

Background: Fungal tyrosinase maturation involves multiple processes of the dinuclear copper assembly and proteolytic activation.
Results: Structural examinations and mutational studies of the pro-tyrosinases revealed that three endogenous cysteines contribute to the copper incorporation.
Conclusion: The three highly flexible cysteines are essential for assembly of the active site across the protein shell.
Significance: Elucidation of such a copper incorporation process provides useful insights into metal homeostasis.
Keywords: Copper, Copper Transport, Post-translational Modification, Protein Cross-linking, Tyrosinase, Copper Chaperone, Protein-derived Cofactor, Zymogen
Abstract
Tyrosinase, a dinuclear copper monooxygenase/oxidase, plays a crucial role in the melanin pigment biosynthesis. The structure and functions of tyrosinase have so far been studied extensively, but the post-translational maturation process from the pro-form to the active form has been less explored. In this study, we provide the crystal structures of Aspergillus oryzae full-length pro-tyrosinase in the holo- and the apo-forms at 1.39 and 2.05 Å resolution, respectively, revealing that Phe513 on the C-terminal domain is accommodated in the substrate-binding site as a substrate analog to protect the dicopper active site from substrate access (proteolytic cleavage of the C-terminal domain or deformation of the C-terminal domain by acid treatment transforms the pro-tyrosinase to the active enzyme (Fujieda, N., Murata, M., Yabuta, S., Ikeda, T., Shimokawa, C., Nakamura, Y., Hata, Y., and Itoh, S. (2012) ChemBioChem. 13, 193–201 and Fujieda, N., Murata, M., Yabuta, S., Ikeda, T., Shimokawa, C., Nakamura, Y., Hata, Yl, and Itoh, S. (2013) J. Biol. Inorg. Chem. 18, 19–26). Detailed crystallographic analysis and structure-based mutational studies have shown that the copper incorporation into the active site is governed by three cysteines as follows: Cys92, which is covalently bound to His94 via an unusual thioether linkage in the holo-form, and Cys522 and Cys525 of the CXXC motif located on the C-terminal domain. Molecular mechanisms of the maturation processes of fungal tyrosinase involving the accommodation of the dinuclear copper unit, the post-translational His-Cys thioether cross-linkage formation, and the proteolytic C-terminal cleavage to produce the active tyrosinase have been discussed on the basis of the detailed structural information.
Introduction
Tyrosinase (EC 1.14.18.1), a dinuclear copper monooxygenase/oxidase, is a key enzyme in the melanin biosynthesis and is widely distributed in mammals, plants, fungi, and bacteria. The dinuclear copper center is involved in the catalytic reactions of tyrosinase, the hydroxylation of phenolic substrates (phenolase activity, Scheme 1), and the subsequent oxidation of catechols to o-quinones (catecholase activity, Scheme 1) (1–4). A similar dinuclear copper center, so-called type 3 copper, also plays important roles in catechol oxidase catalyzing exclusively the latter reaction (catecholase activity) and in hemocyanin, which serves as the dioxygen carrier in the hemolymph of mollusks and arthropods (5). In the crystal structures of these proteins, six histidine residues, which are provided by a four-helix bundle in the copper-binding domain, coordinate to the two copper ions (three histidine imidazoles for each copper ion, CuA and CuB) in the active site (Fig. 1, A and B) (6–12). In mushroom tyrosinase (12), sweet potato catechol oxidase (8), and octopus hemocyanin (7), one of the histidine imidazole groups coordinating to CuA is covalently bound to nearby cysteine via an unusual thioether linkage (Fig. 1), although its function still remains obscure.
SCHEME 1.
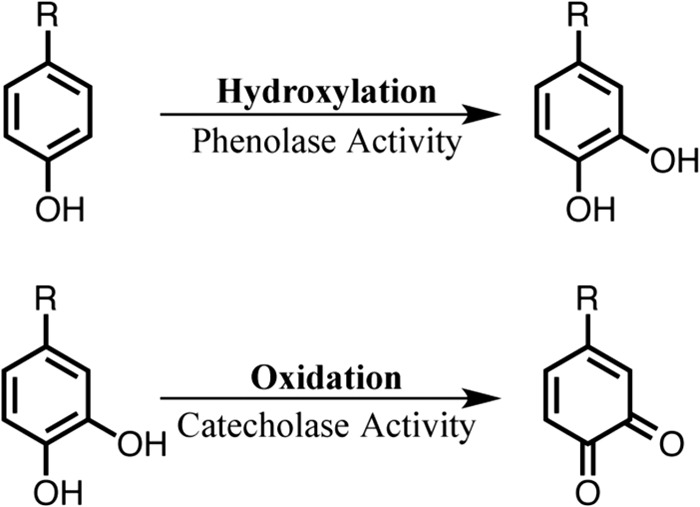
FIGURE 1.
Three-dimensional structures of the dinuclear copper sites containing a His-Cys cross-linkage. A, potato catechol oxidase (PDB code 1BT1); B, octopus hemocyanin (PDB code 1JS8); C, ChemDraw structure of 2-cysteinyl-histidine (His-Cys) cross-linkage.
In other copper-containing proteins, such as blue copper proteins, multicopper oxidases, cytochrome c oxidase, dopamine β-hydroxylase, peptidylglycine α-hydroxylating monooxygenase, particulate methane monooxygenase, quercetin 2,3-dioxygenase, copper amine oxidase, galactose oxidase, and Cu,Zn-SOD, the copper cofactors also play crucial roles to dictate electron transfer, two- and/or four-electron reduction of O2, alkane hydroxylation, dehydrogenation of alcohols and amines, and superoxide dismutation (13). However, copper is known to be highly toxic to cells due to its undesirable property in yielding the deleterious hydroxyl radical (14). Thus, the copper content inside the cell is severely regulated by copper membrane transporter proteins, which transport copper from the extracellular medium to the cytoplasm (15). Therefore, the amount of free copper ions available in the cytoplasm of the cell is very limited. To survive under such copper-depleted conditions, copper chaperones are often employed to deliver copper ions into the active site of copper proteins. In this case, copper ions are accommodated by using multiple cysteine and/or methionine units from the conserved motifs CXnC (n = 1–3) and/or MXmM (m = 1, 2), which are crucial for copper chaperone activity in the proteins (15, 16).
For mammalian and yeast tyrosinases, it is thought that copper ions are transferred into the active site by Atox1 copper chaperone and copper-transporting ATPase (ATP7A), which catalyze copper movement across the membranes in the Golgi apparatus on the secretion process (15, 17). However, detailed mechanism of the copper incorporation process has yet to be elucidated. Recently, a copper chaperone, the so-called caddie protein (ORF378), was crystallized in the complex with the bacterial tyrosinase from Streptomyces castaneoglobisporus (9, 18), where ORF378 binds to the active site of tyrosinase to help the copper trafficking. This result provides some insights into the molecular mechanism of the copper incorporation process of type 3 copper proteins.
In the case of octopus hemocyanin, however, the dinuclear copper site is completely covered by an additional shielding domain (C-terminal domain) (7), thus prohibiting the direct access of a copper chaperone to the active site. The situation may be similar with fungal tyrosinase and plant catechol oxidase, which also involve a C-terminal shielding domain as mentioned below. In these cases, copper chaperone-like machinery may be equipped in the same protein to facilitate the copper incorporation. This has been proved in this study using fungal tyrosinase from Aspergillus oryzae.
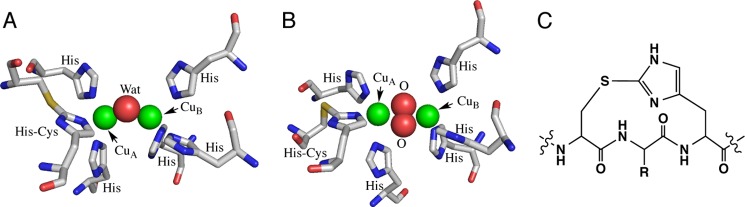
With regard to the C-terminal domain, it may play an important role in regulating the enzyme activity. Namely, the C-terminal domain prohibits substrate access to the enzyme-active site and blocks the oxidase/oxygenase activity to avoid undesirable intracellular reactions of highly reactive quinonoid products. In support of this notion, several lines of evidence have indicated that a proteolytic enzyme in vivo cleaves a part of the protein of the fungal pro-tyrosinase and plant pro-catechol oxidase to induce the enzymatic activity (19–21). Furthermore, the cloned cDNA clearly indicates the existence of such an additional extension in the C-terminal domain, although the isolated active forms of mushroom tyrosinase (PPO3)3 (12) and sweet potato catechol oxidase (8) lack the C-terminal domains in the crystal structures. On the basis of these results, it has been suggested that C-terminal domain of pro-tyrosinase is cleaved off by a proteolytic enzyme to produce the active form (Fig. 2, B to C) after copper ions are transferred into the active site through the protein shell (Fig. 2, A to B). However, the structural information of pro-tyrosinase has yet to be available, so that little is known about the details of the maturation processes, including the copper incorporation and the proteolytic activation.
FIGURE 2.
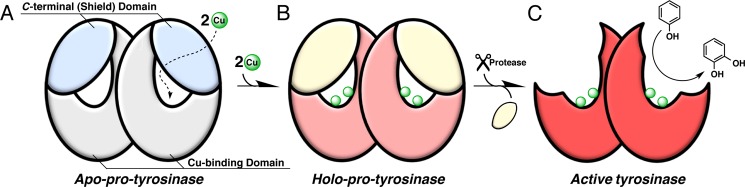
Schematic representation of fungal tyrosinase maturation process.
We have recently reported that the apo-form of recombinant pro-tyrosinase from A. oryzae (inactive precursor) can be overproduced from Escherichia coli by using the full-length of cDNA (melB; 27% identity to the full-length sequence of PPO3), and reconstitution of this apo-pro-tyrosinase with copper ion under aerobic conditions induces autocatalytic formation of His94–Cys92 cross-linkage in the enzyme-active site (22). The holo-pro-tyrosinase thus formed has no catalytic activity, but the trypsin treatment converts the holo-pro-form into the active form of the enzyme, in which the C-terminal domain (Gly464–Ala616) is absent (cleaved) (23).
Here, we successfully determined the crystal structures of the holo-pro- and the apo-pro-forms of melB tyrosinase from A. oryzae at a 1.39- and a 2.05-Å resolution, respectively, to provide the first detailed structural information about the fungal tyrosinase containing the C-terminal domain. The structural data of holo-pro-form provides important insights into the role of the C-terminal domain not only as a reactivity regulation domain but also as a copper chaperone-like machinery. Comparison of the crystal structures between the apo-pro-form and the holo-pro-form together with the mutagenesis studies have elucidated the detailed molecular mechanism of the maturation process of tyrosinase; the incorporation of copper ions and autocatalytic formation of the His-Cys cross linkage.
EXPERIMENTAL PROCEDURES
Expression of Tyrosinases and Selenomethionine-substituted Tyrosinase
melB pro-form of tyrosinase was prepared by using pETH-melB plasmid as described previously (22, 23). As for selenomethionine-substituted tyrosinase, expression plasmids were transformed into E. coli B834 (DE3) pLysS-competent cells. The transformants were grown in 2 liters of a baffled Erlenmeyer flask of 0.4 liters of Se-Met core medium (Wako) broth containing 50 mg/liter kanamycin, 4% glucose, 5 mg/liter vitamin B1, 50 mg/liter l-SeMet, 1.2 g/liter MgSO4, 24.4 mg/liter FeCl3, 50 μl/liter 2 m HCl, 4% glucose, with shaking at 135 rpm at 25 °C until the cultures reached an A600 of 0.8. At this point, isopropyl 1-thio-β-d-galactopyranoside was added to the mixture (0.1 mm after addition), and the resulting mixture was allowed to continue being shaken for an additional 24 h at 18 °C.
Purification of Tyrosinase
If not mentioned otherwise, the same purification protocol was used for all proteins. melB pro-tyrosinase was purified as described previously with some modifications (23). Harvested cells (∼10 g) from 1 liter of culture were resuspended in 40 ml of the binding buffer (0.3 m NaCl, 10 mm imidazole, and 20 mm BisTris, pH 7.2) containing 0.2 mg/ml lysozyme. After 15 min of incubation on ice, the sample was sonicated 120 times for 5 s on ice (20-watt output). After centrifugation at 30,000 × g for 30 min, the clarified supernatant was loaded onto a 5-ml nickel-agarose column (COSMOGEL His-Accept, Nacalai Tesque) previously equilibrated with the binding buffer, and the histidine affinity tag-fused protein was eluted with nickel-Sepharose elution buffer (0.1 m imidazole, 0.3 m NaCl, 0.5 mm tris(2-carboxyethyl)phosphine, and 20 mm BisTris, pH 7.2). The histidine affinity tag-free pro-tyrosinase was released from the fusion protein by incubation with 2 units of HRV3C protease/mg of protein on ice for over 16 h. The protein solution was then dialyzed against 10 mm Tris-HCl buffer, pH 8.2, containing 10 mm imidazole at 4 °C to remove the excess imidazole and NaCl. The dialyzed solution was loaded onto a 5-ml nickel-agarose column previously equilibrated with this dialysis buffer again to remove the released histidine affinity-tagged protein. This eluent was subjected to anion-exchange chromatography by using a Q-Sepharose HP column (volume of 5 ml, GE Healthcare) with a linear gradient elution of NaCl (10–200 mm) in 10 mm Tris-HCl buffer, pH 8.2. The fractions containing pro-tyrosinase were collected and concentrated by ultrafiltration using a VIVA SPIN 20 (Sartorius). Before crystallization, the purified protein solution was desalted using a PD-10 column (GE Healthcare) and changed to 20 mm BisTris buffer, pH 7.2. The protein was stored at −80 °C until use. Purity was checked by SDS-PAGE.
Site-directed Mutagenesis
Oligonucleotide-directed mutagenesis experiments were performed on pETH-melB vector that contained melB cDNA. The pair of about 25-base mutagenic oligonucleotide (Table 1) was obtained from Integrated DNA Technology. Site-directed mutagenesis was carried out by the inverse PCR and following DpnI digestion and self-ligation. For the mutant, the absence of undesired mutations on the melB gene was confirmed by DNA sequence analysis using a 3100 Genetic Analyzer (Applied Biosystems) after the construction of the vector.
TABLE 1.
Oligonucleotides used for site-directed mutagenesis
| Name | Sequence (5′ →3′)a | Length | Use |
|---|---|---|---|
| MelBH332A-f1 | GCGaactgggtgggaggattc | 21 | Forward primer for H332A |
| MelBH332A-r1 | gacgttgttgtgaatagcttctaagttcatc | 31 | Reverse primer for H332A |
| MelBV359A-f1 | ccagtggccgcgtatgatccgattttc | 27 | Forward primer for V359A |
| MelBV359A-r1 | TGcactgctcatatggccagcaccccag | 28 | Reverse primer for V359A |
| MelBF513A-f1 | agcggcagccttgaggacagtaactg | 26 | Forward primer for F513A |
| MelBF513A-r1 | AGCgttgaagacactgcccacaaaattctg | 30 | Reverse primer for F513A |
| MelBC522A-f1 | gacaagtgcgctcagcaagagcaag | 25 | Forward primer for C522A |
| MelBC522A-r1 | TGCgttactgtcctcaaggctgccgctgaag | 31 | Reverse primer for C522A |
| MelBC525A-f1 | gctcagcaagagcaagagggtgttttg | 27 | Forward primer for C525A |
| MelBC525A-r1 | TGCcttgtcacagttactgtcctcaaggctg | 31 | Reverse primer for C525A |
| MelBC522A/C525A-f1 | aagGCAgctcagcaagagcaag | 22 | Forward primer for C522A/C525A |
| MelBC522A/C525A-r1 | gtcTGCgttactgtcctcaaggctgccgctgaag | 34 | Reverse primer for C522A/C525A |
a The changed nucleotides are shown in capitals, and the codons corresponding to the amino acid residues to be changed are underlined.
Quantification of Protein and Copper Content
Protein concentration was determined by a modified Lowry method using a DC protein assay kit (Bio-Rad) according to the supplier's manual. Copper concentration was determined by an ICP-AES on an ICPS-8100 (Shimadzu). The calibration curve of copper ion was made by using a copper ion standard solution (10.0 ppm).
Crystallization of Copper-bound Pro-tyrosinase (Holo-pro-tyrosinase)
The protein sample was concentrated to 6 mg/ml by ultrafiltration using a VIVA SPIN 20 and reserved at −80 °C. Crystals of holo-pro-tyrosinase were obtained by the hanging drop vapor diffusion method. Crystallization droplets were prepared by mixing the precipitant solution containing 18% polyethylene glycol 3350, 50 mm NH4F (1 μl), and 6 mg/ml tyrosinase solution (2 μl). Crystals were obtained at 4 °C in 1 day and were soaked in the solution containing 25% polyethylene glycol 3350, 50 mm NH4F, and 20 mm BisTris at pH 7.2, for cryo-protection.
Crystallization of Copper-depleted Pro-tyrosinase (Apo-pro-tyrosinase)
The protein sample was concentrated to 17 mg/ml by ultrafiltration using a VIVA SPIN 20 and reserved at −80 °C. Apo-pro-tyrosinase was crystallized at 4 °C in a similar condition as holo-pro-tyrosinase using the precipitant solution containing 20% polyethylene glycol 3350, 50 mm KCl (1 μl), and 17 mg/ml tyrosinase solution (2 μl). For cryo-protection, solution containing 22.5% polyethylene glycol 3350, 15% ethylene glycol, 50 mm KCl, 20 mm BisTris at pH 7.2 was used.
Crystallization of Selenomethionine-substituted Pro-tyrosinase
The protein sample was concentrated in a similar condition as holo-pro-tyrosinase and crystallized at 4 °C using the precipitant solution containing 20% polyethylene glycol 3350, 50 mm NH4F (1 μl), and 6 mg/ml tyrosinase solution (2 μl). The same solution as holo-pro-tyrosinase crystals was used for cryo-protection.
X-ray Data Collection
All diffraction data were collected at 100 K on the BL44XU beamline at the SPring-8 synchrotron facility (Harima, Hyogo, Japan). Before flash cooling, crystals were directly soaked in a respective cryoprotectant solution for 20 min. X-ray diffraction images were collected using an MX225HE CCD detector (Rayonix, Evanston, IL) equipped with a Helix Technology cryo-system (Cryo Industries of America, Manchester, NH). All diffraction images were recorded on the CCD camera, and the data were processed and scaled with the HKL-2000 program package. The data collection statistics are summarized in Table 2.
TABLE 2.
Data collection and refinement statistics
| Holo-form (PDB code 3W6W) | Apo-form (PDB code 3W6Q) | Se-SAD | |
|---|---|---|---|
| Data set | |||
| X-ray source | BL44XU | BL44XU | BL44XU |
| Space group | P21 | P21 | P21 |
| Unit cell | a = 52.31 Å, b = 118.09 Å, c = 84.21 Å, β = 97.40° | a = 56.97 Å, b = 108.75 Å, c = 231.24 Å, β = 93.83 ° | a = 54.03 Å, b = 118.16 Å, c = 78.78 Å, β = 91.40 ° |
| Wavelength | 0.90000 Å | 0.90000 Å | 0.97892 Å |
| Resolutiona | 50.00 to 1.39 Å (1.42 to 1.39 Å) | 50.00 to 2.05 Å (2.09 to 2.05 Å) | 50.00 to 2.52 Å (2.56 to 2.52 Å) |
| No. of total/unique reflections | 736,459/198,757 | 606,461/175,066 | 122,994/33,456 |
| Redundancy | 1.9 | 1.3 | 1.9 |
| Completenessa | 98.3% (96.7%) | 97.7% (84.7%) | 98.8% (99.5%) |
| Rmergea | 5.6% (76.3%) | 5.1% (10.8%) | 4.7% (10.6%) |
| I/σa | 28.8 (1.9) | 38.7 (13.3) | 42.8 (23.5) |
| Refinement | |||
| Rwork/Rfree | 17.74%/19.92% | 14.39%/18.29% | |
| No. of protein/solvent atoms | 9815/1095 | 18531/1820 | |
| No. of metal ion atoms | 4 (copper) | 0 | |
| B-factors of protein/solvent | 18.1/27.9 | 24.1/30.8 | |
| B-factors of metal ions | 16.5 | ||
| r.m.s.d. bond/angle | 0.009 Å/1.102° | 0.007 Å/1.004° | |
| Ramachandran favored/allowed | 98.7/1.3% | 98.6/1.4% | |
a Values in parentheses are for the highest resolution shells.
Phasing and Initial Model Building
We attempted to carry out molecular replacement using MOLREP (24). Mushroom tyrosinase (PDB code 2Y9W) was selected for molecular replacement because of its 27% amino acid similarity to melB tyrosinase. However, we were unable to obtain good solutions for the molecular replacement calculations. We also attempted to obtain the phase information using a copper single-wavelength anomalous dispersion and multiple-wavelength anomalous dispersion methods, but we have failed. Finally, the structure of Se-Met pro-tyrosinase was solved by the single-wavelength anomalous dispersion method using Se-Met-substituted pro-tyrosinase. Initial phases were calculated with the program AutoSolve (figure-of-merit, 0.48), and the structure model was continuously built with the program Automodel in Phenix program package (25). The resulting model covered 935 residues of the two subunits in the asymmetric unit with 738 side chains assigned. The initial model thus obtained, of which the R-factors are 0.284 (work) and 0.339 (free), was used as search model for phasing of the holo-pro-tyrosinase crystal.
Structure Determination
The structure of holo-pro-tyrosinase was solved by the molecular replacement method using the model of Se-Met tyrosinase as the search model. Automatic model-building program Autobuild in Phenix program package (25) built the initial model. The structure model was manually rebuilt with the program COOT (26) followed by refinement calculations with the program Phenix.refine, including TLS optimization (27). This procedure was iterated until the model did not further improve. During the refinement, the copper-ligand distances and the distance of His94–Cys92 cross-linkage were loosely restrained with the ReadySet program. For apo-pro-tyrosinase, the structure was solved by molecular replacement using the final model of holo-pro-tyrosinase (chain A) as a search model, and the refinement procedure was the same as the case of holo-pro-tyrosinase. In both structures, Ramachandran analysis with the Molprobity program (28) showed no outliers. Secondary structures were assigned by the DSSP program (29). The atomic coordinates and structure factors for melB holo-pro-tyrosinase and apo-pro-tyrosinase were deposited in the PDB under the accession codes 3W6W and 3W6Q, respectively. All figures of protein structures were prepared using the program PyMOL (Version 1.3 Schrödinger, LLC).
Copper Uptake Assays
Copper binding assays were performed in E. coli BL21(DE3) cytoplasm. Expression of pro-tyrosinase was performed as reported previously (22, 23) except using the ZYM5052 medium (its copper concentration was determined to be <0.1 μm by ICP analysis) containing various concentrations of CuSO4 (1 μm to 3 mm). Harvested cells were washed with 0.85% NaCl aqueous solution to remove the remaining copper ions on the cell surface at least three times. After the purification as described above without the step of anion-exchange chromatography, the copper content of pro-tyrosinase was determined by ICP-AES analysis. This experiment was performed at least two times for each mutant and copper concentration.
RESULTS AND DISCUSSION
Overall Structure of Holo-form of Full-length melB
The crystal structure of melB holo-pro-tyrosinase was determined by molecular replacement using the structure of the selenomethionine-substituted melB apo-pro-tyrosinase as a search model. Holo-pro-form was crystallized with one homodimer in the crystallographic asymmetric unit (space group P21). The final structures were refined to a resolution of 1.39 Å with excellent statistics (Table 2). Two protomers in the asymmetric unit are related by a noncrystallographic 2-fold axis (Fig. 3A), displaying an ellipsoid shape with dimensions of 95 × 65 × 45 Å (Fig. 3A). Elements of secondary structure (α for α-helix, α′ for 310-helix, π for π-helix, and β for β-strand) are identified and numbered sequentially in Fig. 4.
FIGURE 3.

Whole structure and dimer interfaces of melB holo-pro-tyrosinase. A, homodimeric structure in the crystallographic asymmetric unit (upper, top view; middle, side view; bottom, bottom view). Subunit interfaces of the dimeric structure have three regions at the dimer interface, as indicated by red (region I) and green (region II) rectangles. Two same interactions of region II (green) exist in the interface (upper) along a 2-fold axis. The secondary structures are colored differently according to individual chains: chain A, blue; chain B, pink. B, magnified view of the two regions of dimer interface. Residues are shown as sticks. Dotted lines indicate closet contacts between atoms.
FIGURE 4.

Multiple sequence alignment of amino acid sequences of fungal tyrosinase and secondary elements of melB (α for α-helix, α′ for 310-helix, π for π-helix, and β for β-strand). The alignment was generated with ClustalW by using MEGA5 software. The conserved residues are marked with an asterisk and highlighted in rectangles (green, copper-coordinating histidines; red, conserved cysteines; yellow, the place holder; purple, YXY motif; gray, YG motif). The dashed line indicates residues not visible in the electron density. A. oryzae B, melB from A. oryzae (BD165761); A. bisporus, PPO3 from Agaricus bisporus (CAA59432); N. crassa, Neurospora crassa (CAE81941); P. nameko, Philiota nameko (AB275647); P. sanguineus, Pycnoporus sanguineus (AAX46018); L. edodes, Lentinula edodes (BAB71735).
The full-length melB protomer consists of 621 amino acid residues deduced from cDNA sequence. In chain B of the crystal structure, most of the residues were well defined in the final electron density, except for some disordered loops in the flexible regions (Gln81–Pro88, Asn158–Gln160, Ser213–Ser222, Asp304–Gln316, Gly515–Gly532, and Asp589–Asp597, Table 3). Conversely, the electron densities of three loops (Gln81–Pro88, Ser213–Ser222, and Gly515–Gln527) were observed in chain A. On the last loop, the backbone nitrogen atoms of Asp523(A), Lys524(A), and Cys525(A) of chain A were hydrogen bonded to Asp552(B′) and Asn492(B′) of the next protomer (chain B′), the chain B molecule related by the crystallographic symmetry, respectively (Fig. 5). In addition, there is also a hydrogen bond between Asp523(A) and Asp552(B′). These results indicate that these hydrogen bonds fix one of the conformations of this loop (Gly515–Gln527) and further stabilized those of other two loops located in the neighbors (Gln81–Pro88 and Ser213–Ser222).
TABLE 3.
Missing residues and atoms
| Holo-form | ||||
| Missing residues | ||||
| Chain A | Gly (−4)-Gly (−2) | His154–Gln160 | Lys305–Ala316 | Asp589–Asp597 |
| Chain B | Gly (−4)–Gly (−1) | Gln81–Pro88 | Asn158–Gln160 | Ser213–Ser222 |
| Asp304–Gln316 | Gly515–Gly532 | Asp589–Asp597 | ||
| Missing atoms | ||||
| Chain A | Lys215 (Cϵ, Nζ) | Lys395 (Cϵ, Nζ) | Lys413 (Cδ, Cϵ, Nζ) | |
| Lys524 (Cδ, Cϵ, Nζ) | Lys606 (Cϵ, Nζ) | |||
| Chain B | Lys123 (Cϵ, Nζ) | Phe223 (Cγ, Cδ1, Cδ2, Cϵ1, Cϵ2, Cζ) | ||
| Lys319 (Cγ, Cδ, Cϵ, Nζ) | Lys397 (Nζ) | Lys413 (Cδ, Cϵ, Nζ) | ||
| Asp440 (Cγ, Oδ1, Oδ2) | Lys482 (Cϵ, Nζ) | |||
| Apo-form | ||||
| Missing residues | ||||
| Chain A | Lys82–Lys85 | Gly155–Gln160 | Ser213–Phe223 | Asp304--Gln316 |
| Ser516–Gly532 | Arg593–Asp597 | |||
| Chain B | Gly (−4)–Gly (−1) | Gln81–Gly86 | Gly155–His156 | Ser213–Phe223 |
| Asp304–Gln316 | Ser516–Gly532 | Pro591–Asp597 | ||
| Chain C | Gly (−4)–Gly (−1) | Lys82–Gly86 | Gly155–Gln160 | Ser213–Ser222 |
| Asp304–Asp316 | Ser516–Gly532 | Pro591–Gly595 | ||
| Chain D | Gln81–Asn87 | Gly155–Asn158 | Ser213–Ser222 | Asp304–Gln316 |
| Ser516–Gln531 | Pro591–Asp597 | |||
| Missing atoms | ||||
| Chain A | Lys13 (Cϵ, Nζ) | Lys123 (Cδ, Cϵ, Nζ) | His154 (Cγ, Nδ1, Cδ2, Cϵ1, Cϵ2) | |
| Lys395 (Cϵ, Nζ) | Lys413 (Cϵ, Nζ) | |||
| Chain B | Lys13 (Cϵ, Nζ) | Lys319 (Cϵ, Nζ) | Arg345 (Cδ, Nϵ, Cζ, Nη1, Nη2) | |
| Lys397 (Cδ, Cϵ, Nζ) | Lys413 (Cϵ, Nζ) | Asp440 (Cγ, Oδ1, Oδ2) | ||
| Lys606 (Cϵ, Nζ) | ||||
| Chain C | His154 (Cγ, Nδ1, Cδ2, Cϵ1, Cϵ2) | Lys395 (Cϵ, Nζ) | Lys397 (Cδ, Cϵ, Nζ) | |
| Arg443 (Cδ, Nϵ, Cζ, Nη1, Nη2) | Lys553 (Cδ, Cϵ, Nζ) | Ser596 (Cγ) | ||
| Arg601 (Nη1, Nη2) | Glu609 (Cδ,Oϵ1, Oϵ2) | |||
| Chain D | Lys13 (Cϵ, Nζ) | Lys123 (Cδ, Cϵ, Nζ) | Lys395 (Cϵ, Nζ) | |
| Glu531 (Cγ, Cδ, Oϵ1, Oϵ2) | Lys553 (Cϵ, Nζ) | |||
FIGURE 5.
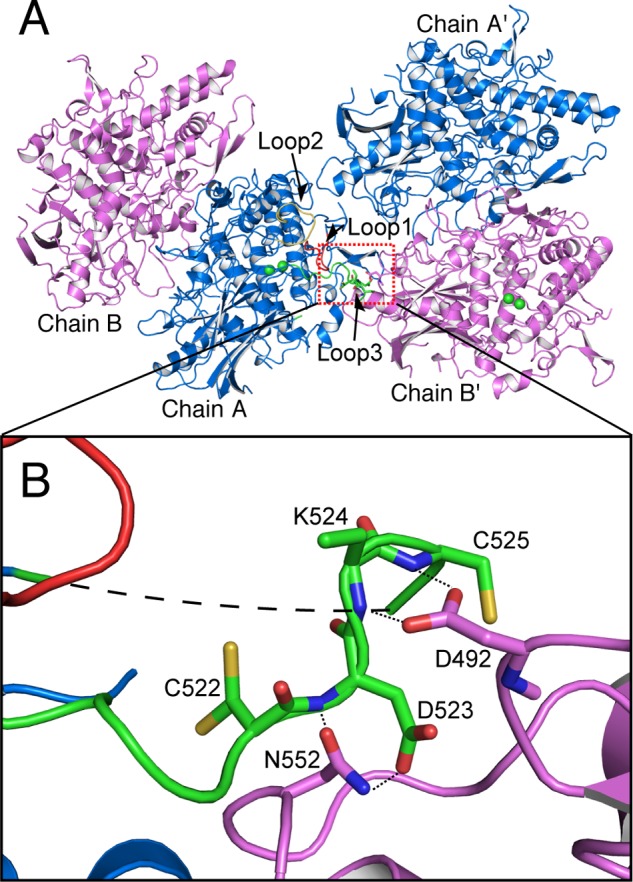
Interaction between the flexible loop and the next protomer related by the crystallographic symmetry. A, two homodimeric structures of holo-pro-form related by the crystallographic symmetry. Colored loops are loop 1 (Lys82–Lys85, red), loop 2 (Ser213–Phe223, yellow), and loop 3 (Ser516–Gly532, green) of holo-pro-form chain A. B, magnified view on the contacting region indicated by dotted rectangle. Dotted lines and small dashed line indicate the closet contacts between atoms, and the long dashed line indicates residues not visible in the electron density.
Dimer Interface
melB pro-tyrosinase was found to exist mainly as a homodimer at physiological condition in our previous study (23). A very tight inter-protomer interaction was observed in the crystal structure of holo-pro-tyrosinase. The homodimer is glued together through extensive hydrophobic and charge-charge interactions, and the accessible surface area buried in the interface is 1925 Å2 (Fig. 3A). Thus, we propose that the structure obtained via tight association is a biologically functional dimer. The dimer of holo-pro-tyrosinase is formed between chains A and B in a side-to-side manner along a 2-fold axis; thus, the substrate-binding pockets face alternate directions so as not to interfere with each other. The dimer interface can be classified into three regions (Fig. 3A, indicated by green and red rectangles). Region I (red region) predominantly involves hydrophobic interactions among leucine clusters (Leu259(A), Leu348(A), Leu350(A), Leu259(B), Leu348(B), and Leu350(B), Fig. 3B, red). The interface of region II (Fig. 3B, green region) is stabilized by hydrogen bonds between Tyr26(A) and Asp245(B), salt bridge between Glu163(A) and Lys349(B), and cation-π interaction between Arg165(A) and Tyr242(B). Furthermore, a tight hydrophobic interaction through van der Waals contacts is situated at the heart of region II (Thr171(A), Trp254(A), Leu255(A), Leu239, Tyr242(B), and Val246(B), Fig. 3B, green). Thus, both polar and hydrophobic interactions stabilize the association of the A and B chains.
Domain Structure of Holo-form of Full-length melB from A. oryzae
Each protomer consists of two structural domains as follows: the N-terminal copper-binding domain (Ser1–Phe463, indicated by blue) and the C-terminal shielding domain (Gly464–Ala616, indicated by red) (Fig. 6A). Because two protomers are nearly identical to each other, we describe the monomer structure by referring mainly to the chain-A molecule. The copper-binding domain includes the central core structure (Arg19–Tyr435) (3) and the peripheral loop region connecting the secondary structure elements (Figs. 4 and 6C). While melB pro-tyrosinase having about 100 extra residues is much larger than mushroom tyrosinase (PPO3, PDB code 2Y9W), the core structure was almost identical to that of the mushroom tyrosinase (root mean square deviation (r.m.s.d.) value for Cα atoms of 351 matched residues is 2.5 Å, Figs. 4 and 6C). A significant structural difference has arisen between the loop regions surrounding the core structures (Fig. 6C). The melB core structure is also similar to those of the other type 3 copper proteins, sweet potato catechol oxidase (PDB code 1BT3, r.m.s.d. 3.3 Å, 261 matched residues) and the functional unit of octopus hemocyanin (PDB code 1JS8, r.m.s.d. 2.9 Å, 258 matched residues) (Fig. 6, E and F). In these proteins, including melB tyrosinase, a four-helix bundle of the core structure provides the highly conserved six histidine residues as the copper ligands (three histidine imidazoles for each copper ion, CuA and CuB) in the active site, and one of these histidines (His94, melB numbering) is covalently bound to nearby cysteine (Cys92, melB numbering) via a thioether linkage (Fig. 6, B, D, and F).
FIGURE 6.

Crystal structure of protomer of melB holo-pro-tyrosinase. A, structure of the chain A molecule. The copper-binding domain (domain I, Ser1–Phe463) is colored in blue and the C-terminal domain (domain II, Gly464–Ala616) in red. Putative proteolytic site (Lys457) is labeled and shown in orange. The N and C termini are indicated as N and C, respectively. The black arrow indicates Phe513 from the C-terminal domain covering the active site. B, active site structure viewed from the C-terminal domain. Phe513 is shown as a stick model colored in yellow. Dotted lines indicate closest contacts between atoms. C, superimposed structure of the copper-binding domain (blue) versus mushroom tyrosinase (PDB code 2Y9W) (gray). D, superimposed structure of active site structure of melB (cyan) versus mushroom tyrosinase (PDB code 2Y9W) (gray). E, superimposed structure of whole structure of melB (the copper-binding domain in cyan, and the C-terminal domain in red) versus Octopus hemocyanin (PDB code 1JS8) (gray and yellow). F, superimposed structure of active site structure of melB (cyan) versus Octopus hemocyanin (PDB code 1JS8) (gray). Both green and brown spheres and red spheres indicate copper and water, respectively. Residues are shown as sticks.
The dinuclear copper center of melB tyrosinase is buried in the cleft of the copper-binding domain and is inaccessible to the exterior solvent due to the occlusion by the C-terminal domain as expected. The tip of the C-terminal domain is the Phe513 residue, and its side chain is accommodated just above this dicopper active center as the “placeholder” for phenolic substrates (Fig. 6B). The phenyl ring of Phe513 stacks onto the imidazole ring of one of the CuB ligands (His332) at the distance of ∼3.6 Å, and the edge of the aromatic ring is close to CuB (∼3.9 Å, Fig. 6B). Furthermore, one conformer of the side chain of Val359 residue (Val359(A)) is also at a van der Waals distance (∼3.5 Å) from the aromatic ring of Phe513 residues (Fig. 6B). Notably, the mutant V359A could be expressed in the soluble fraction in E. coli, whereas mutants H332A and F513A were expressed as inclusion body (Fig. 7), indicating that the π-stacking between Phe513 and His332 may strongly contribute to the stabilization of inter-domain interaction in melB pro-tyrosinase (Fig. 6, A and B).
FIGURE 7.

SDS-12.5% PAGE. Precipitation (P) (lanes 1, 3, 5, and 7) and supernatant (S) (lanes 2, 4, 6, and 8) of cell extract of E. coli containing melB expression plasmid. Lane M, marker; lanes 1 and 2, wild type; lanes 3 and 4, V359A; lanes 5 and 6, H332A; lanes 7 and 8, F513A. Arrows indicates the bands of melB pro-tyrosinase.
The C-terminal domain exhibits a seven-stranded antiparallel β-sandwich structure, whose topology is the truncated jellyroll motif (Fig. 8, A and D). The C-terminal domain does not show any structural similarity to the caddie protein of S. castaneoglobisporus tyrosinase (Fig. 8C). By structural similarity search using the DALI server (30), the closest structural homolog was found in the shielding domain of octopus hemocyanin functional unit (Fig. 8B, PDB code 1JS8, r.m.s.d. 2.5 Å, 89 matched residues) despite little sequence identity (7%), the copper-binding domain of which is also structurally similar to that of melB tyrosinase described above. Thus, the protomer can be superimposed to that of octopus hemocyanin not only with the copper-binding domain but also with the C-terminal domain (Fig. 6E). Consistent with the structural similarity, the holo-pro-tyrosinase has no catalytic activity, but the reduced form of holo-pro-tyrosinase produced by the reduction of the as-isolated enzyme with hydroxylamine exhibits a reversible dioxygen-binding ability, and its affinity toward dioxygen is almost equal to that of octopus hemocyanin (23). The Leu2830 residue in the octopus hemocyanin structure occupied a similar position as Phe513 in melB tyrosinase (Fig. 6F), so the aromatic carbons of Phe513 might not have much influence on the dioxygen-binding ability.
FIGURE 8.
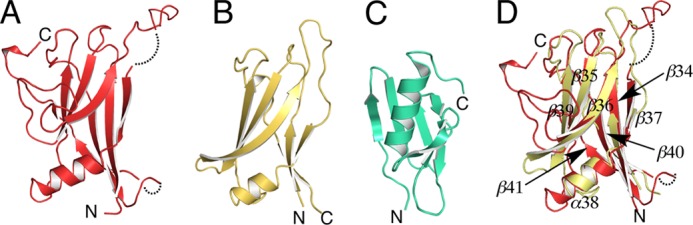
Structure of C-terminal domains and caddie protein. A, structure of the C-terminal domain of melB holo-pro-tyrosinase. B, structure of the shielding domain of the functional unit of Octopus hemocyanin (PDB code 1JS8). C, structure of the caddie protein of S. castaneoglobisporus tyrosinase (PDB code 1WX2). D, superimposed structure of the C-terminal domain of melB (red) versus shielding domain of octopus hemocyanin (PDB code 1JS8) (yellow). The dotted line indicates the residues not visible in the electron density.
The proteolytic digestion of the C-terminal domain having Phe513 placeholder leads to opening the entrance to the enzyme-active site for substrate incorporation (Fig. 2, B to C). In the copper-binding domain, the core region (Arg19–Tyr435) is followed by a linker consisting of 28 residues (Ala436–Lys463), which contains the five short helices (α29, α′30, α31, π32, and α33, see Figs. 4 and 9). This linker region has an inflection point containing conserved Tyr-Gly sequence motif (Tyr452 and Gly453), located in the π-helix (π32, Fig. 9). In the proteolytic activation in our previous study, the C-terminal domain and the last part of this linker (α33) were digested (Glu458–Ala616, see below) (23). The hydroxyl group of Tyr452 has hydrogen bonds to the side chains of the functionally conserved Asp147 and Arg377, enforcing this region of the linker to associate with the tyrosinase core domain (Fig. 9). Thus, this linker moiety is situated on the surface of the monomer, and the α33 helix is bulged out from the surface of the copper-binding domain, so the proteolytic enzyme can easily attack this region. This cleavage site (between Lys457 and Glu458) is also located next to the equivalent C-terminal position in the active mushroom tyrosinase (PPO3).
FIGURE 9.
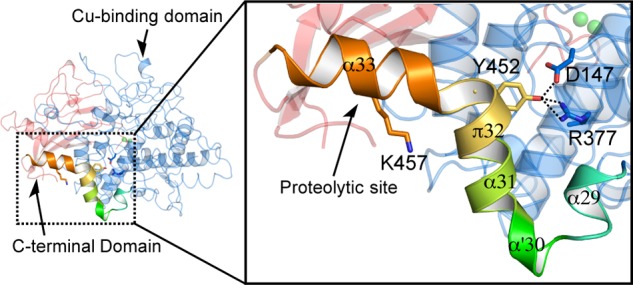
Structure of the linker region between the core and C-terminal domains. Location of the proteolytic site in the molecule. Magnified view of the C-terminal region of the copper-binding domain shows the interactions between YG motif and core region. Dashed lines show hydrogen bonds.
Conserved Cysteines in melB Apo-pro-tyrosinase
As in the case of holo-pro-tyrosinase, melB apo-pro-tyrosinase exhibits a homodimeric structure. The crystal structure of apo-pro-form of melB was determined by molecular replacement using the structure of holo-pro-form as a search model. The final structure of apo-pro-form was refined to a resolution of 2.05 Å with good statistics (Table 2). Apo-pro-form melB tyrosinase was crystallized in the monoclinic space group of P21. Most of the residues are well defined in the final model in each protomer (Fig. 10A), and the crystallographic asymmetric unit has two sets of homodimers (chains A and B and chains C and D). Four protomer molecules are almost identical to each other and very similar to those of the holo-pro-form (Fig. 10B). In chain A, the six disordered loops and peptide side chains were observed (Lys82–Lys85, Gly155–Gln160, Ser213–Phe223, Asp304–Gln316, Ser516–Gly532, and Arg593–Asp597), as is the case of chain B of holo-pro-form structure. The whole structures of apo-pro-form and holo-pro-form closely resemble each other, showing a 0.5-Å r.m.s.d. over all backbone atoms except the disordered region (chain A and B of apo-pro-form crystal versus chain A and B of holo-pro-form crystal).
FIGURE 10.
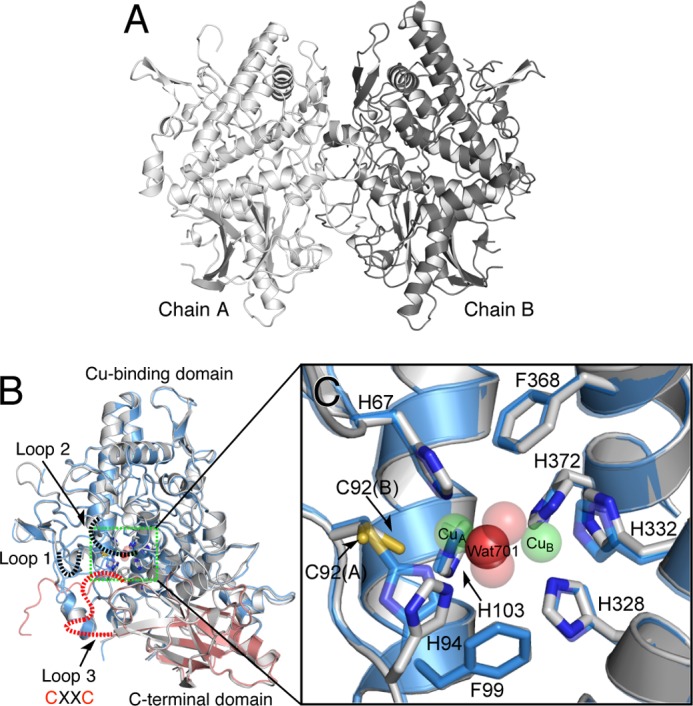
Crystal structure of melB apo-pro-tyrosinase. A, homodimeric structure of the apo-pro-form (gray and dark gray). B, subunit structures of apo-pro-form (chain A, gray) versus holo-pro-form (chain A, cyan, copper-binding domain; red, C-terminal domain). Dotted lines indicate the disordered loops colored in black and red. C, magnified view on the active site region of apo-pro-form (chain A, gray) and holo-pro-form (chain A, transparent cyan and sphere) indicated by the green rectangle. Green and red spheres indicate copper and water, respectively. Residues are shown as sticks and colored by atom type (carbon, as the respective structural element; nitrogen, blue; oxygen, red).
Despite the similarity of the whole structures, the positions of the three-conserved histidine residues (His94, His332, and His372) of apo-pro-form are different from those of the holo-pro-tyrosinase, and a water molecule (Wat701) is present in place of the two copper ions (Figs. 10C and 11, A and B). Wat701 is hydrogen-bonded to three Nϵ2 nitrogen atoms of the imidazole groups of His67, His94, and His103 (<3.0 Å CuA-coordinating), but those of the other histidines (>3.0 Å CuB-coordinating) are not within hydrogen-bonding distance. The side chains of His94 and His372 take different conformations from those of the holo-pro-form and exhibited higher average B-factors (33.3 and 19.5 Å2, respectively) than those of the other four histidines (14.3 Å2). These results suggest that the side chain of these two histidine residues are flexible as compared with those of the others; His94 and His372 imidazoles exhibit no hydrogen-bonding interaction with other protein residues. The positions of side chains His332 and Phe513 deviate somewhat from those in holo-pro-tyrosinase due to the absence of the metal coordination (Fig. 11D). It should be noted that no electron density was found between Cys92 and His94 side chains (Fig. 11B), definitely demonstrating that the His94–Cys92 thioether linkage is absent in the apo-form, and the covalent His-Cys bond is formed after the copper up-take as reported in our previous paper (22). Furthermore, the side chain of Cys92 adopts dual conformations: one (Cys92(A)) is exactly the same as the conformation observed in the holo-form and the other one (Cys92(B)) faces to the direction to the CuA-binding site (Figs. 10C and 11B). In the latter conformation, S atom of Cys92 side chain is within bonding distance to CuA (< 2.5 Å), suggesting a direct interaction of the thiol (or thiolate) group of Cys92 and copper ion(s) during the maturation process.
FIGURE 11.

Structural comparison of the holo-pro-form and apo-pro-form of melB. A, His94–Cys92 linkage of the holo-pro-form, and B, free Cys92 and His94 side chains of the apo-pro-form covered with electron density map (σ level = 1.5). C, superimposition of apo-form chain A (gray) and holo-form chain A (blue). Dashed rectangle indicates the disordered region of apo-pro-form chain A. Colored loops in this rectangle are loop 1 (Lys82–Lys85, red), loop 2 (Ser213–Phe223, yellow), and loop 3 (Ser516–Gly532, green) of holo-pro-form chain A. Dotted lines indicate the residues not visible in the electron density. D, superimposed structure of the active site region of apo-pro-form (chain A, gray) and holo-pro-form (chain A, blue and sphere). Residues are shown as stick models. Dotted lines indicate the closet contacts between atoms. Phe513 id shown as a stick model colored in yellow (holo) and purple (apo). Green spheres indicate copper ions.
It should be noted that the electron density of the surface region enveloping the CuA-binding site is completely disordered in each protomer of apo-pro-tyrosinase. This area is composed of three loops (loops 1–3: Lys82–Lys85, Ser213–Phe223, and Ser516–Gly532, see Figs. 10B and 11C). From the amino acid sequence analysis of these flexible loops, a CXXC motif composed of the conserved two cysteines (Cys522 and Cys525, Figs. 4, 10B, and 11C) are found on loop 3 of the C-terminal domain. Such a motif is seen in the copper chaperones such as Atox1 and Ccc2 in yeast (15). Thus, we consider that the three cysteines (Cys92, Cys522, and Cys525) are involved in the copper uptake process (Fig. 2, A to B).
Assembling Process of Dinuclear Copper Center
To confirm this hypothesis, we have performed a copper uptake experiment from the E. coli cytoplasm by using Cys-to-Ala mutants. The extent of assembly of the dinuclear copper center of as-isolated melB pro-tyrosinase practically depended on the CuSO4 concentration in the medium (Fig. 12, black circles). From this result, it was thought that the influx of the copper(II) ion into the E. coli cytoplasm might increase as the CuSO4 concentration in the medium becomes higher. The redox state of the copper becomes a monovalent cation (copper(I)) in the cytoplasm of E. coli as in the eukaryotic cells (31). Wild type of melB pro-tyrosinase was successfully metalated to full complement even at a low concentration of CuSO4 (∼1 mm). However, C522A and C525A mutants (Fig. 12, blue and green circles, respectively) needed more than 1 order of magnitude higher concentration of CuSO4 in the medium to complete the assembly of the dinuclear copper center, and a full complement of double mutants (C522A/C525A, Fig. 12, orange circle) was not observed within the range of the concentration capable of growing E. coli (≤3 mm). Furthermore, C92A mutants (C92A and C92A/C522A/C525A, Fig. 12, red and purple circle, respectively) have almost no ability to incorporate the copper ions from the cytoplasm of E. coli. Thus, it could be concluded that Cys92 residue as well as the 522CXXC525 motif contributes to the copper incorporation of melB apo-pro-tyrosinase (Fig. 2, A to B).
FIGURE 12.
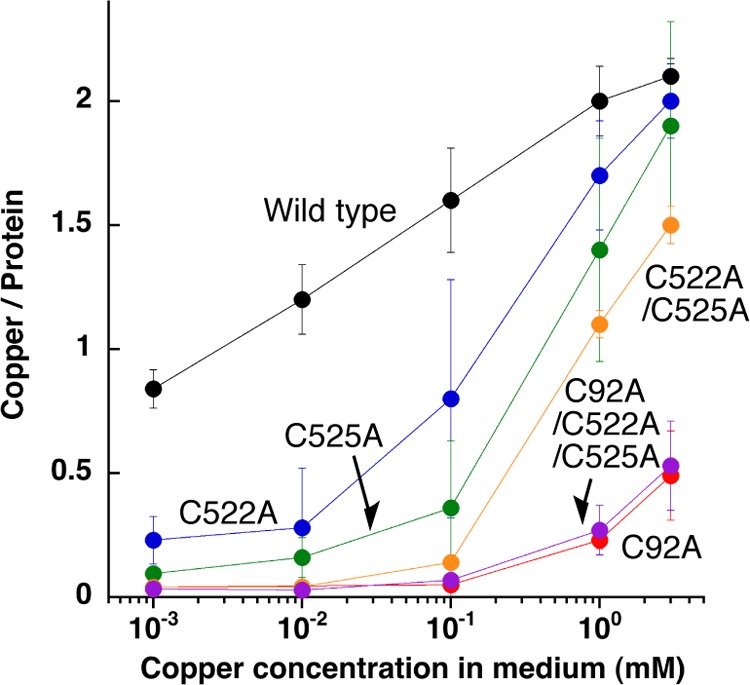
Copper content versus CuSO4 concentration. Copper content of purified pro-tyrosinase from E. coli cultivated in the medium containing various concentrations of CuSO4. Wild type, black; C92A, red; C522A, blue; C525A, green; C522A/C525A, orange; C92A/C522A/C525A, purple.
From these results, we concluded that the C-terminal shielding domain functions not only to control the enzyme activity by prohibiting the substrate access but also to induce the copper incorporation into the active site like copper chaperone. The completely disordered region of this shielding domain (loop 3) exists near CuA site and contains the two highly flexible cysteines (Cys522 and Cys525) within the distance range of about 5–15 Å from the sulfur atom of Cys92. This structural feature is reminiscent of the action of copper chaperones. The 522CXXC525 motif may act as the linear bidentate ligand to copper(I) (Fig. 13B) and serve as a copper shuttle to Cys92, where the copper(I) ion may be transiently ligated by three cysteine residues (Fig. 13, B to C). Another copper(I) ion may be incorporated in the similar manner to produce a dicopper(I) form of the reduced enzyme (Fig. 13, D to E). During this process, Cys92 may switch the conformation between Cys92(A) and Cys92(B), enhancing the copper(I) transport into a CuA binding site (Fig. 13, C to E). From the dicopper(I) species thus constructed (Fig. 13E), (μ-η2/η2-peroxido)dicopper(II) species is generated by the reaction with dioxygen (Fig. 13F). As demonstrated in our previous study, the His-Cys cross-linkage formation proceeds autocatalytically involving the formation of this active species as a key intermediate to produce the Met-pro-tyrosinase (dicopper(II), Fig. 13G) (22). As a result, two copper ions can be transported into the active site by the contribution of these cysteines (Cys92, Cys522, and Cys525).
FIGURE 13.

Schematic representation of putative copper incorporation process into pro-tyrosinase.
Maturation Process of melB Tyrosinase
In this study, we have obtained the detailed structural information of fungal pro-tyrosinase containing the shielding C-terminal domain, which covers the entrance of the active site. We have confirmed that three cysteines (Cys92, Cys522, and Cys525) play significant roles in the copper incorporation process. Fungal tyrosinase exploits the thiol group of Cys92 for the metal incorporation as well as enhancement of the enzymatic activity as described in our previous paper (22). In this context, the His-Cys-forming cysteines found in octopus hemocyanin and sweet potato catechol oxidase may also participate in the copper incorporation process (Fig. 1, A and B). Although CXXC motifs like fungal tyrosinase are not conserved on the C-terminal domain of these proteins, we were able to find a disulfide bond in the proximity of their His-Cys cross-linkage (within 10 Å distance) on the copper-binding domain in each crystal structure (octopus hemocyanin, Cys2549–Cys2559, and sweet potato catechol oxidase, Cys27–Cys89), indicating that the free thiol form of these disulfides may have similar functions to that of 522CXXC525 motif of melB tyrosinase. Besides Cys92, two cysteines (Cys522 and Cys525) on the C-terminal domain are highly conserved among any fungal tyrosinases (Fig. 4), whereas the specific partner protein for tyrosinase as a copper chaperone has not been found in the fungi yet. Therefore, the fungal tyrosinase may assemble the dinuclear copper center by itself by using the internal three cysteines, directly transferring from copper permeases on the cytoplasmic membrane such as Ctr1/3 homologs (15) without the mediation by the external copper chaperones (Fig. 2, A to B). However, the exact interaction between tyrosinase and such copper permeases is not known; therefore, the detailed molecular mechanism of the copper transfer between these proteins needs further investigation.
We have examined that the trypsin treatment induces the activation of melB holo-pro-tyrosinase and explored its activation mechanism (Fig. 2, B to C); the tryptic digestion induced cleavage of the C-terminal domain (Glu458–Ala616) while keeping the dimeric structure of the enzyme (23). As mentioned above, the interactions between subunits are located in the copper-binding domain predominantly, indicating that the dimeric structure is retained after cleavage of the C-terminal domain involving the activation process. The core structure of copper-binding domain is almost identical to that of the active form of mushroom tyrosinase (PPO3) as well as catechol oxidase. Therefore, we could conclude that this cleavage process induces little conformational change of the active site but simply causes the removal of Phe513 from the active site (Fig. 6, A and C). To our knowledge, no protease specific to tyrosinase has been discovered yet, but the proteases with trypsin-like activity are common in fungi (32). Thus, such an enzyme may be involved in this proteolytic activation process of fungal tyrosinase (Fig. 2, B to C).
Acknowledgments
We thank E. Yamashita, A. Higashiura, M. Suzuki, and A. Nakagawa of SPring-8 BL-44XU for their support during x-ray data collection.
This work was supported in part by Grant-in-aid for Scientific Research on Innovative Areas (Molecular Activation Directed toward Straightforward Synthesis) 22105007 (to S. I.) and Grant-in-aid for Challenging Exploratory Research 20038784 from the Ministry of Education, Culture, Sports, Science and Technology. Japan (to N. F.).
The atomic coordinates and structure factors (codes 3W6Q and 3W6W) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PPO3
- polyphenol oxidase 3
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- SeMet
- selenomethionine
- ICP-AES
- inductively coupled plasma-atomic emission spectrometry.
REFERENCES
- 1. Solomon E. I., Sundaram U. M., Machonkin T. E. (1996) Multicopper oxidases and oxygenases. Chem. Rev. 96, 2563–2606 [DOI] [PubMed] [Google Scholar]
- 2. Itoh S., Fukuzumi S. (2007) Monooxygenase activity of type 3 copper proteins. Acc. Chem. Res. 40, 592–600 [DOI] [PubMed] [Google Scholar]
- 3. Flurkey W. H., Inlow J. K. (2008) Proteolytic processing of polyphenol oxidase from plants and fungi. J. Inorg. Biochem. 102, 2160–2170 [DOI] [PubMed] [Google Scholar]
- 4. Itoh S. (2003) In Comprehensive Coordination Chemistry II (McCleverty J. A., Meyer T. J., eds) Vol. 8, pp. 369–393, Elsevier, Amsterdam [Google Scholar]
- 5. van Holde K. E., Miller K. I. (1995) Hemocyanins. Adv. Protein Chem. 47, 1–81 [DOI] [PubMed] [Google Scholar]
- 6. Hazes B., Magnus K. A., Bonaventura C., Bonaventura J., Dauter Z., Kalk K. H., Hol W. G. (1993) Crystal structure of deoxygenated Limulus polyphemus subunit-II hemocyanin at 2.18-angstrom resolution–Clues for a mechanism for allosteric regulation. Protein Sci. 2, 597–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cuff M. E., Miller K. I., van Holde K. E., Hendrickson W. A. (1998) Crystal structure of a functional unit from octopus hemocyanin. J. Mol. Biol. 278, 855–870 [DOI] [PubMed] [Google Scholar]
- 8. Klabunde T., Eicken C., Sacchettini J. C., Krebs B. (1998) Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Biol. 5, 1084–1090 [DOI] [PubMed] [Google Scholar]
- 9. Matoba Y., Kumagai T., Yamamoto A., Yoshitsu H., Sugiyama M. (2006) Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 281, 8981–8990 [DOI] [PubMed] [Google Scholar]
- 10. Sendovski M., Kanteev M., Ben-Yosef V. S., Adir N., Fishman A. (2011) First structures of an active bacterial tyrosinase reveal copper plasticity. J. Mol. Biol. 405, 227–237 [DOI] [PubMed] [Google Scholar]
- 11. Li Y., Wang Y., Jiang H., Deng J. (2009) Crystal structure of Manduca sexta prophenol oxidase provides insights into the mechanism of type 3 copper enzymes. Proc. Natl. Acad. Sci. U.S.A. 106, 17002–17006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ismaya W. T., Rozeboom H. J., Weijn A., Mes J. J., Fusetti F., Wichers H. J., Dijkstra B. W. (2011) Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 50, 5477–5486 [DOI] [PubMed] [Google Scholar]
- 13. Karlin K. D. (1993) Metalloenzymes, structural motifs, and inorganic models. Science 261, 701–708 [DOI] [PubMed] [Google Scholar]
- 14. Gaetke L. M., Chow C. K. (2003) Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 189, 147–163 [DOI] [PubMed] [Google Scholar]
- 15. Robinson N. J., Winge D. R. (2010) Copper metallochaperones. Annu. Rev. Biochem. 79, 537–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis A. V., O'Halloran T. V. (2008) A place for thioether chemistry in cellular copper ion recognition and trafficking. Nat. Chem. Biol. 4, 148–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang N., Hebert D. N. (2006) Tyrosinase maturation through the mammalian secretory pathway: Bringing color to life. Pigment Cell Res. 19, 3–18 [DOI] [PubMed] [Google Scholar]
- 18. Matoba Y., Bando N., Oda K., Noda M., Higashikawa F., Kumagai T., Sugiyama M. (2011) A molecular mechanism for copper transportation to tyrosinase that is assisted by a metallochaperone, caddie protein. J. Biol. Chem. 286, 30219–30231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kupper U., Niedermann D. M., Travaglini G., Lerch K. (1989) Isolation and characterization of the tyrosinase gene from Neurospora crassa. J. Biol. Chem. 264, 17250–17258 [PubMed] [Google Scholar]
- 20. Halaouli S., Record E., Casalot L., Hamdi M., Sigoillot J. C., Asther M., Lomascolo A. (2006) Cloning and characterization of a tyrosinase gene from the white-rot fungus Pycnoporus sanguineus, and overproduction of the recombinant protein in Aspergillus niger. Appl. Microbiol. Biotechnol. 70, 580–589 [DOI] [PubMed] [Google Scholar]
- 21. Kawamura-Konishi Y., Tsuji M., Hatana S., Asanuma M., Kakuta D., Kawano T., Mukouyama E. B., Goto H., Suzuki H. (2007) Purification, characterization, and molecular cloning of tyrosinase from Pholiota nameko. Biosci. Biotechnol. Biochem. 71, 1752–1760 [DOI] [PubMed] [Google Scholar]
- 22. Fujieda N., Ikeda T., Murata M., Yanagisawa S., Aono S., Ohkubo K., Nagao S., Ogura T., Hirota S., Fukuzumi S., Nakamura Y., Hata Y., Itoh S. (2011) Post-translational His-Cys cross-linkage formation in tyrosinase induced by copper(II)-peroxo species. J. Am. Chem. Soc. 133, 1180–1183 [DOI] [PubMed] [Google Scholar]
- 23. Fujieda N., Murata M., Yabuta S., Ikeda T., Shimokawa C., Nakamura Y., Hata Y., Itoh S. (2012) Multifunctions of MelB, a fungal tyrosinase from Aspergillus oryzae. ChemBioChem. 13, 193–201 [DOI] [PubMed] [Google Scholar]
- 24. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 28. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure-Pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 30. Holm L., Rosenström P. (2010) Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rensing C., Grass G. (2003) Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 27, 197–213 [DOI] [PubMed] [Google Scholar]
- 32. Yike I. (2011) Fungal proteases and their pathophysiological effects. Mycopathologia 171, 299–323 [DOI] [PubMed] [Google Scholar]