

Background: Telomerase requires the interaction of conserved protein and RNA motifs.
Results: Site-directed hydroxyl radical probing and mutagenesis identified an essential region of RNA-protein interactions.
Conclusion: The conserved CP2 protein motif is proximal to a conserved telomerase RNA motif.
Significance: Understanding how telomerase protein and RNA subunits interact allows us to begin constructing a more complete mechanistic model of telomerase function.
Keywords: Ribonuclear Protein (RNP), RNA, RNA-binding Protein, Telomerase, Telomeres
Abstract
The ends of linear chromosomes are extended by telomerase, a ribonucleoprotein complex minimally consisting of a protein subunit called telomerase reverse transcriptase (TERT) and the telomerase RNA (TER). TERT functions by reverse transcribing a short template region of TER into telomeric DNA. Proper assembly of TERT and TER is essential for telomerase activity; however, a detailed understanding of how TERT interacts with TER is lacking. Previous studies have identified an RNA binding domain (RBD) within TERT, which includes three evolutionarily conserved sequence motifs: CP2, CP, and T. Here, we used site-directed hydroxyl radical probing to directly identify sites of interaction between the TERT RBD and TER, revealing that the CP2 motif is in close proximity to a conserved region of TER known as the template boundary element (TBE). Gel shift assays on CP2 mutants confirmed that the CP2 motif is an RNA binding determinant. Our results explain previous work that established that mutations to the CP2 motif of TERT and to the TBE of TER both permit misincorporation of nucleotides into the growing DNA strand beyond the canonical template. Taken together, these results suggest a model in which the CP2 motif binds the TBE to strictly define which TER nucleotides can be reverse transcribed.
Introduction
Eukaryotic cells must distinguish the natural ends of chromosomes from double-stranded DNA breaks that are a mark of DNA damage. To do so, chromosome ends are capped by chromatin structures called telomeres, comprising repetitive DNA sequences that recruit specific DNA-binding proteins (1). Telomeres are shortened with each round of cell division and so require the enzyme telomerase to maintain telomere length in actively dividing cells. Telomerase dysfunction is associated with diseases that affect proliferative tissues, such as dyskeratosis congenita and aplastic anemia (2). On the other hand, aberrant telomerase overexpression can help to confer proliferative potential to cells and is associated with 90% of human cancer cell lines, making telomerase an attractive target for cancer therapies (3).
Telomerase is a RNA-protein complex that includes both the protein telomerase reverse transcriptase (TERT)2 and telomerase RNA (TER). TER contains a region complementary to the telomeric DNA sequence, known as the template. The template region of TER is repetitively reverse transcribed by TERT to extend telomere DNA (4).
TERT contains four domains: an N-terminal domain, an RNA binding domain (RBD), a reverse transcriptase domain, and a C-terminal extension (5). TERs also have several conserved structural motifs required for the function of the telomerase holoenzyme. TERs from ciliates, yeasts, and mammals all contain an essential RNA pseudoknot proximal to the template (6). In addition, TERs universally possess a template boundary element (TBE) that defines the region of TER to be reverse transcribed by TERT (7, 8). The high fidelity of template definition established by RBD-TBE interactions is a central feature of the telomerase catalytic cycle because incorporation of a single nontemplate nucleotide will prevent synthesis of subsequent telomere DNA repeats.
Recent studies in the Tetrahymena thermophila model system have shown that the RBD domain of TERT is primarily responsible for interactions with TER, is sufficient to bind TER with high affinity, and includes several conserved protein motifs involved in RNA binding (9–13). However, it is still not known which portions of the RBD interact with which conserved RNA motifs. An x-ray crystal structure of a large C-terminal fragment of the T. thermophila RBD has been solved (14), and comparison with the Tribolium castaneum TERT structure suggests that the conserved T motif forms a β-strand hairpin near the position of the template RNA (15). Furthermore, the conserved CP motif was shown to be adjacent to the T motif in an electropositive groove, perhaps positioned to bind the TBE (14). However, a co-crystal with the RBD bound to RNA was not attained, and so it could not be definitively shown which protein motifs interact with which RNA domains. A third conserved domain, the CP2 motif, was not included in the RBD construct that was solved by x-ray crystallography.
In the present work, we employed site-directed Fe(II)-EDTA hydroxyl radical probing to map RNA-protein interactions in telomerase, as done previously for the ribosome (16, 17). Our results demonstrated the CP2 motif and the TBE of Tetrahymena TER are in close proximity. To explore further to role of the CP2 motif in TER binding, we generated CP2 point mutants and measured their affinity for TER. Electrophoretic mobility shift assays (EMSAs) indicated that deletion of the entire CP2 motif severely compromised TER binding, and many single-point mutations in the CP2 motif result in a reduced affinity for TER, consistent with the CP2 motif being an important determinant of RBD binding. Among the single amino acid mutants analyzed, the strongest binding defect was observed with a mutation to Arg237, a residue previously shown to play a role in telomerase activity and template definition (8, 10). Quantitative EMSAs demonstrated that a single amino acid CP2 point mutant (R237A) showed an approximate 7-fold reduction in TER affinity compared with WT RBD.
Finally, we verified that telomerase harboring an R237A mutation is severely knocked down in telomerase activity. Interestingly, the assembly protein p65 can partially rescue telomerase activity in R237A TERT; however, p65 does not rescue the previously reported R237A template boundary defect. Taken together, our results demonstrate that the CP2 motif is essential for a functional interaction between TERT and TER and is a critical protein determinant of template definition.
EXPERIMENTAL PROCEDURES
PCR Mutagenesis
Plasmids containing the T. thermophila RBD fused to an N-terminal His tag were PCR-mutagenized using custom DNA primers. Linear plasmid amplicons were treated with T4 polynucledotide kinase (NEB) and T4 DNA ligase to generate circular plasmids, which were used to transform Escherichia coli 10β cells. Clones were sequenced to confirm the presence of the desired mutation. The Cys-lite RBD had the following mutations: C232A, C300S, C331A, C359A, C387A, C424A.
Protein Expression and Purification
TERT RBD was expressed in E. coli BL21 (DE3) cells induced at 21 °C with 0.8 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h. Cells were harvested by centrifugation and lysed by sonication in buffer containing 20 mm Tris, pH 8.0, 500 mm NaCl, 1 mm MgCl2,1 mm PMSF, 10% glycerol, and 5 mm β-mercaptoethanol. Cell lysate was centrifuged to remove precipitates and cell debris, and supernatant protein was purified by nickel affinity chromatography. Purified protein was eluted into a buffer containing 20 mm Tris, pH 8.0, 200 mm NaCl, 1 mm MgCl2, 500 mm imidazole, and 5 mm β-mercaptoethanol and flash frozen with liquid nitrogen for later use.
For quantitative EMSAs, proteins underwent additional rounds of purification. Following nickel affinity chromatography, the eluate was diluted to a buffer containing 20 mm Tris, pH 7.0, 50 mm NaCl, 1 mm MgCl2, 10% glycerol, and 5 mm β-mercaptoethanol and purified on an ion exchange source-S column (GE Healthcare) using a gradient from 50 mm to 1 m NaCl. The protein eluted at approximately 300 mm NaCl and was concentrated in a centrifugal concentrator (Millipore) with a 30-kDa molecular mass cut-off. Protein was flash frozen with liquid nitrogen and later purified on a Sephadex-200 (GE Healthcare) sizing column in 20 mm Tris, pH 8.0, 200 mm NaCl, 1 mm MgCl2, 10% glycerol, and 5 mm β-mercaptoethanol. Protein was concentrated off of the sizing column and flash frozen for later use. All protein constructs eluted as a single monomeric peak off of the sizing column, consistent with an ∼40-kDa protein. The percentage activity of WT and R237A protein constructs were determined using stoichiometric gel shifts. 2.5 μm cold TER was incubated with 4 nm end-labeled 32P-labeled TER and 2–16 μm RBD constructs in 20 mm Tris, pH 8.0, 100 mm NaCl, 1 mm MgCl2, 10% glycerol, and 5 mm β-mercaptoethanol. WT and R237A RBD demonstrated approximate percentage binding activities of ∼30 and ∼27%, respectively (data not shown).
Fe-BABE Labeling
Protein constructs containing only a single cysteine at the desired labeling site were dialyzed into a buffer lacking reducing agent (20 mm Tris, pH 8.0, 200 mm NaCl, 1 mm MgCl2, 10% glycerol) overnight, then switched to fresh dialysis buffer and dialyzed for an additional 2 h. The concentration of Tris in the buffer was then raised to 80 mm, and the dialyzed protein was incubated with a 4-fold molar excess of Fe-BABE for 3.5 h at room temperature in the dark. Next, the protein was dialyzed overnight against fresh dialysis buffer to remove excess Fe-BABE, and again for an additional 2 h. Fe-BABE-labeled RBD was quantified by Bradford assay and flash frozen in liquid nitrogen for later use. Mock-labeled Cys-lite RBD was treated identically to labeled protein; however, the protein construct lacked any cysteines to interact with the Fe-BABE moiety.
Hydroxyl Radical Probing
Protein constructs were incubated with 125 ng of in vitro transcribed TER end-labeled with 32P (PerkinElmer Life Sciences). Binding was performed in a buffer containing 20 mm Tris, pH 8.0, 100 mm NaCl, and 1 mm MgCl2, 0.1 mg/ml tRNA, 80 units of RNasin, and 450 nm p65 for 10 min at room temperature in a final volume of 50 μl. 1 μl of 250 mm sodium ascorbate and 1 μl of 1.25% H2O2 were added to the side of each tube and mixed instantaneously by a centrifuge pulse. Reactions were incubated for 10 min on ice and then quenched with 10 μl of 20 mm thiourea, and a radiolabeled DNA recovery control was added. The reactions were then phenol:chloroform-extracted and ethanol-precipitated. The RNA was run on a 7% sequencing polyacrylamide gel containing 8 m urea, and the gel was dried and imaged using a phosphorimaging screen (GE Healthcare) and a typhoon scanner (GE Healthcare). The T1 ladder was generated using full-length TER and RNase T1 (Ambion).
Hydroxyl Radical Probing Quantification
Hydroxyl radical probing gels were quantified with SAFA (18) as described previously (19). The quantified band intensities were compared between 1000 nm Fe-BABE-labeled protein and 1000 nm mock-labeled protein by dividing the intensity of the band in the labeled protein lane over the intensity of the band in the mock-labeled protein lane.
EMSAs
EMSAs were performed as described previously (19). For the quantitative EMSAs in Fig. 4, 0.4 nm end-labeled RNA was used instead of body-labeled RNA. Band intensities were quantified using Imagequant, and data were plotted and fit in Origin to determine Kd values. Percentage bound complexes were plotted in Origin, and Kd values were determined by fitting to the binding equation F = ((Fmax)(cn))/((Kd)n + (cn)), where F represents the fraction bound, c represents the concentration, Kd is the dissociation constant (the concentration at which 50% of the RNA is bound), Fmax represents the maximal value of F, and n represents the Hill coefficient.
FIGURE 4.
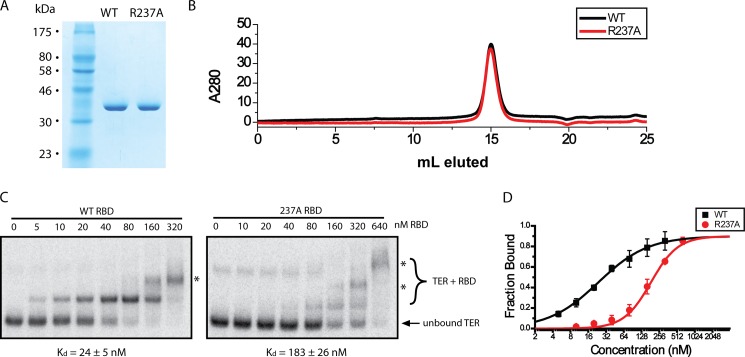
Quantitative EMSAs on highly purified protein demonstrate the CP2 motif is critical for RNA binding. A, SDS-PAGE gel of equal quantities of WT and R237A RBD protein. B, analytical sizing chromatography demonstrating that both proteins elute identically, consistent with a monomeric protein of ∼40 kDa. C, EMSAs for WT and R237A RBD. The dissociation constant (Kd) was determined for all protein variants as well as the S.D. (error bars) from triplicate measurements and is indicated below the gel as Kd ± S.D. D, Kd quantification for WT and R237A RBD. The Kd was determined by measuring the fraction bound at each concentration and fitting the data to the Hill equation. The Hill coefficients obtained were n = 1.0 and n = 1.9 for WT and R237A constructs, respectively. The higher Hill coefficient for R237A protein is likely the result of the increased contribution of higher molecular mass species (asterisks) skewing the fitting.
Telomerase Reconstitution
Prebinding reactions were generated with 62.5 ng of TER with or without 5 pmol of p65 in 20 mm Tris, pH 8.0, 100 mm NaCl, 1 mm MgCl2, and 1 mm dithiothreitol in a final volume of 2.5 μl. Prebinding reactions were incubated for 10 min at room temperature, and telomerase was reconstituted in a 50-μl scale rabbit reticulocyte lysate reaction as described previously (20).
Telomerase Activity Assays
Telomerase activity assays were performed as described previously (21). Some reactions were supplemented with 100 μm dATP as indicated.
RESULTS
Tethered Hydroxyl Radical Probing Demonstrates That the CP2 Motif Is Proximal to the TBE
We initially characterized the structural basis of RBD-TER interactions using site-directed hydroxyl radical probing. In this technique, a single reactive cysteine in the protein of interest is labeled with an Fe(II)-EDTA moiety. The functionalized protein is incubated with RNA in a buffer containing ascorbate and H2O2, generating hydroxyl radicals by the Fenton reaction (22). These hydroxyl radicals cleave any RNA nearby, but can only travel ∼20 Å before quenching. Sites of RNA cleavage can be identified by PAGE, giving a readout of regions of RNA-protein interaction. To facilitate site-specific Fe(II)-EDTA tethering, six endogenous cysteines were mutated out of a plasmid coding for the TERT RBD, generating a Cys-lite RBD construct (Fig. 1A). Single cysteines were then introduced back into the protein, generating a series of constructs containing only a single cysteine at specific regions of interest in the TERT RBD. The proteins were expressed in E. coli, affinity-purified, and labeled with Fe-BABE. Fe-BABE-labeled proteins were incubated with 32P-labeled TER (Fig. 1B), and ascorbate and hydrogen peroxide were added to the RNA-protein complexes to initiate the formation of hydroxyl radicals.
FIGURE 1.

Hydroxyl radical probing of RBD-TER complexes. A, primary structure map of T. thermophila TERT, indicating the position of major domains and conserved motifs. For hydroxyl radical probing studies, an RBD construct was expressed with all endogenous cysteines mutated to alanine or serine, and a single cysteine reintroduced at residue 232 in the CP2 motif. B, secondary structure of T. thermophila TER, indicating the positions of stems I, II, III, and IV. The template RNA is highlighted in orange and the TBE is highlighted in blue. Box indicates region of interest highlighted in D. C, site-directed hydroxyl radical probing results for Cys232-Cys-lite RBD. Lane 1, TER alone; lane 2, RNase T1 digestion ladder of TER indentifying position of guanine residues; lanes 3–7, hydroxyl radical probing performed in the presence of 62.5, 125, 250, 500, and 1000 nm Cys232-Fe-BABE Cys-lite RBD, respectively; lane 8, 1000 nm Cys-lite RBD mock-labeled with Fe-BABE and hydroxyl radical probing performed; lane 9, 1000 nm unlabeled Cys232 TERT RBD hydroxyl radical probing; lane 10, 1000 nm Cys232-Fe-BABE-labeled Cys-lite RBD incubated with RNA without adding ascorbate and hydrogen peroxide. The chromatogram on the right demonstrates the intensity of cleavage products, comparing lane 7 (black) and lane 8 (red). D, hydroxyl radical probing performed using 1000 nm Cys232-Fe-BABE Cys-lite RBD and 1000 nm mock-labeled Cys-lite RBD. The experiment was repeated in triplicate and the -fold increase in cleavage was calculated at each residue using the gel quantifying program SAFA (18). Values represent the mean ± S.D. (error bars), with values above 1 indicating increased cleavage due to Fe-BABE-induced hydroxyl radical probing at residue 232 and values below 1 indicating decreased cleavage. Most residues showed no change; however, there was a significant increase in cleavage at the base of stem II. The white horizontal dashed line indicates no change in cleavage compared with the mock-labeled Cys-lite RBD construct. The inset represents the mean values in D plotted against the secondary structure, with the size of the shapes scaled to represent the degree of cleavage or protection.
The majority of amino acid labeling sites showed no significant difference between Fe-BABE-labeled protein and unlabeled protein (Table 1). However, RBD labeled at residue Cys232 showed a considerable increase in cleavage in TER nucleotides 17–20 and 37–41 (Fig. 1C). Titrating increasing concentrations of Cys232-Fe-BABE RBD resulted in a corresponding increase in the intensity of the cleavage products (Fig. 1C, lanes 3–7), whereas incubation of the RNA with mock-labeled Cys-lite RBD, unlabeled Cys232 RBD, or Cys232-Fe-BABE RBD in the absence of hydrogen peroxide and ascorbate resulted in no cleavage at this site (Fig. 1C, lanes 8–10, respectively). A chromatogram of the lane intensity profiles of a lane with Cys232-FeBABE (lane 7) and a mock-labeled control (lane 8) clearly demonstrates the extent of the increase in cleavage (Fig. 1C, right).
TABLE 1.
Most protein labeling sites show no site-specific hydroxyl radical cleavage
The table displays the domain location, the motif, and the amino acid number of cysteine labeling sites used in site-directed hydroxyl radical probing experiments. Cleavage products of site-directed hydroxyl radical probing experiments were compared with those obtained from unlabeled protein and scored based on the observed increase in the intensity of cleavage products (− indicates no observed increase in intensity, ++ indicates moderate to strong cleavage products). Labeling sites in the N-terminal (TEN) domain were obtained with a construct containing residues 1–516 of TERT. We note that a negative probing result does not necessarily preclude the possibility of RNA interactions at this site. A specific labeling site may suffer from poor accessibility to Fe-BABE labeling, or the label itself could prevent the RNA interaction it was intended to measure.
| Location | Labeling site amino acid number | Strength of hydroxyl radical cleavage |
|---|---|---|
| TEN | 61 | − |
| TEN | 77 | − |
| TEN | 129 | − |
| TEN T2 motif | 134 | − |
| TEN T2 motif | 173 | − |
| TEN | 187 | − |
| RBD CP2 motif | 232 | ++ |
| RBD | 260 | − |
| RBD | 283 | − |
| RBD CP motif | 331 | − |
| RBD | 342 | − |
| RBD | 443 | − |
| RBD T motif | 486 | − |
| RBD | 500 | − |
To demonstrate the reproducibility of the result, the experiment was repeated three times, and the intensities of the cleavage products were quantified using the gel quantification program SAFA (18). The intensity of the cleavage product was measured at each individual residue and compared between RNA incubated with 1000 nm Cys232-FeBABE Cys-lite RBD and RNA incubated with 1000 nm mock-labeled Cys-lite RBD (Fig. 1D). The experiment demonstrates a highly reproducible cleavage pattern, with the most hydroxyl radical-induced cleavage observed at the base of stem II, most notably at residue 17. Plotting the mean cleavage intensity against the secondary structure of the RNA demonstrates the extent to which the pattern of cleavage mirrors the RNA secondary structure, peaking at the base of stem II and diminishing along the base-paired residues (Fig. 1D, inset).
The observed sites of cleavage at the base of TER stem II are part of a previously described conserved RNA motif known as the template boundary element. Mutations to the RNA in this region show errors in template definition, allowing the nontemplate residue U42 to be aberrantly reverse transcribed by TERT in the presence of dATP (8, 12). Furthermore, this region was shown to be required for high affinity TERT interaction, leading to the model that the TBE is bound tightly by the RBD and that this interaction prevents the TERT reverse transcription domain from reverse transcribing nontemplate TER nucleotides (8, 12).
The site of protein labeling, Cys232, is part of an evolutionarily conserved motif, known as the CP2 motif. The CP2 motif is found only in ciliate TERT, and previous mutational studies demonstrated that mutations to many of the residues in the CP2 motif have a significant defect in telomerase activity (10). Interestingly, one CP2 mutant showed the same defect in template definition as observed with a TBE mutant, a result that is consistent with the observed physical proximity of these two domains in our hydroxyl radical probing experiments.
EMSAs Demonstrate That the CP2 Motif Contributes to TER Binding
Although our hydroxyl radical probing results indicate that the CP2 motif is physically proximal to the TBE in the RNA-protein complex, they do not directly demonstrate that CP2 plays an essential role in RNA binding. To explore further the role of the CP2 motif in mediating RNA-protein interactions, we first expressed an RBD construct that lacks the CP2 motif entirely (ΔCP2) (Fig. 2A) and compared its RNA binding activity with the full-length RBD protein by EMSA. The ΔCP2 RBD construct was readily expressed and purified (Fig. 2B) but demonstrated considerably diminished RNA binding activity compared with WT protein (Fig. 2C). The high ΔCP2 RBD protein concentrations required to bind TER in our EMSA experiments, together with the propensity to form aggregates at protein concentrations above 320 nm, precluded our ability to determine a dissociation constant accurately. However, these results strongly suggest that the evolutionarily conserved CP2 motif is necessary for efficient and specific binding to TER.
FIGURE 2.
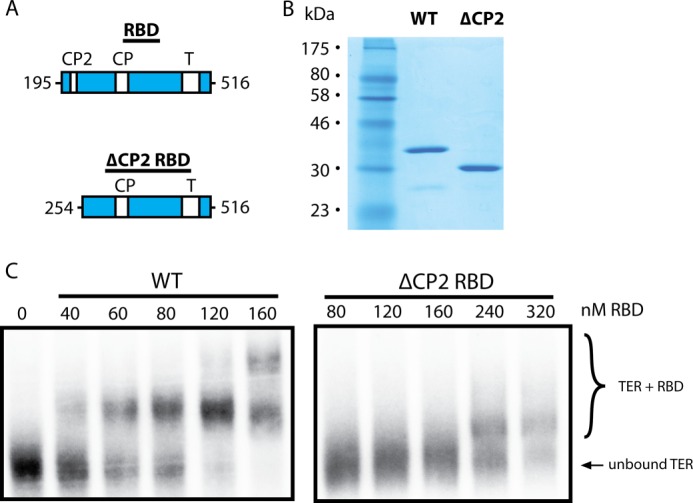
An RBD construct lacking the CP2 motif has a highly reduced affinity for TER. A, primary structure map of T. thermophila TERT wild-type and ΔCP2 RBD. B, SDS-PAGE gel of purified wild-type RBD and the ΔCP2 RBD mutant. C, EMSAs performed using full-length TER and increasing concentrations of either wild-type RBD (left) or an RBD construct lacking the CP2 motif (ΔCP2, right). The bottom band represents unbound TER and the top bands represent RBD-TER complexes.
The CP2 motif is a 12-amino acid sequence conserved across various ciliate species, containing many hydrophobic and positively charged residues potentially involved in RNA binding (Fig. 3A). To further dissect the contribution of the CP2 motif to TER binding we next performed an alanine scan through the CP2 region of TERT. To simplify the purification procedure for a semiquantitative initial screen, we only purified the proteins with one round of nickel affinity chromatography. Wild-type RBD and proteins encoding mutants Y231A, C232A, H234A, and R237A showed robust expression and purified to high levels of purity as determined by SDS-PAGE (Fig. 3B, left). Mutations R226A, I229A, and F230A showed a modest decrease in protein expression (Fig. 3B, right) and consequently co-purified with increased levels of contaminating protein, confounding accurate determination of protein concentrations by Bradford assays. Instead, the protein concentrations of these constructs were estimated by comparing Coomassie Blue staining of only the RBD band on SDS-PAGE gels (Fig. 3B, arrow).
FIGURE 3.

CP2 motif mutants demonstrate weaker TER affinity by EMSA. A, sequence conservation map of the CP2 motif, comparing TERT sequences of five ciliate species. Numbering is for the residue numbers in the T. thermophila TERT sequence. B, left panel, SDS-PAGE gel of WT RBD as well as equal quantities of point mutants Y231A, C232A, H234A, and R237A. B, right panel, SDS-PAGE gel of purified WT RBD as well as R226A, I229A, and F230A protein. These constructs showed greater contamination, therefore the protein concentration was estimated by comparing the Coomassie Blue intensity at the RBD band (arrow) against the known WT RBD concentration. C, EMSAs for WT RBD (as shown in Fig. 2C) as well as all mutagenesis constructs. The bottom band represents unbound TER, and the top bands represent RBD-TER complexes.
This initial screen with partially purified protein indicated an approximate dissociation constant, or the concentration of protein where 50% of the RNA is bound, for the WT protein of ∼60 nm (Fig. 3C). The CP2 point mutations F230A and Y231A appeared to have no effect on RNA binding, as gels for these constructs were virtually identical to the WT protein. Mutants I229A, C232A, and H234A appeared to have a modest defect in RNA binding, as evidenced by the slightly increased concentrations of protein required to bind the RNA (Fig. 3C). However, the largest and most obvious defect was observed with the mutants of the flanking arginines (R226A and R237A). In addition to the reduced affinity, the higher molecular mass complexes observed with the R237A mutant appeared as a diffuse smear on the gel, likely due to partial RBD binding or weakened complex formation, indicative of perhaps a further defect (Fig. 3C).
To further study the contribution of the CP2 motif to RNA binding, we followed our initial screen with quantitative EMSAs using fully purified protein constructs. For these measurements we focused on a comparison of WT RBD with one point mutant that showed a significant defect in our screen (R237A). These protein constructs were purified using nickel affinity chromatography, ion exchange chromatography, and sizing chromatography before use in EMSAs.
SDS-PAGE indicated that both WT and R237A proteins were purified to homogeneity (Fig. 4A). In addition, analytical sizing chromatography demonstrated that both proteins eluted at the same elution volume, consistent with a monomeric protein of ∼40 kDa (Fig. 4B), demonstrating that the mutation did not predispose the R237A mutant to aggregation. The quantitative EMSAs on these protein constructs confirmed the results of the initial screen. Under the conditions of our assay, the highly purified WT RBD, demonstrated a Kd of 24 ± 5 nm, a value that is slightly better than observed in our initial EMSA screens. The binding defect observed for the R237A mutant persisted, exhibiting a considerable decrease to 183 ± 26 nm compared with WT RBD (Fig. 4, C and D). The effect of the mutation is not due to a general folding defect, as both proteins showed similar specific activities in stoichiometric binding assays (see “Experimental Procedures”).
Under higher protein concentrations, EMSAs tended to reveal higher molecular mass complexes, likely corresponding to multiple RBD constructs binding to a single RNA (Fig. 4C, asterisks). A recent electron microscopy structure of Tetrahymena telomerase confirms that T. thermophila TERT and TER bind in a 1:1 ratio (23). Therefore, the higher order RBD-TER complexes we observe are likely artifacts of using purified protein constructs at high relative protein concentrations and do not reflect a physiologically relevant complex. These higher molecular mass complexes were nevertheless included in the quantification as it is likely that they represent naturally bound complexes augmented with an additional protein interaction. Although this may subtly shift the equilibrium between bound and unbound forms, this analysis is sufficient to make a meaningful comparison between the affinities of the WT and R237A protein constructs.
Previous work has suggested multiple sites of interaction between TERT and TER (12, 24). This is likely a large source of the residual binding activity in the ΔCP2 mutant (Fig. 2). It is likely that different parts of the RBD maintain separate binding sites on TER, increasing the overall affinity through an avidity effect (19, 24–26). Our results are consistent with the CP2 motif constituting one of these points of RNA interaction and specifically implicate Arg226 and Arg237 as being critical residues in mediating TER binding.
Mutations to the CP2 Motif Reduce Telomerase Activity and Are Partially Rescued by the Assembly Protein p65
The R237A mutation that showed the most dramatic defect in RBD affinity has been previously identified as a mutation that causes a template definition defect in full-length telomerase (8, 10). A previous study determined that the T. thermophila assembly protein p65 can rescue deleterious TERT mutations, presumably by suppressing defects that arise from weakened TERT-TER interactions (27). We set out to test the effect of p65 on R237A mutant telomerase activity. Full-length TERT was reconstituted with TER in the absence or presence of purified p65 in rabbit reticulocyte lysate, and the catalytic activity of the telomerase complexes was assessed using a direct telomere DNA primer extension assay. As expected, wild-type telomerase shows robust extension activity in both the presence and absence of p65 (Fig. 5). However, R237A telomerase displayed a dramatic decrease in telomerase activity, with virtually no telomerase activity observed in the absence of p65 (Fig. 5). We note that a previous study of the R237A TERT in the absence of p65 reported detectable telomerase activity, albeit at a significantly reduced level compared with wild-type TERT (8). We expect the inability of our present experiments to detect activity with the R237A mutant in the absence of p65 likely reflects small differences in the protocols used to reconstitute and purify the telomerase complex. Nevertheless, our results are qualitatively consistent with the previous report in that the R237A mutation results in a marked reduction in the efficiency of telomerase reconstitution.
FIGURE 5.
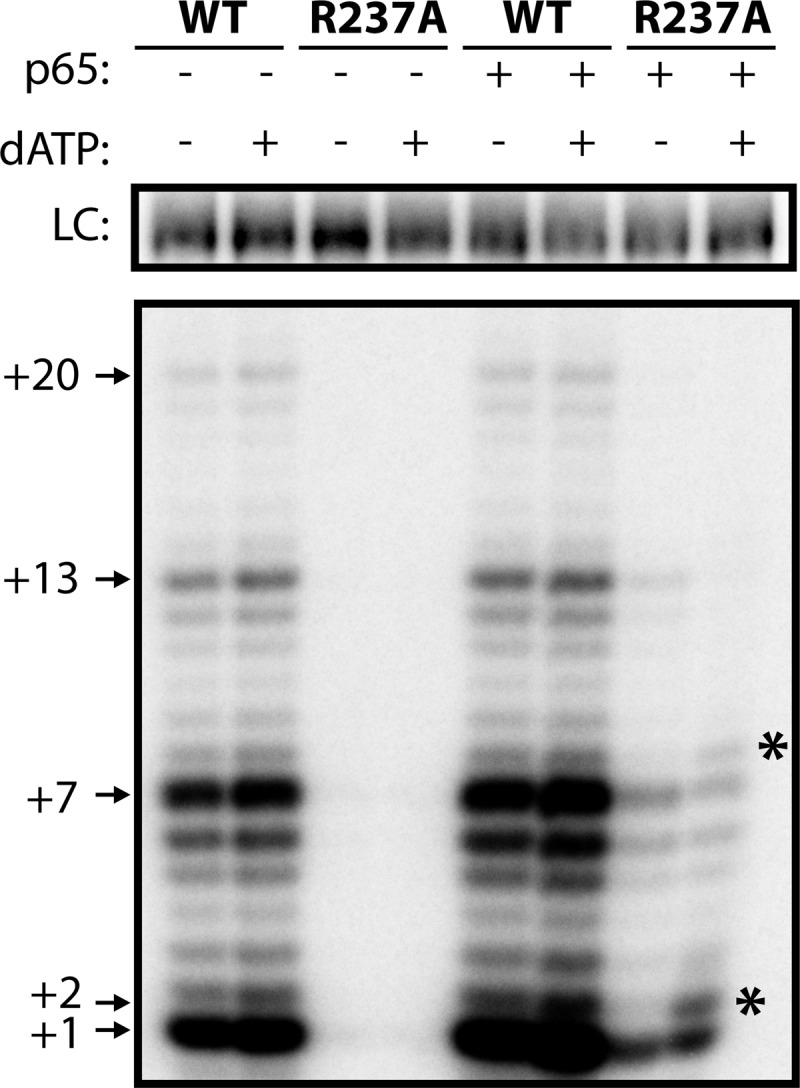
A CP2 mutant demonstrates telomerase activity defects. Telomerase activity assays performed from rabbit reticulocyte lysate-reconstituted telomerase, using either WT TERT or TERT harboring a CP2 mutation (R237A). Assays were performed in the presence or absence of the assembly protein p65 and in the presence or absence of dATP as indicated. Numbers (+1, +2, +7, etc.) indicate the number of nucleotides incorporated by telomerase at the indicated band. R237A TERT displayed a template boundary defect as indicated by an increase in the +2 product upon addition of dATP (asterisks). LC, loading control.
The presence of p65 improved R237A telomerase activity, although there was still considerably less extension than wild-type telomerase. Previous studies have shown that the R237A mutation inhibits TERT-TER assembly (8). Because p65 is an assembly co-factor, we conclude that p65 likely rescues telomerase activity in the CP2 mutant by stabilizing the interaction between R237A TERT and TER.
We also tested R237A telomerase for template boundary defects. In the presence of a template boundary defect, residue U42 of TER aberrantly enters the TERT active site. This results in dATP incorporation into the telomeric DNA primer, which is observed as an additional band one nucleotide above the canonical repeat addition band. In our assay, the repeat addition band is at the +1 site; therefore, template boundary defects can be observed by an increase in intensity of the +2 band when telomerase activity reactions are supplemented with dATP. Wild-type telomerase showed no increase at the +2 position in the presence of dATP, indicative of robust wild-type template definition (Fig. 5). Due to the undetectable level of catalytic activity in the absence of p65, R237A mutants could not be assessed for template boundary defects under these conditions. However, in the presence of p65, there was a considerable increase in template read-through when dATP was present (Fig. 5, asterisks). Taken together, our results indicate that the CP2 motif is important for TERT-TER assembly and telomerase activity and that the assembly protein p65 can partially rescue telomerase activity in a CP2 mutant. Furthermore, the CP2 motif is essential for proper template definition as reported previously (8, 10), and template definition defects persist in a CP2 mutant even in the presence of p65.
DISCUSSION
The CP2 Motif Is Proximal to the TBE and Contributes to Template Definition
Our site-directed hydroxyl radical probing results reveal that the CP2 motif is adjacent to the TBE in the TERT-TER complex, and gel shift assays verify that the CP2 motif is essential for proper RNA binding. Furthermore, the residues most strongly implicated in CP2-TER interaction (Arg226 and Arg237) are basic residues that may contact the RNA backbone through an electrostatic interaction. Finally, telomerase activity assays on a CP2 mutant confirm that the motif is critical for telomerase activity, showing both a severe assembly defect and a template boundary defect. Taken together, our experiments favor a model in which the CP2 motif binds the TBE to establish the template boundary. Nevertheless, we note that it remains a formal possibility that the CP2 motif is proximal to the TBE and influences RNA binding through an indirect interaction mediated by other amino acids.
A Model for TBE Interaction with the TERT RBD
Comparison of x-ray crystal structures of the T. castaneum TERT in complex with its DNA substrate and the T. thermophila RBD show significant structural homology (14, 15, 28). Alignment of the two structures reveals the likely position of the T. thermophila RBD with respect to the reverse transcriptase domains and the RNA template (Fig. 6A). In this structure, the 5′ end of the template is positioned adjacent to the T motif. Because the TBE begins three nucleotides upstream of this position, this puts a constraint on the position of the TBE within the structure. Based on this alignment, the CP motif is optimally positioned to interact with the TBE in an electropositive groove adjacent to the template (14). The evolutionary conservation of the CP motif, previous biochemical experiments (13), and its position within the RBD structure suggest that the CP motif plays some part in binding the TBE. We therefore propose a structural model based on the solved crystal structures of the T. thermophila RBD, the T. castaneum full-length TERT, and NMR structures of stem II of T. thermophila TER which places the base of stem II adjacent to the CP motif (14, 15, 29) (Fig. 6B).
FIGURE 6.

Model of TERT-TER interactions. A, structural alignment of T. castaneum FL TERT (gray, PDB ID 3KYL) and T. thermophila RBD (purple, PDB ID 2R4G). The positions of the conserved T and CP motifs are highlighted in red. The position of the 5′ end of the template (TER residue 43) near the T motif places a constraint on the position of the TBE (residues 15–40), likely placing the TBE near the CP motif in the RBD structure. B, model of telomerase structure. The reverse transcriptase domain and C-terminal extension motifs from the T. castaneum structure are modeled in gray, and the T. thermophila RBD is in purple as in A. Based on the structural alignment in A, the CP motif likely binds the base of stem II RNA, placing stem II (PDB ID 2FRL) within the existing structural model. Our results definitively implicate the CP2 motif in stem II binding, suggesting that the CP2 motif extends toward stem II to cooperatively bind stem II with the CP motif.
At present, there is no structural information on the CP2 motif. Nevertheless, we can use our hydroxyl radical probing results to place the CP2 motif adjacent to the TBE in our structural model (Fig. 6B). In this model, the CP and CP2 domains co-operate to bind the base of stem II and maintain the template boundary. This conclusion is strongly supported by our biochemical data as well as telomerase activity assays which previously demonstrated that mutants to both the CP and CP2 domains affect template definition (8).
Comparison of the Tetrahymena CP2 Motif with Vertebrate TERT
The CP2 motif is an area of conservation found only in ciliates. The lack of conservation of the CP2 motif across species is not entirely unexpected, as it is known that there is also a divergence in the RNA sequences of the TBE (7). This raises the possibility that this region of the protein may co-evolve with TER TBEs, accounting for the divergence of this region of the protein across phyla. Interestingly, in an analogous position in vertebrate TERT there is another region of conservation, found only in vertebrates, known as the vertebrate-specific region (30). Our results suggest that the vertebrate-specific region may function in vertebrates in a manner analogous to the CP2 motif in ciliates, promoting TBE interactions and establishing the template boundary.
Acknowledgment
We thank Prof. Kathleen Collins for the RBD plasmid used in this study.
This work was supported, in whole or in part, by National Institutes of Health Grant GM095850 (to M. D. S.) and National Institutes of Health Training Grants T32 GM8646 (to B. M. A.) and T34 GM007910-31 (to A. G.).
- TERT
- telomerase reverse transcriptase
- Fe-BABE
- Fe(III)-1-(p-bromoacetamidobenzyl)-EDTA
- PDB
- Protein Data Bank
- RBD
- RNA binding domain
- TBE
- template boundary element
- TER
- telomerase RNA.
REFERENCES
- 1. Palm W., de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 2. Vulliamy T. J., Dokal I. (2008) Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie 90, 122–130 [DOI] [PubMed] [Google Scholar]
- 3. Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M., Wright W. E., Weinrich S. L., Shay J. W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 4. Greider C. W., Blackburn E. H. (1989) A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337, 331–337 [DOI] [PubMed] [Google Scholar]
- 5. Hengesbach M., Akiyama B. M., Stone M. D. (2011) Single-molecule analysis of telomerase structure and function. Curr. Opin. Chem. Biol. 15, 845–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Theimer C. A., Feigon J. (2006) Structure and function of telomerase RNA. Curr. Opin. Struct. Biol. 16, 307–318 [DOI] [PubMed] [Google Scholar]
- 7. Lin J., Ly H., Hussain A., Abraham M., Pearl S., Tzfati Y., Parslow T. G., Blackburn E. H. (2004) A universal telomerase RNA core structure includes structured motifs required for binding the telomerase reverse transcriptase protein. Proc. Natl. Acad. Sci. U.S.A. 101, 14713–14718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lai C. K., Miller M. C., Collins K. (2002) Template boundary definition in Tetrahymena telomerase. Genes Dev. 16, 415–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai C. K., Mitchell J. R., Collins K. (2001) RNA binding domain of telomerase reverse transcriptase. Mol. Cell. Biol. 21, 990–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller M. C., Liu J. K., Collins K. (2000) Template definition by Tetrahymena telomerase reverse transcriptase. EMBO J. 19, 4412–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bosoy D., Peng Y., Mian I. S., Lue N. F. (2003) Conserved N-terminal motifs of telomerase reverse transcriptase required for ribonucleoprotein assembly in vivo. J. Biol. Chem. 278, 3882–3890 [DOI] [PubMed] [Google Scholar]
- 12. O'Connor C. M., Lai C. K., Collins K. (2005) Two purified domains of telomerase reverse transcriptase reconstitute sequence-specific interactions with RNA. J. Biol. Chem. 280, 17533–17539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bryan T. M., Goodrich K. J., Cech T. R. (2000) Telomerase RNA bound by protein motifs specific to telomerase reverse transcriptase. Mol. Cell 6, 493–499 [DOI] [PubMed] [Google Scholar]
- 14. Rouda S., Skordalakes E. (2007) Structure of the RNA-binding domain of telomerase: implications for RNA recognition and binding. Structure 15, 1403–1412 [DOI] [PubMed] [Google Scholar]
- 15. Mitchell M., Gillis A., Futahashi M., Fujiwara H., Skordalakes E. (2010) Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat. Struct. Mol. Biol. 17, 513–518 [DOI] [PubMed] [Google Scholar]
- 16. Heilek G. M., Marusak R., Meares C. F., Noller H. F. (1995) Directed hydroxyl radical probing of 16S rRNA using Fe(II) tethered to ribosomal protein S4. Proc. Natl. Acad. Sci. U.S.A. 92, 1113–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lancaster L., Kiel M. C., Kaji A., Noller H. F. (2002) Orientation of ribosome recycling factor in the ribosome from directed hydroxyl radical probing. Cell 111, 129–140 [DOI] [PubMed] [Google Scholar]
- 18. Das R., Laederach A., Pearlman S. M., Herschlag D., Altman R. B. (2005) SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11, 344–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akiyama B. M., Loper J., Najarro K., Stone M. D. (2012) The C-terminal domain of Tetrahymena thermophila telomerase holoenzyme protein p65 induces multiple structural changes in telomerase RNA. RNA 18, 653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berman A. J., Akiyama B. M., Stone M. D., Cech T. R. (2011) The RNA accordion model for template positioning by telomerase RNA during telomeric DNA synthesis. Nat. Struct. Mol. Biol. 18, 1371–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu J. Y., Stone M. D., Zhuang X. (2010) A single-molecule assay for telomerase structure-function analysis. Nucleic Acids Res. 38, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Culver G. M., Noller H. F. (2000) Directed hydroxyl radical probing of RNA from iron(II) tethered to proteins in ribonucleoprotein complexes. Methods Enzymol. 318, 461–475 [DOI] [PubMed] [Google Scholar]
- 23. Jiang J., Miracco E. J., Hong K., Eckert B., Chan H., Cash D. D., Min B., Zhou Z. H., Collins K., Feigon J. (2013) The architecture of Tetrahymena telomerase holoenzyme. Nature 496, 187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robart A. R., O'Connor C. M., Collins K. (2010) Ciliate telomerase RNA loop IV nucleotides promote hierarchical RNP assembly and holoenzyme stability. RNA 16, 563–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stone M. D., Mihalusova M., O'connor C. M., Prathapam R., Collins K., Zhuang X. (2007) Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature 446, 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sperger J. M., Cech T. R. (2001) A stem-loop of Tetrahymena telomerase RNA distant from the template potentiates RNA folding and telomerase activity. Biochemistry 40, 7005–7016 [DOI] [PubMed] [Google Scholar]
- 27. Berman A. J., Gooding A. R., Cech T. R. (2010) Tetrahymena telomerase protein p65 induces conformational changes throughout telomerase RNA (TER) and rescues telomerase reverse transcriptase and TER assembly mutants. Mol. Cell. Biol. 30, 4965–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gillis A. J., Schuller A. P., Skordalakes E. (2008) Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature 455, 633–637 [DOI] [PubMed] [Google Scholar]
- 29. Richards R. J., Theimer C. A., Finger L. D., Feigon J. (2006) Structure of the Tetrahymena thermophila telomerase RNA helix II template boundary element. Nucleic Acids Res. 34, 816–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moriarty T. J., Huard S., Dupuis S., Autexier C. (2002) Functional multimerization of human telomerase requires an RNA interaction domain in the N terminus of the catalytic subunit. Mol. Cell. Biol. 22, 1253–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]