMSO content of the reaction did not exceed 10%. To remove the excess unreacted dye, Bid was subjected to nickel-affinity chromatography and washed with at least 25 ml of wash buffer (10 mm Hepes, pH 7.3, 300 mm NaCl, 0.5 mm EDTA, 10% glycerol, and 0.5% CHAPS). The labeled Bid was then eluted with elution buffer (wash buffer supplemented with 200 mm imidazole). The labeling efficiency of Bid at a given residue was measured by dividing the dye concentration (measured by absorbance) by the protein concentration (measured by Bradford assay). Only if the labeling efficiency was ∼70% or higher was the protein used.
Purified unlabeled WT Bid or labeled Bid proteins (2–4 mg/ml) were incubated with 500 units of caspase 8 (Enzo Life Sciences) with a final concentration of 1 unit/μl for ∼40 h at room temperature to generate cBid. The cleavage efficiency was checked by Coomassie Blue staining of an SDS-PAGE gel, and cBid was dialyzed against storage buffer. To generate recombinant tBid, cBid (labeled or otherwise) was subjected to nickel-affinity chromatography, tBid was isolated using 2% (w/v) octylglucoside (Sigma) in the wash buffer to remove tBid from the bound p7 fragment, and the eluent was dialyzed against storage buffer. WT Bax and Bcl-XL were purified as described previously (12, 37, 38).
Liposome Preparation
Liposomes 100 nm in diameter with a lipid composition made up of phosphatidylcholine (48%), phosphatidylethanolamine (28%), phosphatidylinositol (10%), dioleoyl phosphatidylserine (10%), and tetraoleoyl cardiolipin (4%) (all from Avanti Polar Lipids) were prepared as described previously (37), with the exception that the assay buffer used was 10 mm Hepes, pH 7.3, 200 mm KCl, and 1 mm MgCl2. The liposome concentration was estimated as described (37) using the fact that there are ∼84,000 lipids per liposome.
Mitochondria Preparation
Mitochondria were isolated from the livers of bak−/− (38) and mtch2+/+ and mtch2−/− (25) mice and frozen in trehalose according to the literature (40). Briefly, mouse liver was prepared in AT buffer: 300 mm trehalose (Sigma), 10 mm Hepes-KOH, pH 7.5, 10 mm KCl, 1 mm EGTA, 1 mm EDTA, 0.1% (w/v) BSA (Bioshop). The isolated mitochondria were frozen at 50 mg/ml final protein concentration (Bradford assay) in 10-μl aliquots and stored at −80 °C until use. When ready to use, mitochondria were rapidly thawed and washed in AT/KCl buffer at 1 mg/ml: 300 mm trehalose, 10 mm Hepes-KOH, pH 7.7, 80 mm KCl, 1 mm EGTA, 1 mm EDTA, 0.1% (w/v) BSA. The mitochondria were resuspended in regeneration buffer at 1 mg/ml final protein concentration (300 mm trehalose, 10 mm Hepes-KOH, pH 7.7, 80 mm KCl, 1 mm EGTA, 1 mm EDTA, 0.1% (w/v) BSA, 5 mm succinate, 2 mm ATP, 10 μm phosphocreatine, 10 μg/ml creatine kinase (added fresh)) and used immediately.
Membrane Permeabilization Assays
Membrane permeabilization assays with liposomes encapsulating ANTS (8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt) and DPX (p-xylene-bis-pyridinium bromide) were carried out as described previously (37, 38). Outer membrane permeabilization of mitochondria was assessed by measuring cytochrome c release after incubating the mitochondria with the proteins for 30 min at 37 °C. Mitochondria were isolated from the reaction by centrifugation, and cytochrome c was detected in the supernatant and pelleted fractions by immunoblotting. The cytochrome c polyclonal antibody was produced in our laboratory and was used at a dilution of 1:5000. The secondary antibody conjugated to horseradish peroxidase (Bio-Rad) was used at a dilution of 1:20,000. Immunoblots were analyzed using ImageJ. The curves were fit using Prism 5 (GraphPad).
Liposome Binding Assay
20 nm cBid 126C or tBid 126C labeled with Alexa Fluor 488 C5 maleimide (Invitrogen) was incubated with different concentrations of liposomes labeled with 0.008 mol% DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt; Invitrogen). After incubation for 15 min at 37 °C to reach binding equilibrium, the reactions were then transferred to 96-well Griener Senseoplates (Sigma), and fluorescence correlation spectroscopy measurements were performed on an Insight spectrometer (Evotec) as described (37). The fluorescence of cBid or tBid labeled with Alexa488 and DiD-labeled liposomes was excited at 488 and 635 nm respectively, and the emission was collected in separate channels. Measurements consisting of 8 individual 30-s runs were performed at room temperature.
The fraction of protein bound to liposomes was calculated by analyzing the autocorrelation functions for two independent species, the unbound protein and the protein bound to liposomes, as previously described (41). Then the dissociation constant, Kd, was extracted from the titration curve by assuming that the protein in solution (P) and the protein bound to membrane (P*) were in equilibrium. The dissociation constant, Kd is,
 |
where L represents the available lipids. Because lipids were always present in excess, [L] can be considered to be the total lipid concentration, so that the bound protein fraction is,
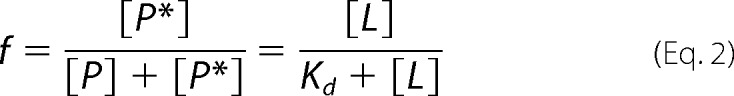 |
To present the Kd in terms of liposomes, lipid concentration was converted to liposome concentration in Eq. 1, as explained above.
Separation of p7·tBid
To prepare double-labeled cBid, purified Bid ΔK 126C was first labeled with the acceptor Alexa Fluor 647 C2 maleimide (Invitrogen) at position 126C and then with the donor Alexa Fluor 550 carboxylic acid, succinimidyl ester (Invitrogen), at the free amino group at the N terminus. As a control, Bid ΔK 126C was labeled with the donor only. The labeled proteins were then cleaved with caspase 8 to generate cBid ΔK 126C Alexa550-Alexa647 (+A) and cBid ΔK 126C Alexa550 (−A). The rate of separation of the two fragments was measured by recording the loss of fluorescent resonance energy transfer (FRET) signal between the two dyes at 37 °C with constant stirring. For this, the sample was excited at a wavelength of 550 nm (2-nm bandwidth), and the emission was recorded at a wavelength of 572 nm (10-nm bandwidth) to measure the intensity of the donor. Cleaved protein was added to assay buffer placed into a quartz cuvette coated with dioleoylphosphatidylglycerol to obtain a final concentration of 20 nm concentrations of protein. A small increase in the intensity of the donor was observed possibly due to fragment separation upon dilution. The intensity of the donor stabilized 1 min after adding the protein and was used as the initial donor fluorescence in the presence of the acceptor, F+A (t = 0). Liposomes were then added to the reaction, and the change in the intensity of the donor, F+A (t), was recorded over time. A parallel reaction was prepared with cBid ΔK 126C Alexa555 where the fluorescence of the donor in the absence of the acceptor, F−A (t) was recorded. In the end the change in % FRET Efficiency was assessed by considering the quantity (1 − (F+A (t)/F−A (t))) ×100.
The reactions with mitochondria with and without Mtch2 were carried out as described above with mitochondria resuspended in regeneration buffer at 1 mg/ml. However, in this case 2 nm cleaved protein was added directly to mitochondria, as no change in donor intensity was observed in the regeneration buffer.
Bid to Membrane FRET
For liposomes, the assay was carried out as described previously (11). Briefly, cBid and tBid single cysteine mutants were labeled with the donor, DACM (N-(7-dimethylamine-4-methylcoumarin-3-yl) maleimide (Invitrogen). Liposomes were prepared with 1 mol% of PE lipids labeled with the acceptor NBD either at the head (16:0 NBD PE 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2–1,3-benzoxadiazol-4-yl)) or at the tail (16:0–12:0 NBD PE 1-palmitoyl-2-{12-[(7-nitro-2–1,3-benzoxadiazol-4-yl) amino]dodecanoyl}-sn-glycero-3-phosphoethanolamine) (Avanti Polar Lipids). Excitation at 380 nm (2 nm bandwidth) and emission at 460 nm (10 nm bandwidth) was used to measure the change in the intensity of the donor at 37 °C with constant stirring. The fluorescence intensity recorded in the sample containing cBid DAC and liposomes NBD was noted F+A (t). A reaction with unlabeled liposomes was carried out in parallel to obtain the fluorescence intensity in the absence of acceptor, F−A (t). The change in FRET was monitored by calculating % FRET Efficiency (1 − (F+A (t)/F−A (t))) × 100.
For experiments containing mitochondria, cBid 126C was labeled with the donor Alexa Fluor 488 C5 maleimide (Invitrogen). To label mitochondria with the acceptor, 10 μm DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; Invitrogen) was incubated with 1 mg/ml mitochondria suspended in regeneration buffer for 30 min at 37 °C. Mitochondria were pelleted to remove unincorporated DiI and resuspended in regeneration buffer. 4 nm cBid 126C Alexa488 was incubated with 1 mg/ml mitochondria with and without Mtch2 for 30 min at 37 °C. Excitation at 493 nm (5 nm bandwidth) and emission at 516 nm (5 nm bandwidth) was used to measure the change in the intensity of the donor. The fluorescence intensity recorded in the sample containing cBid 126C Alexa488 and mitochondria-DiI was noted F+A (t). A reaction with unlabeled mitochondria was carried out in parallel to obtain the fluorescence intensity in the absence of acceptor, F−A (t). The change in FRET was monitored by calculating % FRET Efficiency (1 − (F+A (t)/F−A (t))) × 100.
Environment Change Measurements
Bid single cysteine mutants were labeled with the environment-sensitive dye, N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine (IANBD) (Invitrogen). Changes in environment of the NBD dye were assessed by recording changes in either fluorescence intensity or lifetime.
For fluorescence intensity measurements, single cysteine cBid mutants labeled with NBD were added to either liposomes or mitochondria with and without Mtch2, and the change in the steady state intensity of NBD was recorded over time at 37 °C with constant stirring. The excitation wavelength used was 475 nm (2 nm bandwidth), and the emission wavelength used was 530 nm (10 nm bandwidth). The NBD fluorescence, F(t), was normalized by the fluorescence at time t = 0, F0. The quantity F(t)/F0 is a measure of the hydrophobicity of the environment in the vicinity of the residue labeled with NBD.
The fluorescence lifetime experiments were conducted using frequency-domain lifetime measurements. Before the measurement, 20 nm cBid NBD was incubated with 7.2 nm liposomes for 15 min at 37 °C to reach binding equilibrium. A 470-nm laser with a 450/65-nm filter was used as the excitation source, and the emission was collected through a 525/40-nm filter. The excitation filter was used to block harmonics from the laser. Solutions of fluorescein in 0.1 n NaOH (τ = 4.1 ns) and coumarin in 100% ethanol (τ = 2.5 ns) were used as standards. The measurements were performed at room temperature. Uncertainty values of 0.2° for phase decay and 0.004 for modulation ratio were used for the fitting of the curves. The data were analyzed using a three-component model with the third lifetime to account for noise (τ = 0.2–0.4 ns).
Mitochondria Binding Assay
2 nm cBid 30C or 126C labeled with Alexa Fluor 647 C2 maleimide (Invitrogen) were incubated with 1 mg/ml mitochondria with and without Mtch2 for 30 min at 37 °C. The reactions were then transferred to 96-well Griener Senseoplates (Griener), and fluorescence intensity distribution analysis measurements were performed on an Insight spectrometer (Evotec). The sample was excited at 635 nm while circularly scanning the observation volume over a 45-μm radius circle with a frequency of 0.354 Hz to avoid photo-bleaching (37). For each sample, a series of 10 measurements, each lasting 30 s, were recorded.
The data were analyzed assuming the presence of two distinct protein species; one with a specific brightness fixed to that of a single cBid molecule (corresponding to unbound monomeric protein) and the other with a specific brightness left to vary and always found to be higher than that of a single cBid molecule (corresponding to mitochondria with several cBid bound to it). The percent bound protein to mitochondria was calculated as described in Satsoura et al. (37).
Relocalization of YFP-Bax and Bid in Wild-type and Mtch2 Knockdown HeLa Cells
HeLa cells expressing YFP-Bax under a doxycycline promoter were a gift from Dr. Richard Mosser (42). Using shRNA against human Mtch2 (a gift from Dr. Robert Screaton), we depleted Mtch2 protein from HeLa cells as verified by an immunoblot against Mtch2 (data not shown). For automated high throughput cell experiments, WT and Mtch2 knockdown (KD) cells were distributed in 384-well μClear-plates (Greiner), and YFP-Bax expression was induced. Two days after plating, cells were treated with either 10 μg/ml cycloheximide (CHX) alone or 1 ng/ml TNFα and 10 μg/ml CHX for different time periods as indicated. At the end of the treatment, the cells were fixed and immunostained for Bid using a primary anti-Bid antibody (3C5) (Abcam) at a 1:200 dilution and a secondary Alexa Fluor 594-AffiniPure Donkey Anti-Mouse IgG antibody (Jackson ImmunoResearch) at a 1:200 dilution. Thirty minutes before imaging the cells were stained with DRAQ5 (Biostatus) at a 5 μm final concentration. Images were acquired with an Opera High Content Screening System (PerkinElmer Life Sciences) using a 40× water immersion objective. For each well 15 different images were acquired, each containing ∼25 cells. In each image, cells were identified by segmentation using nuclear and cytoplasmic DRAQ5 intensity. Cell features were extracted using a texture analysis script written for Acapella. The cells were then ranked by decreasing YFP-Bax intensity, and the top 200 cells per well were analyzed. Relocalization from a uniform cytoplasmic location to discrete foci representing mitochondria was measured using a Threshold Adjacency Statistics feature (43).
RESULTS
A Pronounced Initial Lag Phase in Bax-mediated Membrane Permeabilization Occurs with cBid but Not tBid
Cleavage of full-length Bid is necessary for the induction of its apoptotic function. Therefore, we generated cBid from purified full-length Bid through proteolysis by recombinant caspase 8. For comparative purposes, tBid was obtained by dissociating the two cBid fragments with the non-ionic detergent octylglucoside (Fig. 1A). Both cBid and tBid are pro-apoptotic and have been used in previously published mechanistic studies. Thus, to study the entire process of activation post cleavage, we compared the extent and kinetics of membrane permeabilization using mitochondria isolated from bak−/− mice. These mitochondria lack both Bak and Bax, as Bax is a cytoplasmic protein in mouse liver and does not detectably co-purify with mitochondria. To permeabilize these mitochondria, they were incubated with Bax and either tBid or cBid. Membrane permeabilization was measured by immunoblotting mitochondrial (pelleted) and released (supernatant) proteins to detect the release of cytochrome c at different time points (Fig. 1B). In a similar assay system that allows for control of lipid constituents, release of liposome-encapsulated fluorophore and quencher (ANTS and DPX) was measured during incubation with Bax and either tBid or cBid without stirring. Membrane permeabilization was followed by the increase in the ANTS fluorescence that occurs when the released fluorophore and quencher are diluted in the incubation buffer (Fig. 1C). Although ultimately achieving similar end points, membrane permeabilization by tBid-activated Bax was faster than that by cBid-activated Bax in both mitochondrial and liposomal systems (Fig. 1, B and C). The delay in membrane permeabilization using cBid-activated Bax was primarily due to a pronounced initial lag phase noted in both assays. To quantify the length of the initial lag phase and the kinetics of the following release phase in liposomes, we fit the data with a model where a no-release phase (lag phase) is followed by a single exponential kinetics release. In reactions using cBid, the initial lag phase lasted 8–10 min and was followed by an exponential phase that was slightly slower than that for tBid irrespective of whether liposomes or mitochondria were used as the target membrane.
Affinity of tBid and cBid for Liposomes
To characterize precisely any difference in the interaction between tBid and cBid with membranes, we measured the apparent dissociation constant (Kd) of tBid and cBid with liposomes. For these experiments tBid and cBid were labeled at the endogenous cysteine at position 126 with Alexa Fluor 488 and incubated with a range of liposome concentrations for analysis by fluorescence correlation spectroscopy. This method can distinguish between fluorescent species of different size based on their diffusion coefficient, and return their concentration (41). Free proteins in solution (∼5-nm diameter) have a high diffusion coefficient compared with proteins bound to liposomes (∼100-nm diameter). The percentage of tBid or cBid bound to liposomes could, therefore, be calculated from fluorescence correlation spectroscopy data (Fig. 2). Analysis of the data with a simple binding model gave an apparent Kd of 0.5 nm for the binding of tBid to liposomes (44 μm when considering the binding of tBid to lipids) and 0.9 nm for the binding of cBid to liposomes (76 μm for cBid and lipids), suggesting that tBid binds only slightly more avidly to the membrane than cBid.
FIGURE 2.
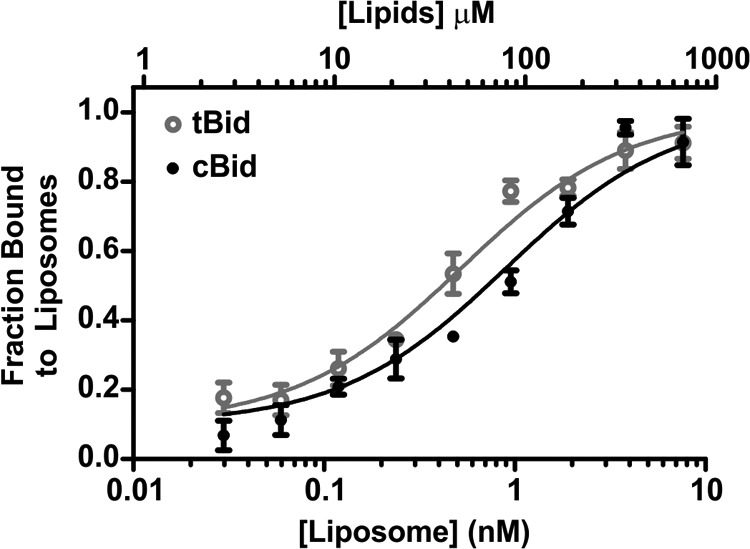
The proteins tBid and cBid have a very similar apparent dissociation constant for binding to liposomes. 20 nm tBid (gray, open circles) and cBid (black, closed circles) labeled with Alexa Fluor 488 were incubated with different concentrations of DiD-labeled liposomes for 15 min. The x axes indicates the concentration of liposomes in nm and the corresponding concentration of lipids in μm. The liposome-bound fractions as measured by fluorescence correlation spectroscopy (8 measurements/experiment) are shown for tBid and cBid. The data (dots) are fit with Equation 2 (line), (mean ± S.E., n = 3 independent experiments).
The Initial Lag Phase Is Independent of Protein Concentration
To examine the difference in kinetics observed between tBid and cBid in more detail, we used the ANTS/DPX liposomal assay to determine the concentration dependence of permeabilization rates for tBid and cBid (Fig. 3, Ai and Aii). There was a clear dose dependence in membrane permeabilization for both proteins, and the reaction end points were similar (Fig. 3B). The curves did not reach 100% because we used detergent-solubilized liposomes as a reference for 100% release, and the ANTS fluorescence further increased in detergent due to a more hydrophobic environment. This resulted in an underestimate of the liposome permeabilization. Next, we obtained the duration of the lag phase and the half-time for the exponential permeabilization phase as a function of the concentration of tBid or cBid (Fig. 3, C and D, respectively) by fitting the data in Fig. 3, Ai and Aii. The duration of the initial lag phase displayed only a small decrease with increasing protein concentration (less than 5 min); therefore, the large difference in the lag phase durations observed for tBid and cBid remained throughout the concentration range studied (Fig. 3C). By contrast, the half-times for permeabilization decreased significantly with increasing protein concentration but remained comparable for the two proteins for the range of concentrations explored (Fig. 3D). The kinetics and the extent of membrane permeabilization both reached a plateau above a concentration of ∼5 nm tBid or cBid. This concentration corresponds to a ratio of ∼10 tBid/cBid molecules per liposome with 100 nm Bax. These results are consistent with the well known catalytic nature of Bax activation by substoichiometric amounts of Bid. Taken together, these data suggest that cBid can cause the same amount of Bax-mediated permeabilization as tBid but that this follows an initial inactive phase (lag phase) that is significantly longer for cBid than for tBid.
FIGURE 3.

The lag in membrane permeabilization is due to slow activation of cBid at the membrane. A, ANTS release was observed when adding varying concentrations of (i) tBid or (ii) cBid to 0.5 nm liposomes containing ANTS/DPX and 100 nm Bax. One representative series of measurements from three independent experiments is shown. Solid lines are fit using a function accounting for a lag phase followed by a single exponential. B, ANTS release after 180 min for each reaction plotted against the concentration of tBid or cBid. Points are the mean ± S.E. n = 3. Solid lines are best fit using a single exponential. t/cBid indicates tBid or cBid. Shown are duration of the lag phase (C) and the half-time of the subsequent exponential phase obtained by fitting the data shown in i and ii (lines) as a function of tBid (gray, open circles) and cBid (black, closed circles) concentration (mean ± S.E., n = 3; D). Linear (C) and Exponential lines (D) of best fit are shown. t/cBid indicates tBid or cBid. E, shown is membrane permeabilization obtained after preincubating cBid with the liposomes for different lengths of time. Liposomes encapsulating ANTS/DPX (0.5 nm) were incubated with 20 nm cBid for the times indicated to the right of the data points, and then 100 nm Bax was added at time = 0 (mean, n = 6). Shown is duration of the lag phase (F) and the half-time of the subsequent exponential phase (G) as a function of the cBid preincubation time with liposomes (mean ± S.E., n = 6). Exponential (F) and linear lines (G) of best fit are shown.
The Initial Lag Phase in Membrane Permeabilization with cBid Is Due to the Time Required for Its Activation at the Membrane
To investigate the lag phase further, we pre-incubated cBid for different periods of time with liposomes encapsulating ANTS/DPX before adding Bax at time t = 0 min (Fig. 3E). All reactions had a similar end-point independent of the preincubation time of cBid with the liposomes. Strikingly, reactions with longer cBid-liposome preincubation times displayed a shorter initial lag phase that approached the short duration of the lag phase observed with tBid (Fig. 3F). By contrast, the kinetics of the exponential phase of ANTS release remained constant over the explored range of preincubation periods (Fig. 3G). This is consistent with the lag phase perhaps being the result of tBid unfolding or changing conformation in some other way at the membrane. Thus, cBid needs to interact with lipid membranes before it activates Bax and causes membrane permeabilization. This rate-limiting step in cBid activation most likely involves the dissociation of the p7·tBid complex and/or the unfolding and insertion of the tBid fragment into the membrane.
Fast Separation of the Cleaved Fragments of Bid in the Presence of a Membrane
To determine if the dissociation of the p7·tBid complex contributes to the cBid lag phase, we examined the localization of the two fragments after contact with membranes using both enriched mitochondria and liposomal assays. For experiments with mitochondria, cBid or tBid was incubated with mitochondria isolated from the livers of bak−/− mice, and the membrane-bound protein fraction was separated from the unbound protein by pelleting. Proteins bound to mitochondria (pelleted) and unbound proteins in the supernatant were identified by immunoblotting with antibodies specific to each fragment (Fig. 4A). As expected, the tBid fragment was present in the membrane-bound fraction, whereas the p7 fragment was only seen in the free-protein fraction. Similar results were obtained when 20 nm cBid was incubated with liposomes, and the membrane-bound protein was separated from the free proteins using size-exclusion chromatography. Immunoblots of different fractions revealed that the tBid fragment eluted in the membrane-bound fractions, whereas the p7 fragment eluted in the free-protein fractions (Fig. 4B). Thus there is complete dissociation of the p7·tBid complex after binding of the tBid fragment to 7.2 nm liposomes.
FIGURE 4.

The time required for separation of the p7 and tBid fragments of cBid does not account for the lag in membrane permeabilization.A, localization of the p7 and tBid fragments of cBid is shown. Mitochondria lacking Bak (1 mg/ml) were incubated with 4 nm tBid or cBid, and separation of the fragments was assayed by pelleting the mitochondria and immunoblotting for the bound protein in the pellet (P) and free protein in the supernatant (S). The approximate position of the 17-kDa molecular mass marker is indicated on the left. The specificity of the antibody used to probe the immunoblots (IB) is indicated to the right. B, targeting of the fragments of cBid to liposomes is shown. Membrane-bound protein was separated from soluble protein by size exclusion chromatography. Membrane binding was assayed for 20 nm cBid incubated with 7.2 nm liposomes for 15 min by chromatography using CL2B resin. Elution fractions corresponding to the excluded volume containing liposomes (fractions 3–4) and the included fractions to the bed volume containing unbound proteins (fractions 8–12) were analyzed by immunoblotting. The approximate position of the 17-kDa molecular mass marker is indicated to the left. The specificity of the antibody used to probe the immunoblots (IB) is indicated to the right. C, shown is a schematic of cBid ΔK 126C labeled as in Fig. 1, highlighting the positions of the FRET donor (Alexa Fluor 555, yellow-green star) and acceptor (Alexa Fluor 647, orange star) dyes at the N terminus and the endogenous cysteine residue at position 126, respectively. The positions of the lysine residues converted to arginine are indicated with blue lines. D, representative data show that the p7 fragment rapidly separates from the tBid fragment in the presence of membranes. The decrease in % FRET efficiency indicates the separation of the p7·tBid fragments of cBid ΔK 126C in the presence of the indicated concentrations of liposomes (black triangles, 0.74 nm; gray squares, 0.37 nm; gray circles, 0.19 nm). % FRET Efficiency was calculated by: (1 − (F+A (t)/F−A (t))) × 100. F+A (t) is the fluorescence of 20 nm cBid ΔK 126C labeled at the N terminus with Alexa Fluor 555 (donor) and at 126C with Alexa Fluor 647 (acceptor). F−A (t) is the fluorescence of a parallel reaction with 5 nm cBid ΔK 126C labeled only at the N terminus with Alexa Fluor 555 (mean, n = 4). A single exponential line of best fit used to determine the end point and half-time of fragment separation is shown. E, end points for % FRET efficiency data similar to that shown in D including the additional liposome concentrations tested indicates that at the end point fragment separation varies linearly with liposome concentration (mean ± S.E., n = 4). F, half-times associated with the separation of the two fragments (black) suggests that the rate of fragment separation (measured as decreased FRET) varies as a power of the liposome concentration. Half-times were obtained from fitting the % FRET efficiency data for each liposome concentration to a single exponential (mean ± S.E., n = 4). The minimum half-time detectable by the instrument is shown as a.
To examine if the separation of the two fragments was responsible for the initial lag phase associated with cBid activation, we designed a cBid mutant for use in an intramolecular FRET assay. To permit efficient and unambiguous labeling of the protein at two different and unique positions, we used a free thiol group for labeling with one dye and an amine group for labeling with the other dye. Therefore, we replaced the lysine residues in cBid 126C with arginine residues, generating Bid ΔK 126C. We labeled the cysteine at position 126 with the acceptor dye Alexa Fluor 647 and the free amino group of the N terminus with the donor dye Alexa Fluor 555. Cleavage by caspase 8 then produced cBid ΔK 126C Alexa555-Alexa647 (Fig. 4C). As a control for any change in the intensity of the donor not due to FRET, we also prepared cBid ΔK 126C Alexa555. Both cBid ΔK 126C Alexa555 and cBid ΔK 126C Alexa555-Alexa647 retained functional activity in the liposome based membrane permeabilization assay (data not shown).
To obtain the rate of separation of the two fragments after incubation with liposomes, we calculated % FRET efficiency as explained under “Experimental Procedures.” Loss of FRET resulted from the separation of the two fragments (Fig. 4D). These data demonstrate that separation of the two fragments can be fit with a single-exponential function and require only a few seconds as shown for three sample concentrations (Fig. 4D). Therefore, this process (and by inference membrane binding as membranes are required for fragment separation) does not account for the delayed kinetics of activation of cBid at the membrane. By using additional liposome concentrations, we show that the extent of separation of the p7·tBid fragments increased linearly with increasing liposome concentration (Fig. 4E). This result suggests that the efficiency of fragment separation depends on the availability of a membrane. Furthermore, the FRET efficiency approaches 0 (indicating complete separation of the fragments) at ∼6 nm liposomes. This value is in good agreement with the titration of cBid binding to liposomes data presented in Fig. 2.
Similarly, the half-time determined from the exponential phase of fragment separation decreased with increased concentrations of liposomes (Fig. 4F). The time it takes for sample addition and mixing may limit our ability to measure the rate of separation, suggesting that the rate of membrane-mediated fragment separation that we measured (k ∼ 0.25 s−1 nm−1) is consistent with a diffusion-limited process (k ∼ 1 s−1 nm−1) (44). Together, these results suggest that the separation of the two fragments of cBid is spontaneous and rapid, requiring only the presence of a membrane. Thus, fragment separation is not sufficient to account for the lag phase preceding membrane permeabilization (Fig. 3F). This indicates that there is one or several activation steps required for tBid on the membrane after fragment separation and before membrane permeabilization can occur.
The tBid Fragment Undergoes a Conformational Change after Binding to Membranes
Given that the difference in tBid and cBid activation of MOMP is not explained by the separation of the p7·tBid fragments, we used a protein to membrane FRET assay to measure the rate at which proteins integrated into liposome membranes (11). For these experiments, tBid and cBid were labeled with the donor fluorophore DAC at position 126C, and PE labeled with NBD at the tail was incorporated into the liposomes as the acceptor. An increase in % FRET efficiency was used to measure binding of tBid/cBid to liposome-containing NBD-PE. Using this approach, we previously reported a rapid reaction half-time of <5 s between tBid and liposomes (11); however, here we discovered that the reaction half-time of cBid and liposomes is significantly longer: ∼50 s (Fig. 5B). We obtained similar 40–80 s half-times for dyes at other positions in the protein by using single cysteine mutants in protein to liposome FRET assays (data not shown). This result is consistent with the half-time measurements reflecting integration/binding of the protein into the membrane rather than a conformation change resulting in quenching of the dye at position 126.
FIGURE 5.

tBid undergoes a conformational change after binding to membranes. A, shown is a schematic labeled as in Fig. 1 illustrating the locations of the single-cysteine mutations (green stars) in Bid used for labeling with NBD as an environment-sensitive probe. Numbers below the schematic indicate the positions of the cysteine residues. B, shown is binding of tBid and cBid to membranes measured by FRET. An increase in % FRET efficiency indicates binding of protein to liposomes. 20 nm cBid 126C (black) or tBid 126C (gray) labeled with DAC (donor) was incubated with liposomes containing NBD-PE (acceptor), F+A (t), and without NBD-PE, F−A (t). As a measure of % FRET efficiency, (1 − (F+A (t)/F−A (t))) × 100 was calculated (data points are the means, n = 3). Connecting lines are included as a visual guide only. C, shown are changes in NBD fluorescence reflecting changes in the environment for some representatives of the different residues of tBid (black) and cBid (gray) upon binding to membranes. An increase in the NBD fluorescence indicates the residue moving to a more hydrophobic environment. The numbers to the right indicate the residue labeled. Each experiment contained 20 nm labeled protein and 1.56 nm liposomes. NBD fluorescence, F(t), was normalized by F0, the fluorescence at time = 0 (mean ± S.E., n = 3). Solid lines are single-exponential fit. D, shown are end point fluorescence changes due to cBid binding to membranes for NBD fluorescence for all of the tested residues, assayed as in C (t = 300 s) (mean ± S.E., n = 3). The numbers below the bars indicate the residue number. The labels at the bottom indicate the region of the protein. Helices are indicated as in Fig. 5A. The N-terminal region (N) and the loop between helices 6 and 7 (L6–7) are also indicated. E, shown is the reaction half-times for the change in environment of each residue derived by the single-exponential fit from C upon binding to membranes (mean ± S.E., n = 3). F, shown is change in the end point fluorescence for cBid 163C and cBid 126C labeled with NBD as a function of liposome concentration (t = 300 s) (mean ± S.E., n = 3). G, half-times for the conformational changes in cBid 163C and cBid 126C labeled with NBD were not affected by liposome concentration (mean ± S.E., n = 3).
The half-time of binding of the protein to membranes was greatly accelerated in these experiments compared with those shown in Figs. 1 and 3 because they were carried out with constant stirring. Stirring the sample improved the signal-to-noise ratio of the measurements but also resulted in faster kinetics observed compared with the previous experiments (Figs. 1 and 3). Therefore, to relate the kinetics of protein insertion in the membrane to the kinetics of the lag phase observed in membrane permeabilization (Fig. 3), we repeated the FRET without stirring and for the same liposome concentration as the membrane permeabilization assay. In these experiments the half-time for membrane insertion of cBid from the FRET assay was ∼4 min instead of 50 s (data not shown), which is entirely consistent with the observed lag phase of ∼7 min in membrane permeabilization, as the latter also incorporates the time required for Bax activation and oligomerization. Taken together, these data demonstrate that it is the insertion of tBid in membranes that causes the initial lag phase in cBid-Bax mediated membrane permeabilization.
It is likely that insertion into the membrane results in or from a conformational change in the tBid fragment of cBid. We, therefore, hypothesized that a slow structural conformation change in cBid occurs upon binding to the membrane, which is required for functional activity and is responsible for the lag phase we observed for cBid. To detect membrane dependent conformational changes in cBid, we created a series of single cysteine mutants spanning key regions of the protein (Fig. 5A). These mutants were labeled with the fluorophore NBD, a small uncharged dye that shows an increase in steady state emission intensity and lifetime in a hydrophobic environment. These features make NBD an excellent tool to study changes in the environment of specific residues of a labeled protein (11, 45). To ensure that the labeled protein accurately reflected the behavior of the WT protein, we used only the mutants that had a labeling efficiency of ∼70% or higher and also retained membrane permeabilization activity (data not shown). Low labeling efficiencies prevented the analysis of additional residues spanning the membrane binding helices. To measure the kinetics of the conformational changes at or in the membrane, we added NBD-labeled single cysteine mutants of cBid to liposomes and recorded normalized fluorescence of NBD. Control experiments demonstrated that using the first measureable time point as F0 was as accurate as, but less cumbersome than, using a separate sample without liposomes.
The time course for the change in the emission of NBD after membrane binding for selected, labeled cysteine residues in cBid is shown in Fig. 5C. The curve generated using tBid 163C NBD is shown for comparison (Fig. 5C, black). The endpoints of the change in the normalized fluorescence intensity of cBid NBD for the entire series of mutants studied are summarized in Fig. 5D and show a significant increase in hydrophobicity for residues situated in and around helices α5 and α6. For most cBid NBD mutants (cBid NBD 106, 111, 126, 136, 142, 163, and 165) the fluorescence intensity curves could be well fit with a single-exponential function, yielding a single half-time for the environmental change of the corresponding residue (Fig. 5E). For other mutants (cBid NBD 66, 186, and 190) a small but consistent non single-exponential behavior was observed that points to a progressive and complex conformational change. For these cBid mutants only the half-time corresponding to the faster exponential change in environment is shown (Fig. 5E). For mutants in the proximity of the BH3 region (cBid NBD 86, 91, and 93) the changes were too small to accurately determine the kinetics. The reaction half-time does not correlate with the extent of the change in NBD fluorescence for residues 163 and 165 (compare Fig. 5, D and E), demonstrating that longer reaction half-times are not necessarily related to the extent to which the residue becomes buried in the bilayer.
For residues in the BH3 region there was no measureable increase in hydrophobicity. Indeed for residue 91 there is a slight decrease in hydrophobicity. This result suggests that the BH3 region remains accessible to aqueous solution even after other regions of tBid have inserted into the lipid bilayer.
Due to the fast half-times of residues in the beginning of helices α4–5 and α8 of Bid, we propose that those regions of tBid interact with the membrane first, whereas the slow kinetics of the residues close to the end of helices α4–5 and the loop between helices 6–7 (L 6–7) indicate that this region is involved in the last step of the ordered conformational change (Fig. 5D).
tBid Conformational Change Occurs after Membrane Binding
Because the conformational change occurs in the membrane, we predicted the kinetics of the NBD fluorescence to be independent of liposome concentration. Therefore, we chose two NBD-labeled cBid mutants, one with faster kinetics (126C; a residue in α helix 5) and the other with slower kinetics (163C; a residue in the loop between helices 6 and 7), and incubated both with a range of liposome concentrations. As expected, the extent to which the fluorescence of the dye increased correlated with increasing liposome concentration (Fig. 5F) due to an increased fraction of the proteins binding to liposomes (Fig. 2). In contrast, the kinetics of the process captured by NBD fluorescence remained unchanged for the range of liposome concentrations explored (Fig. 5G). This result directly contrasts what was observed for the rate of separation of the fragments (Fig. 4F). These results support our hypothesis that the separation of the two fragments occurs concurrently with the initial binding of cBid to liposomes and is followed by a major conformational change in tBid as the protein is inserting into the membrane. This latter event is, therefore, independent of liposome concentration as long as there are sufficient liposomes to provide binding sites for all of the cBid molecules in the reaction.
At the Membrane tBid Adopts Multiple Conformations
The changes measured in the fluorescence intensity of NBD for the different mutants after the addition to liposomes are relative to that observed for the initial environment of the dye on the protein. Therefore, to obtain a more complete picture of the hydrophobicity of the dye-labeled residues at different positions in cBid before and after the protein interacts with membranes, we measured the fluorescence lifetime of the probes on the proteins. Furthermore, lifetime measurements indicate whether the signal is composed of one, two, or more species on the membrane, which likely represent different conformers. To enhance our ability to detect multiple lifetimes of membrane-bound conformers, the signal from unbound protein was minimized by using a high liposome concentration (7.4 nm) at which most of the protein was bound to membranes (Fig. 2).
Most residues in soluble proteins are not in a single unique environment. Due to thermal and other motions of the protein, exposure to different environments can be modeled as a combination of hydrophilic and hydrophobic environments. The hydrophilic environment is dominated by exposure of the residue to the aqueous milieu, whereas the nature of the hydrophobic environment depends on interactions with hydrophobic residues in the protein, lipid molecules in a membrane, or both. Therefore, we fitted the curves generated from cBid NBD in solution (without liposomes) with a model that took into account the lifetime and the corresponding fraction of NBD in a hydrophilic (τ1, f1, open bars) or hydrophobic environment (τ2, f2, solid bars) (Fig. 6). Experimental noise (τ ∼ 0.3 ns) accounted for the remaining signal (typically less than 20%) and can be observed as the distance from the top of the bars to 1.0 on the scale.
FIGURE 6.

tBid exists in two major conformations at the membrane. The fluorescence lifetime of samples of 20 nm cBid labeled with NBD at different residues with and without 7.2 nm liposomes was measured. The data were analyzed using a model that included a hydrophilic (τ1) and a hydrophobic lifetime (τ2) and noise. The fractions corresponding to each lifetime (f1 and f2) are shown as open and solid bars, respectively, for the residues indicated below the bars, with (+) and without (−) liposomes (Lipos). The various regions probed are indicated at the bottom as in Fig. 5. The values of lifetimes (ns) are indicated on the bars (mean ± S.E., n = 3–4) and roughly divide the protein into the three regions (N terminus, membrane inserting, and C terminus) as indicated at the top.
As expected for cBid in solution, τ1 was relatively constant between 1.3 and 2.8 ns, whereas τ2 varied between 6.8 and 9.7 ns for all of the residues tested. These values of τ1 and τ2 are in general agreement with the lifetime of NBD in water and in chloroform, respectively. The fraction of protein associated with each lifetime, however, varied between different mutants, and the fraction of noise was higher in mutants that had a low hydrophobic fraction in solution (cysteine residues at positions 111, 136, 163, and 165) due to the increased uncertainty in measuring short lifetimes.
To analyze the curves from samples containing both cBid NBD and liposomes, we fixed the value of τ1 to that obtained for each particular residue in solution. This accounts for changes in τ1 due to local differences in amino acid sequence. We then obtained τ2, f1, and f2 from the fit of the lifetime data of the sample with liposomes. The comparison of f2 before and after the addition of liposomes is in general agreement with the changes observed in steady state intensities for NBD fluorescence (Figs. 5D and 6). For example, f1 varied from >50% for residue 111, a residue that appears to be surface-exposed, to 28% for residues such as 126 in the hydrophobic region of the protein (Fig. 6).
Taken together, the results from the steady state intensity and lifetime measurements indicate that after binding to the liposome membranes, the N-terminal region from 66–106 becomes slightly more solvent-exposed, residues 111–165 enter a more hydrophobic environment except for 126 that is always in a hydrophobic environment, and the C-terminal region from 186–190 remains unchanged. Furthermore, f2 is the predominant fraction for cBid mutants with a dye in the region found between residues 111 and 165, suggesting that this region is likely inserted into the membrane. By contrast, both the N- and the C-terminal regions of tBid have intermediate fractions of protein with τ1 and τ2, indicating that these regions are in more than one conformation at the membrane. Taken together, these results indicate that the conformational change in tBid triggered by binding to membranes is a complex process that involves unfolding of the N- and the C- terminal regions of the protein and the insertion of helices α 4–7 into the membrane.
Mtch2 Facilitates tBid Binding to Membranes by Accelerating the Conformational Change
The MOM protein Mtch2 has emerged as an important factor regulating the ability of tBid to induce apoptosis (25, 46). Mtch2 is a novel protein integral to the MOM and is related to the members of the mitochondrial carrier protein family. Previous work suggested limited binding of tBid to mitochondria lacking Mtch2 when assessed by differential centrifugation (25). To determine the exact step that is facilitated by Mtch2, we compared cBid binding to mitochondria isolated from mtch2+/+ and −/− mouse livers (40). As anticipated, the mitochondria lacking Mtch2 were less susceptible to tBid-mediated MOMP by a process that was inhibited by Bcl-XL (Fig. 7A).
FIGURE 7.

Mtch2 facilitates the conformational change of tBid required for its activation upon binding to mitochondria. A, mitochondria lacking Mtch2 are less susceptible to cBid-induced MOMP. cBid (1 nm) and/or 50 nm Bcl-XL (indicated below the blots) were incubated for 30 min at 37 °C with 1 mg/ml mitochondria with and without Mtch2 as indicated to the left. Cytochrome c release was assayed by isolating the mitochondria by centrifugation and immunoblotting (IB) the bound protein in the pellet (P) fractions and free protein in the supernatant (S) fractions. B, separation of the p7·tBid fragments is very rapid when incubated with mitochondria isolated from the livers of mtch2+/+ or −/− mice. Mitochondria (1 mg/ml) with (black) and without (gray) Mtch2 were incubated with 2 nm cBid ΔK 126C labeled with Alexa Fluor 555 donor and Alexa Fluor 647 acceptor dyes. The decrease in % FRET Efficiency indicates the separation of the p7·tBid fragments of cBid ΔK 126C. % FRET efficiency was calculated by (1 − (F+A (t)/F−A (t))) × 100. F+A (t) is the fluorescence of cBid ΔK 126C labeled with Alexa Fluor 555 (donor) and Alexa Fluor 647 (acceptor). F−A (t) is the fluorescence of a parallel reaction with cBid ΔK 126C labeled only with Alexa Fluor 555 (donor). C, FRET measurements suggest cBid binds to but does not insert into mitochondria from mtch2−/− mice. 4 nm cBid 126C labeled with Alexa Fluor 488 (donor) was added to 1 mg/ml mitochondria from mtch2+/+ or mtch2−/− mice labeled with DiI (acceptor). An increase in % FRET efficiency indicates cBid binding to mitochondria. % FRET Efficiency was calculated by: (1 − (F+A (t)/F−A (t))) × 100. F+A (t) is the fluorescence of cBid 126C Alexa Fluor 488 in the presence of mitochondria labeled with DiI. F−A (t) is the fluorescence of a parallel reaction with cBid 126C Alexa Fluor 488 in the presence of unlabeled mitochondria. Ci, end points for the % FRET efficiency of the reaction after 30 min are shown. Cii, after 30 min of incubation of cBid 126C Alexa Fluor 488 with mitochondria, the samples were centrifuged, and the pelleted mitochondria were resuspended. The fluorescence of cBid 126C Alexa Fluor 488 in resuspended mitochondria is shown (mean ± S.E., n = 3). D, shown is binding of p7 and tBid fragments to mitochondria from mtch2+/+ and mtch2−/− mouse liver. 2 nm cBid 30C (p7, white bars) or 126C (tBid, black bars) labeled with Alexa Fluor 647 was incubated with 1 mg/ml mitochondria with or without Mtch2 (as indicated) for 20 min at 37 °C. The percentage of cBid bound to mitochondria was measured using fluorescence intensity distribution analysis by comparing the concentration of objects with a specific brightness equal to that of a single Bid molecule (presumably free protein), with the concentration of objects with a higher specific brightness (mitochondria with more than one bound fluorescent cBid molecule). Because a small number (1–4) of cBid molecules on a mitochondrion results in a specific brightness close to or equal to that of a single Bid and is considered as “free protein,” this analysis inherently underestimates the amount of bound protein. E, the conformational change in cBid upon binding to mitochondria is delayed in the absence of Mtch2. cBid 163C NBD (2 nm) was added to mitochondria (1 mg/ml) from mtch2+/+ or mtch2−/− mice, and the change in NBD fluorescence that occurs when the protein binds to membranes is expressed as F(t)/F0 (mean ± S.E., n = 3).
To further elucidate the molecular mechanism involved, we used the fluorescence-based assays described above to compare the molecular mechanisms of cBid binding to and permeabilizing membranes from mitochondria with and without Mtch2. Potential differences in the separation of the two fragments of cBid in these mitochondria were assessed using the FRET assay described above (Fig. 4). Although the rate and the extent of fragment separation in mitochondria lacking Mtch2 was slightly compromised compared with mitochondria with Mtch2 (detected as higher % FRET efficiency between the dyes in the two fragments) (Fig. 7B), the separation of p7 and tBid was extended by only about ∼10 s. Therefore, neither initial interaction with the membrane nor fragment separation accounts for the functional defect in MOMP observed in the absence of Mtch2 (Fig. 7A).
To observe binding of cBid to mitochondria directly rather than by using biochemical methods, we measured protein binding to mitochondria by FRET. As an acceptor, the MOM was labeled with the dye DiI, and cBid 126C labeled with Alexa Fluor 488 was used as a donor. cBid binding proceeded to a lesser extent to the MOMs without Mtch2 compared with Mtch2 (Fig. 7Ci), but the overall decrease in binding was relatively small compared with the decrease in MOMP (Fig. 7A). Furthermore, using fluorescence intensity distribution analysis to calculate protein binding to mitochondria, we observed ∼50–70% binding of tBid and ∼5% binding of the p7 fragment labeled with Alexa Fluor 647 to mitochondria with and without Mtch2 (Fig. 7D). However, when the mitochondria were pelleted and then resuspended, the amount of cBid 126 Alexa488 that remained bound to mitochondria lacking Mtch2 was very small (Fig. 7Cii). The simplest explanation for these data is that in the absence of Mtch2 cBid binds to but does not insert into the MOM to the same extent.
Therefore, we investigated the relative changes in the conformation of tBid upon binding to the MOM by adding cBid 163C NBD to mitochondria with and without Mtch2 and monitoring changes in NBD intensity over time. With mitochondria containing Mtch2, we observed an increase in cBid 163C NBD fluorescence indicative of the protein inserting residue 163 into the membrane. However, with mitochondria lacking Mtch2, there was a small initial decrease due to a decrease in the hydrophobicity of the residue, perhaps due to the separation of the p7 fragment. This is followed by fluctuations in the signal indicating that during the first 15 min, residue 163 of tBid did not move to a more hydrophobic environment (Fig. 7E). A slow increase in NBD fluorescence was visible after that, indicating tBid binding to mitochondria independent of Mtch2. Thus in mitochondria, Mtch2 greatly accelerates the conformational change in tBid shown here to be required for insertion into the MOM and essential for the activation of tBid.
Knockdown of Mtch2 Causes a Significant Delay between Bid and Bax Relocalization in Cells
Our data suggest that when cBid binds to membranes, the two fragments separate rapidly, but the Mtch2-facilitated conformational change that is required for tBid to recruit Bax is much slower. Thus, a clear prediction of our results is that in cells, knockdown of Mtch2 would delay recruitment of Bax to mitochondria without significantly affecting relocalization of Bid. To study the effect of Mtch2 on tBid and Bax relocalization in cells during apoptosis, we knocked down Mtch2 protein expression using an shRNA against Mtch2 in HeLa cells stably expressing YFP-Bax. Apoptosis was induced in WT and Mtch2 KD HeLa cells expressing YFP-Bax by treatment with TNFα and CHX for 8 h, as it is well established that this cell death pathway proceeds via activation of Bid by caspase 8-mediated cleavage. YFP-Bax can be imaged directly by fluorescence microscopy; however, to visualize endogenous Bid, treated cells were fixed and immunostained. Representative fluorescence images showing the localization of Bid and YFP-Bax in untreated (CHX alone) and treated (TNFα and CHX) cells are shown in Fig. 8, Ai and Aii. Using automated high content image acquisition in separate channels for YFP-Bax and Bid, >2000 cells per time point were analyzed to yield objective and quantitative measurements of changes in the distribution of the proteins over time. A threshold adjacency statistic feature that gave the largest relative change between untreated and treated WT cells while retaining equivalent background values for the untreated cell lines (WT and Mtch2 KD) was visually verified to report the relocalization of Bid and Bax in cells. The values of this Threshold Adjacency Statistics feature increase as the protein localization progresses from a uniform distribution (corresponding to a cytoplasmic protein) to a punctuated pattern (corresponding to mitochondria-bound protein) (Fig. 8A). Graphic representation of the relocalization of Bid and Bax in WT and Mtch2 KD HeLa cells is shown in Fig. 8, B and C, respectively. It is apparent in WT HeLa cells that as time proceeds, the Bid and Bax fluorescence signals become more punctuated indicative of mitochondrial localization simultaneously (Fig. 8B). This is more evident for YFP-Bax relocalization as indicated by the differences in the y axis scales shown in Fig. 8, B and C. Notably, in Mtch2 KD cells, Bid relocalization is as efficient as it is in the WT cells, whereas Bax relocalization is significantly delayed (Fig. 8C). This observation in intact cells is entirely consistent with results from isolated mitochondria (Fig. 7). When directly comparing the relocalization of Bid and Bax, the rate of change for both is synchronized in the presence of Mtch2 indicated by the linear positive correlation between Bid:Bax relocalization, whereas Bax relocalization is relatively delayed in the absence of Mtch2 (Fig. 8D). This supports the concept that the recruitment of tBid to membranes is not sufficient for the activation of tBid in cells, and that subsequent to cBid binding to MOM, Mtch2 accelerates tBid activation. Based on our in vitro results, we ascribe tBid activation to a conformational change in tBid.
FIGURE 8.

Down-regulation of Mtch2 delays Bax relocalization subsequent to Bid relocalization in cells. A, representative fluorescence images show the relocalization of Bid and YFP-Bax in WT and Mtch2 KD HeLa cells. Bid-mediated apoptosis was induced by treatment of the cells with TNFα and CHX in WT and Mtch2 KD cells. Treated cells were fixed at the end of the experiment and were immunostained for the endogenous Bid. Panel Ai shows the untreated cells (10 μg/ml CHX alone) at the 8-h time point, and panel Aii shows treated cells (1 ng/ml TNFα and 10 μg/ml CHX) at a 4-h time point. Scale bar = 10 μm. B and C. Relocalization of tBid was unaffected, but Bax relocalization was delayed in Mtch2 KD HeLa cells. WT (B) and Mtch2 KD HeLa cells (C) stably expressing YFP-Bax were treated as in A. Using high throughput image-based screening of images recorded in the YFP and the Alexa Fluor 595 channel of both cell lines, >2000 cells were analyzed for each TNFα + CHX treatment time. Cells were treated with CHX alone for 8 h. Relocalization of Bid (black) and Bax (gray) was measured by using a Threshold Adjacency Statistics feature that gave the largest relative change between the untreated and treated WT cells while retaining equivalent background values for both untreated cell lines (WT and Mtch2 KD). Connecting lines are included as a visual aid only. Each point is an average of 800 cells: 200 cells/independent experiment (error bars, S.E., n = 4). D, Bax relocalization is delayed relative to tBid relocalization in Mtch2 KD (gray) compared with WT (black) HeLa cells. The black dotted line is a linear line fit to the WT data.
DISCUSSION
We have examined the sequence of conformational changes that occurs when cBid interacts with membranes and propose a model for cBid activation subsequent to cleavage between the p7 and the tBid fragments (Fig. 9). In the first step subsequent to cleavage, interaction with a membrane (liposome or mitochondrial) results in rapid separation of the fragments (Figs. 4, 7B, and 9A). After the initial binding to membranes, tBid undergoes an elaborate conformational change (Figs. 5, 6, 7E, and 9, B and C), facilitated by Mtch2 (Figs. 7 and 8) that results in the protein anchoring in the membrane. Although it is possible that tBid adopts a single unique conformation on the membrane, our data favor a situation in which at least two main conformers are in equilibrium on the membrane (Fig. 6). We show that the conformational change is a prerequisite for activation of Bax or Bak to elicit membrane permeabilization (including MOMP) (Figs. 1, 3, and 8). Therefore, Bax must be binding to the membrane-inserted conformation of tBid. This result is consistent with previous data showing that only the membrane-bound form of tBid binds Bax (apparent Kd 25 nm (10)).
FIGURE 9.

Proposed activation mechanism for tBid. Upon receiving an apoptotic signal via a death receptor, caspase 8 is activated and cleaves full-length Bid. In solution, the two fragments of cBid remain bound as a complex due to strong hydrophobic interactions. At the MOM, the p7 and the tBid fragments separate very quickly upon the initial binding of tBid to the MOM (A). Mtch2 acts as a receptor for tBid and catalyzes a conformational change in the protein (from the transient intermediate form B to the membrane-integrated form) (C) by anchoring the membrane binding helices in the membrane. As suggested by NBD lifetime data, two co-existing membrane-bound conformers are shown, although more different conformations may exist. The position of helices with respect to the membrane in B and C is putative but is in agreement with our data and with previously published studies (32, 39). The regions of tBid that are the first to interact with the membrane in the absence of Mtch2 (highlighted in red) also interact with Mtch2 (B). The conformational change in tBid leads to two plausible membrane-bound tBid conformers that are in equilibrium on the MOM, as suggested by the curved arrows (C). It is not clear whether Mtch2 remains bound to one or both conformers at the membrane.
The fast binding of cBid to the MOM (leading to the separation of the p7·tBid fragments) and the conformational change of tBid at the MOM recapitulated what we observed with liposomes (Fig. 7). Although cBid binding to mitochondria was independent of Mtch2 in enriched mitochondria and intact cells, the conformational change and tBid-dependent recruitment of Bax were delayed in the absence of Mtch2 (Figs. 7 and 8). Therefore, tBid undergoes a conformational change after binding to the MOM that is necessary for tBid activation and subsequent recruitment of Bax to the MOM leading to MOMP.
Previous studies have suggested that specific post-translational modifications are required for the activation of tBid, such as myristoylation of the tBid fragment (22) and “unconventional” ubiquitination and degradation of the p7 fragment (43). Using our in vitro approach with defined components, our results suggest that such modifications are not obligatory for the activation of tBid at the MOM. Furthermore, the very rapid spontaneous separation of the two fragments of cBid argues that post-translational modifications of tBid are unlikely to contribute to the separation of the fragments in a biologically significant manner.
Examining the kinetics with which individual cysteine residues at various locations in tBid interact with the lipid bilayer leads us to postulate that the unfolding of tBid at the membrane begins with the interaction of α-helices 4, 5, and 8 with the membrane (Fig. 9B) and is followed by insertion of α-helices 6 and 7 in the hydrophobic core of the membrane (Figs. 5D and 9C). Furthermore, the fluorescence lifetimes obtained for the NBD labels in the N- and C-terminal region of cBid with liposomes are well fit by a model containing substantial fractions of molecules with both an aqueous and a hydrophobic lifetime for NBD (Fig. 6). When the protein bound to membranes, the longer lifetime and its corresponding fraction both dropped substantially for several of the residues in the N-terminal region consistent with this part of the protein unfolding (Fig. 6). However, under these conditions, our binding studies suggest that most of the protein is bound to the membrane (Fig. 2). Therefore, the simplest explanation for these data is that for these residues only half of the membrane-bound protein is in a hydrophobic environment that indicates fluctuations in the structure of the protein at the membrane. Together these results suggest that the labeled tBid exists as two different membrane-bound conformers. Although this is the simplest explanation, our data are also compatible with tBid having a large number of conformations on the membrane. However, we propose the simplest possible scenario where two membrane-inserted conformers of tBid are in equilibrium; one where the N- and C- terminal regions are present at the interface of the membrane (solvent-exposed), and the other where they interact with the membrane (hydrophobic) (Fig. 9C). Clearly, more extensive characterizations of this equilibrium will be required to understand the potential functional differences between the two different conformations of tBid at the membrane. Furthermore, the exact arrangement of the helices with respect to the membrane remains elusive.
One obvious possibility is that the fast membrane insertion of purified tBid is due to the incubation in octylglucoside, which is required for isolating tBid from the p7 fragment, substitutes for the time required for this transformation to occur in situ. Aptly, as a detergent, octylglucoside may mimic the environment of a membrane to facilitate the conformational change necessary for full activation of tBid.
Recently the regions of tBid and Mtch2 that mediate this protein-protein interaction have been identified (42). Peptides corresponding to residues close to the N terminus of tBid (59–73), surrounding the loop between α-helices 4 and 5 (111–125), and in α-helix 8 (181–191) bound to peptides from two separate regions of Mtch2 (Fig. 9, highlighted by red regions). Remarkably, the NBD kinetics of the single cysteine mutants (106, 126C, 186C, and 190C) close to these identified regions show the fastest half-times, suggesting that these sites are also the first to interact with the membrane. In addition, the finding that p13/p11 N-terminal cleaved product tBid is only generated in mtch2+/+ but not in mtch2−/− cells (24) strongly supports that Mtch2 is essential for the conformational change in tBid during which the N terminus becomes solvent-exposed (Fig. 5) and accessible for cleavage (44). These results are completely consistent with our experiments in cells demonstrating that cBid migration to the MOM during apoptosis is not affected by Mtch2 knockdown, whereas Bax recruitment was significantly delayed. Taken together, our data indicate that the conformational change of tBid at the MOM is required for tBid activation and subsequent recruitment of Bax to the MOM. Whether Mtch2 binds to all tBid conformers or is reciprocal with binding of other Bcl-2 family proteins remains to be determined.
Our data suggest that α helices 4–7 of tBid become embedded in the membrane during the conformational change. We along with others have observed a similar change in the analogous helices of Bax that occurs during activation (7), further suggesting that Bid is more similar to Bax than to other BH3 proteins. Additionally, it is clear that tBid and Bim also recruit cytosolic anti-apoptotic proteins such as Bcl-XL to the MOM, where these proteins mutually sequester each other (37). Competitive binding between different members of the Bcl-2 family proteins, therefore, causes reciprocal changes in conformations between tBid and both other classes of Bcl-2 proteins. We propose that having multiple conformers is one way that a small protein can expose different sites to bind to different protein partners. Moreover, we propose that the effect of membrane binding on the conformation of tBid may extend to other Bcl-2 family proteins including those that are intrinsically disordered (e.g. Bim and Bad). For example, the unstructured BH3 region of Bim adopts a helical conformation upon binding to Bcl-XL (47). Studies to test these hypotheses and the functional consequences of the different conformations of tBid on the regulation of MOMP will be the subject of future detailed investigations.
We have identified three individual steps of activation of tBid on the MOM using two in vitro model systems (Fig. 9). Our results highlight the different steps that can potentially be regulated by post-translational modifications or small molecules during the initiation of MOMP by the activator tBid. After cleavage, the p7 and tBid fragments quickly separate at the MOM, where the conformational change in tBid is facilitated by its interaction with Mtch2. Because this conformational change is a prerequisite to Bax activation and MOMP, its modulation opens a new avenue for small molecule targeting to promote or inhibit MOMP.
Acknowledgments
We thank Dr. Robert Screaton (Department of Pediatrics, Department of Cellular and Molecular Medicine, University of Ottawa) for providing the shRNA against Mtch2.
This work was supported by Canadian Institute of Health Research Grants FRN86657 (to C. F.) and FRN12517 (to D. W. A. and B. L.).
- MOM
- mitochondrial outer membrane
- MOMP
- MOM permeabilization
- cBid
- cleaved Bid
- tBid
- truncated Bid
- BH
- Bcl-2 homology
- ANTS
- 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt
- DPX
- p-xylene-bis-pyridinium bromide
- PE
- phosphatidylethanolamine
- CHX
- cycloheximide
- KD
- knockdown
- DAC
- N-(7-dimethylamino-4-methylcoumarin-3-yl)
- NBD
- 12-(N-methyl-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl))
- DiI
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate.
REFERENCES
- 1. Adams J. M. (2003) Ways of dying. Multiple pathways to apoptosis. Genes Dev. 17, 2481–2495 [DOI] [PubMed] [Google Scholar]
- 2. Wang X. (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15, 2922–2933 [PubMed] [Google Scholar]
- 3. Leber B., Lin J., Andrews D. W. (2010) Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene 29, 5221–5230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shamas-Din A., Kale J., Leber B., Andrews D. W. (2013) Mechanisms of action of bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 5, a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hardwick J. M., Youle R. J. (2009) SnapShot. BCL-2 proteins. Cell 138, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kvansakul M., Yang H., Fairlie W. D., Czabotar P. E., Fischer S. F., Perugini M. A., Huang D. C., Colman P. M. (2008) Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 15, 1564–1571 [DOI] [PubMed] [Google Scholar]
- 7. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. (2001) Proapoptotic BAX and BAK. A requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Annis M. G., Soucie E. L., Dlugosz P. J., Cruz-Aguado J. A., Penn L. Z., Leber B., Andrews D. W. (2005) Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 24, 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wei M. C., Lindsten T., Mootha V. K., Weiler S., Gross A., Ashiya M., Thompson C. B., Korsmeyer S. J. (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14, 2060–2071 [PMC free article] [PubMed] [Google Scholar]
- 10. Kim H., Tu H. C., Ren D., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J., Cheng E. H. (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., Andrews D. W. (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084 [DOI] [PubMed] [Google Scholar]
- 12. Yethon J. A., Epand R. F., Leber B., Epand R. M., Andrews D. W. (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J. Biol. Chem. 278, 48935–48941 [DOI] [PubMed] [Google Scholar]
- 13. Kim P. K., Annis M. G., Dlugosz P. J., Leber B., Andrews D. W. (2004) During apoptosis Bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol. Cell 14, 523–529 [DOI] [PubMed] [Google Scholar]
- 14. Peng J., Tan C., Roberts G. J., Nikolaeva O., Zhang Z., Lapolla S. M., Primorac S., Andrews D. W., Lin J. (2006) tBid elicits a conformational alteration in membrane-bound Bcl-2 such that it inhibits Bax pore formation. J. Biol. Chem. 281, 35802–35811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dlugosz P. J., Billen L. P., Annis M. G., Zhu W., Zhang Z., Lin J., Leber B., Andrews D. W. (2006) Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 25, 2287–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shamas-Din A., Brahmbhatt H., Leber B., Andrews D. W. (2011) BH3-only proteins. Orchestrators of apoptosis. Biochim. Biophys. Acta 1813, 508–520 [DOI] [PubMed] [Google Scholar]
- 17. Yin X. M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K. A., Korsmeyer S. J. (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400, 886–891 [DOI] [PubMed] [Google Scholar]
- 18. Li H., Zhu H., Xu C. J., Yuan J. (1998) Cleavage of BID by Caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501 [DOI] [PubMed] [Google Scholar]
- 19. Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481–490 [DOI] [PubMed] [Google Scholar]
- 20. Gross A., Yin X. M., Wang K., Wei M. C., Jockel J., Milliman C., Erdjument-Bromage H., Tempst P., Korsmeyer S. J. (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 274, 1156–1163 [DOI] [PubMed] [Google Scholar]
- 21. Chou J. J., Li H., Salvesen G. S., Yuan J., Wagner G. (1999) Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 96, 615–624 [DOI] [PubMed] [Google Scholar]
- 22. Kudla G., Montessuit S., Eskes R., Berrier C., Martinou J. C., Ghazi A., Antonsson B. (2000) The destabilization of lipid membranes induced by the C-terminal fragment of Caspase 8-cleaved Bid is inhibited by the N-terminal fragment. J. Biol. Chem. 275, 22713–22718 [DOI] [PubMed] [Google Scholar]
- 23. Zha J., Weiler S., Oh K. J., Wei M. C., Korsmeyer S. J. (2000) Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290, 1761–1765 [DOI] [PubMed] [Google Scholar]
- 24. Tan K. O., Tan K. M., Yu V. C. (1999) A novel BH3-like domain in BID is required for intramolecular interaction and autoinhibition of pro-apoptotic activity. J. Biol. Chem. 274, 23687–23690 [DOI] [PubMed] [Google Scholar]
- 25. Zaltsman Y., Shachnai L., Yivgi-Ohana N., Schwarz M., Maryanovich M., Houtkooper R. H., Vaz F. M., De Leonardis F., Fiermonte G., Palmieri F., Gillissen B., Daniel P. T., Jimenez E., Walsh S., Koehler C. M., Roy S. S., Walter L., Hajnóczky G., Gross A. (2010) MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat. Cell Biol. 12, 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson A. J., Kunji E. R., Gross A. (2012) Mitochondrial carrier homolog 2 (MTCH2). The recruitment and evolution of a mitochondrial carrier protein to a critical player in apoptosis. Exp. Cell Res. 318, 1316–1323 [DOI] [PubMed] [Google Scholar]
- 27. Suzuki M., Youle R. J., Tjandra N. (2000) Structure of Bax. Coregulation of dimer formation and intracellular localization. Cell 103, 645–654 [DOI] [PubMed] [Google Scholar]
- 28. Muchmore S. W., Sattler M., Liang H., Meadows R. P., Harlan J. E., Yoon H. S., Nettesheim D., Chang B. S., Thompson C. B., Wong S. L., Ng S. L., Fesik S. W. (1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381, 335–341 [DOI] [PubMed] [Google Scholar]
- 29. Hinds M. G., Smits C., Fredericks-Short R., Risk J. M., Bailey M., Huang D. C., Day C. L. (2007) Bim, Bad and Bmf. Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 14, 128–136 [DOI] [PubMed] [Google Scholar]
- 30. McDonnell J. M., Fushman D., Milliman C. L., Korsmeyer S. J., Cowburn D. (1999) Solution structure of the proapoptotic molecule BID. A structural basis for apoptotic agonists and antagonists. Cell 96, 625–634 [DOI] [PubMed] [Google Scholar]
- 31. Hu X., Han Z., Wyche J. H., Hendrickson E. A. (2003) Helix 6 of tBid is necessary but not sufficient for mitochondrial binding activity. Apoptosis 8, 277–289 [DOI] [PubMed] [Google Scholar]
- 32. Oh K. J., Barbuto S., Meyer N., Kim R. S., Collier R. J., Korsmeyer S. J. (2005) Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding. J. Biol. Chem. 280, 753–767 [DOI] [PubMed] [Google Scholar]
- 33. Wang K., Yin X. M., Chao D. T., Milliman C. L., Korsmeyer S. J. (1996) BID. A novel BH3 domain-only death agonist. Genes Dev. 10, 2859–2869 [DOI] [PubMed] [Google Scholar]
- 34. Youle R. J., Strasser A. (2008) The BCL-2 protein family. Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 35. Billen L. P., Shamas-Din A., Andrews D. W. (2008) Bid. A Bax-like BH3 protein. Oncogene 27, S93–S104 [DOI] [PubMed] [Google Scholar]
- 36. Polster B. M., Basañez G., Young M., Suzuki M., Fiskum G. (2003) Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J. Neurosci. 23, 2735–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Satsoura D., Kučerka N., Shivakumar S., Pencer J., Griffiths C., Leber B., Andrews D. W., Katsaras J., Fradin C. (2012) Interaction of the full-length Bax protein with biomimetic mitochondrial liposomes. A small-angle neutron scattering and fluorescence study. Biochim. Biophys. Acta 1818, 384–401 [DOI] [PubMed] [Google Scholar]
- 38. Billen L. P., Kokoski C. L., Lovell J. F., Leber B., Andrews D. W. (2008) Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 6, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Veresov V. G., Davidovskii A. I. (2007) Monte Carlo simulations of tBid association with the mitochondrial outer membrane. Eur. Biophys. J. 37, 19–33 [DOI] [PubMed] [Google Scholar]
- 40. Yamaguchi R., Andreyev A., Murphy A. N., Perkins G. A., Ellisman M. H., Newmeyer D. D. (2007) Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ. 14, 616–624 [DOI] [PubMed] [Google Scholar]
- 41. Middleton E. R., Rhoades E. (2010) Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 99, 2279–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stankiewicz A. R., Lachapelle G., Foo C. P., Radicioni S. M., Mosser D. D. (2005) Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J. Biol. Chem. 280, 38729–38739 [DOI] [PubMed] [Google Scholar]
- 43. Hamilton N. A., Pantelic R. S., Hanson K., Teasdale R. D. (2007) Fast automated cell phenotype image classification. BMC Bioinformatics 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berg O. G., von Hippel P. H. (1985) Diffusion-controlled macromolecular interactions. Annu. Rev. Biophys. Biophys. Chem. 14, 131–160 [DOI] [PubMed] [Google Scholar]
- 45. Alder N. N., Jensen R. E., Johnson A. E. (2008) Fluorescence mapping of mitochondrial TIM23 complex reveals a water-facing, substrate-interacting helix surface. Cell 134, 439–450 [DOI] [PubMed] [Google Scholar]
- 46. Katz C., Zaltsman-Amir Y., Mostizky Y., Kollet N., Gross A., Friedler A. (2012) Molecular basis of the interaction between proapoptotic truncated BID (tBID) protein and mitochondrial carrier homologue 2 (MTCH2) protein. Key players in mitochondrial death pathway. J. Biol. Chem. 287, 15016–15023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu X., Dai S., Zhu Y., Marrack P., Kappler J. W. (2003) The structure of a Bcl-xL/Bim fragment complex. Implications for Bim function. Immunity 19, 341–352 [DOI] [PubMed] [Google Scholar]