

Background: How Fgfr1 mutations cause cleft palate is unclear.
Results: Deleting Fgfr1 in neural crest cells caused defects in both palate shelf epithelium and mesenchyme and led to cleft palate.
Conclusion: FGFR1 signaling in cranial neural crest (CNC) cells regulates palate shelf growth and fusion during palatogenesis.
Significance: The finding for the first time demonstrates how FGF signaling in CNC cells regulates palatogenesis.
Keywords: Craniofacial Development, Fibroblast Growth Factor (FGF), Fibroblast Growth Factor Receptor (FGFR), Transgenic Mice, Tyrosinase
Abstract
Cleft palate is a common congenital birth defect. The fibroblast growth factor (FGF) family has been shown to be important for palatogenesis, which elicits the regulatory functions by activating the FGF receptor tyrosine kinase. Mutations in Fgf or Fgfr are associated with cleft palate. To date, most mechanistic studies on FGF signaling in palate development have focused on FGFR2 in the epithelium. Although Fgfr1 is expressed in the cranial neural crest (CNC)-derived palate mesenchyme and Fgfr1 mutations are associated with palate defects, how FGFR1 in palate mesenchyme regulates palatogenesis is not well understood. Here, we reported that by using Wnt1Cre to delete Fgfr1 in neural crest cells led to cleft palate, cleft lip, and other severe craniofacial defects. Detailed analyses revealed that loss-of-function mutations in Fgfr1 did not abrogate patterning of CNC cells in palate shelves. However, it upset cell signaling in the frontofacial areas, delayed cell proliferation in both epithelial and mesenchymal compartments, prevented palate shelf elevation, and compromised palate shelf fusion. This is the first report revealing how FGF signaling in CNC cells regulates palatogenesis.
Introduction
Cleft palate is the most common congenital craniofacial defect in human, which is often associated with cleft lip. It affects about 0.1% newborns in America according to the Center for Diseases Control and Prevention. The palate is formed by fusion of the primary and secondary palates. It consists of two structural parts, the anterior bony hard palate and the posterior muscular soft palate. The primary palate is derived from the posterior protrusion of the medial nasal process, and the secondary palate develops from bilateral outgrowth of the maxillary process (1, 2). Palate development is a multistep process that involves bilateral palatal shelf growth beside the tongue, elevation of the palatal shelves that grow toward the midline, fusion of palatal shelves at the midline to form the medial edge epithelium (MEE),5 degeneration of the MEE, and disappearance of the midline epithelial seam (1, 3). The maxillary shelves are mainly composed of the cranial neural crest (CNC)-derived ecto-mesenchymal cells surrounded by a thin layer of pharyngeal ectoderm-derived epithelial cells (4–7). The CNC cells are a subset of neural crest cells (NCCs) that are pluripotent and are derived from the lateral ridges of the neural plate during early stages of embryogenesis. During craniofacial development, CNC cells migrate ventrolaterally and populate in the branchial arches, which then contribute extensively to the formation of head and neck mesenchymal structures, including the palate. Defects either in facial mesenchyme patterning and growth or in epithelium fusion result in cleft palate. Several reciprocal epithelial-mesenchymal signaling axes, including the fibroblast growth factor (FGF), bone morphogenetic protein (BMP), Sonic hedgehog (SHH), and Wnt signaling pathways, have been show to play important roles in the migration and proliferation of CNC cells (1, 8–10).
The FGF family consists of 18 tyrosine kinase receptor-mediated members that regulate a broad spectrum of cellular activities (11). FGF elicits its regulatory signals via binding and activating the FGF receptor (FGFR) tyrosine kinases encoded by four highly homologous genes. In the palate, FGF and FGFR isoforms are expressed in a spatiotemporally specific manner and constitute a directional regulatory axis between the stromal and epithelial compartments, regulating palate development (12). In palate shelves, Fgf10 expression is restricted to the mesenchyme and Fgfr2b expression to the overlying epithelium. Germ line ablation of Fgf10 or epithelium-specific deletion of Fgfr2b leads to cleft palate with impaired palatal shelf outgrowth (13, 14). Cell proliferation in both epithelium and mesenchyme compartments is reduced in the absence of either Fgf10 or Fgfr2b, suggesting that ablation of the FGF10-FGFR2 signaling axis causes loss of signals from the epithelium back to the underlying mesenchyme, which regulate mesenchymal proliferation. In addition, the FGF signaling intensity during palatogenesis is delicately balanced. Expression of a constitutively active FGFR2 mutant in the epithelium increases palatal shelf mesenchyme proliferation and delays elevation of the shelves, resulting in cleft palate and other craniofacial disorders (15, 16). Disruption of Sprouty 2 (Spry2), a negative feedback regulator of FGF signaling pathways, results in abnormal palate formation (17). In contrast, ablation of Fgfr1 in the epithelium with K14 promoter driven Cre does not cause major craniofacial defects (18). The major cell population in the palate shelf mesenchyme is derived from CNC cells, which expresses Fgfr1 (19). Although it has been reported that embryos with Fgfr1 ablation in NCCs have cleft palate (20), no detailed characterization has been done on how ablation of Fgfr1 in NCCs leads to cleft palate. As FGFR1 is important for patterning in the pharyngeal region (20), its mutations and haploinsufficiency in humans are associated with cleft palate (21–23).
To investigate how mesenchymal FGFR1 regulates craniofacial development, Fgfr1 alleles were tissue-specifically ablated in NCCs by crossing mice bearing floxed Fgfr1 (Fgfr1flox) and Wnt1-Cre (Wnt1Cre) transgenic alleles. Ablation of Fgfr1 in NCCs led to cleft palate, cleft lip, and other severe craniofacial defects. Detailed characterization revealed that ablation of Fgfr1 in NCCs did not abrogate CNC cell contribution to the palate shelf mesenchyme. However, it upset cell signaling in the medial nasal process and maxillary process areas, and it delayed cell proliferation in both the mesenchyme and epithelium of palatal shelves. The mutant palate shelves failed to elevate during palatogenesis. In addition, although it did not fully prevent the fusion process, it compromised the deterioration of the MEE. Together with the report that loss of the mesenchymal-epithelial FGF10-FGFR2IIIb signaling axis affects cell proliferation in both epithelium and mesenchyme (13), the results indicate that the reciprocal FGF signaling axis between the palate mesenchyme and epithelium is important for the growth and elevation of palate shelves. This is the first report on the mechanism by which mesenchymal FGFR1 signaling regulates palatogenesis.
MATERIALS AND METHODS
Animals and Isolation of Tissues
All animals were housed at the Program of Animal Resources, Institute of Biosciences and Technology, Texas A&M Health Science Center, and were handled in accordance with the principles and procedures in the Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Institutional Animal Care and Use Committee. Mice carrying the Wnt1Cre transgenic alleles (24), ROSA26lacZ (25) reporter allele, and Fgfr1flox allele (26) were maintained and genotyped as described previously.
Dissection and in Vitro Culture of Palate Shelves
Palatal shelves were dissected from E13.5 embryos. Two palatal shelves were placed on 8-μm-pore size transwell culture plates (BD Biosciences) with their MEE placed in close apposition without apparent distortion of their tissue shape. The paired palate shelves were cultured for 2 days at 37 °C in DMEM supplemented with 1% penicillin/streptomycin (27). Similar ex vivo cultures were carried with heads without the tongue and mandible from E13.5 embryos (28).
Histological and Immunohistochemical Analyses
Prenatal mouse heads were fixed in 4% paraformaldehyde solution for 2 h at 4 °C. The fixed tissues were serially dehydrated with ethanol, embedded in paraffin, and sectioned at 5-μm thickness according to standard procedures. Immunohistochemical analyses were performed on paraffin sections mounted on Superfrost/Plus slides (Fisher). Antigens were retrieved by boiling in citrate buffer (10 mm) for 20 min at 100 °C or as suggested by the manufacturers. All sections were incubated with primary antibodies diluted in PBS at 4 °C overnight. The mouse anti-p63 (1:200) and mouse anti-pan-cytokeratin (1:200) antibodies were purchased from Santa Cruz Biotechnology, and rabbit anti-E-cadherin (1:200), rabbit anti-pSmad1/5/8 (1:200), and rabbit anti-vimentin (1:200) antibodies were purchased from Cell Signaling Technology. Specifically bound antibodies were detected with FITC-conjugated secondary antibodies (Invitrogen) and visualized with a Zeiss LSM 510 confocal microscope. To-Pro3 was used for nuclear counterstaining.
For LacZ staining, the tissues were first lightly fixed with 0.2% glutaraldehyde for 30 min and then incubated overnight with 1 mg/ml X-Gal at room temperature. The tissues were post-fixed with 4% paraformaldehyde at room temperature for 1 h after a 10-min PBS wash. The tissues were then dehydrated, paraffin-embedded, and sectioned according to regular procedures.
Gene Expression Analysis
Total RNA was extracted with the RiboPure RNA isolation reagent (Ambion, TX). Reverse transcription was carried out with SuperScript III (Invitrogen) and random primers. Real time PCR was performed on Mx3000 (Stratagene), using the SYBR Green JumpStart Taq ReadyMix (Sigma) with pairs of primers specific for each transcript and following the manufacturer's protocol. The ratio between expression levels in the two samples was calculated by relative quantification, using β-actin as a reference transcript for normalization. The primer sequences are listed in Table 1.
TABLE 1.
Primer sequences for real time RT-PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Shh | 5′-ACATCCACTGTTCTGTGAAAGCA-3′ | 5′-TCTCGATCACGTAGAAGACCTTCTTG-3′ |
| Ptch | 5′-TCAACCCAGCCGACCCAGATT-3′ | 5′-CCCTGAAGTGTTCATACATTTGCTTG-3′ |
| Wnt1 | 5′-CATTTGCACTCTTGGCGCAT-3′ | 5′-AAGATCGTCAACCGAGGCTG-3′ |
| Notch1 | 5′-CCCACTGTGAACTGCCCTAT-3′ | 5′-CACCCATTGACACACACACA-3′ |
| Gli1 | 5′-GAAGGAATCCGTGTGCCATT-3′ | 5′-GGATCTGTGTAGCGCTTGGT-3′ |
| Gli2 | 5′-GGCACCAACCCTTCAGACTA-3′ | 5′-CTGAGCTGCTCCTGGAGTTG-3′ |
| Gli3 | 5′-GTCAGCCCTGCGGAATACTA-3′ | 5′-GGAACCACTTGCTGAAGAGC-3′ |
| BMP2 | 5′-AAGGAGGAGGCGAAGA-3′ | 5′-CTGAGTGCCTGCGGTACAGAT-3′ |
| BMP4 | 5′-AGGAGGAGGAGGAAGAGCAG-3′ | 5′-TGTGATGAGGTGTCCAGGAA-3′ |
| BMP7 | 5′-ACCTGGGCTTACAGCTCTCTG-3′ | 5′-CGGAAGCTGACGTACAGCTCATG-3′ |
| TGFβ | 5′-CTAATGGTGGACCGCAACAA-3′ | 5′-GTACAACTCCAGTGACGTCA-3′ |
| CK14 | 5′-CTTCCCAATTCTCCTCATCC-3′ | 5′-GGGCTCTTCCAGCAGTATCT-3′ |
| Jagged1 | 5′-GCACCCGCGACGAGTGTGAT-3′ | 5′-TCCCAGGCCTCCACCAGCAA-3′ |
| Wnt3a | 5′-CGATCTGGTGGTCCTTGGCTGT-3′ | 5′-AGCGGAGGCGATGGCATGGA-3′ |
| Msx1 | 5′-CTCTCGGCCATTTCTCAGTC-3′ | 5′-TACTGCTTCTGGCGGAACTT-3′ |
| Ptx1 | 5′-CTCAACGCTTGCCAGTACAA-3′ | 5′-GGGTCTGGAAAAAGCAAACA-3′ |
| Etv5 | 5′-GGGAGAGACAAAAACCACCA-3′ | 5′-ATGGGTGTGCAGTTTCTTCC-3′ |
For in situ hybridization, paraffin-embedded tissue sections were rehydrated followed by digestion with 20 μg/ml protease K for 7 min at room temperature. After prehybridization at 65 °C for 2 h, the hybridization was carried out by overnight incubation at 65 °C with 0.5 μg/ml digoxigenin-labeled RNA probes for the indicated genes. Nonspecifically bound probes were removed by washing four times with 0.1× digoxigenin washing buffer at 60 °C for 30 min. Specifically bound probes were later detected using alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche Applied Science).
The antisense riboprobe of FGFR1 was generated by in vitro transcription using T3 RNA polymerase with the XbaI-linearized pKS+ΔFGFR1 vector (generous gift from Dr. Juha Partanen) (26). The antisense riboprobe of TGFβ3 was generated by in vitro transcription using T3 RNA polymerase with the EcoRI-linearized pSK-TGFβ3 vector (kindly provided by Dr. Rulang Jiang) (29).
RESULTS
Ablation of Fgfr1 in NCCs Causes Cleft Palate
Mutations in Fgfr1 have been found to be associated with craniofacial malformation, including cleft palate in human. Fgfr1 is expressed in CNC-derived mesenchymal cells during palatogenesis (19). To investigate how FGFR1 signaling in CNC cells affected craniofacial development, the Fgfr1 alleles were tissue-specifically ablated in NCCs by crossing the mice bearing Fgfr1flox and Wnt1Cre alleles. Although they appeared to be morphologically normal at E10.5, Fgfr1 conditional knock-out (Fgfr1cKO) mutant embryos had a slightly reduced frontonasal prominence and maxillary processes (Fig. 1A). LacZ staining of the Fgfr1cKO and wild type embryos bearing the ROSA26lacZ reporter demonstrated that, compared with the Fgfr1 wild type control at this stage, the CNC cell participation in the frontonasal prominence was significantly reduced in mutant embryos (Fig. 1B), which was in line with the report that FGFR1 is required for CNC cell patterning (20). In contrast, LacZ staining of coronal sections of E10.5 embryos showed no difference in ROSA26lacZ reporter activation between wild type and mutant embryos (Fig. 1B, insets), indicating neural tube formation was not affected in mutant embryos.
FIGURE 1.
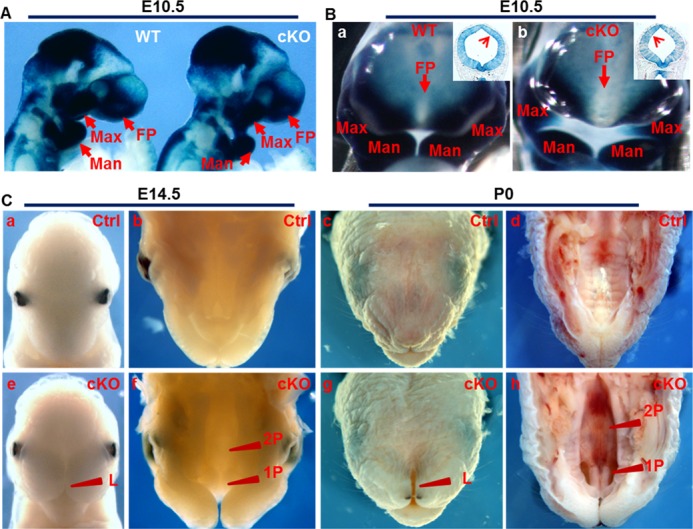
Ablation of Fgfr1 in NCCs cells leads to cleft palate. A and B, whole mount lacZ staining showed distribution of Wnt1Cre expressing NCCs in the head of embryonic (E) day 10.5 embryos bearing the ROSA26lacZ reporter and Wnt1Cre alleles. B, insets in coronal sections of E10.5 embryos bearing the ROSA26 reporter, Wnt1Cre, and the indicated Fgfr1 alleles were LacZ-stained and eosin-counterstained. C, mouse heads were collected at E14.5 or postnatal (P) day 0 for frontal facial morphology analyses. 1P, primary palate; 2P, secondary palate; L, lip; Man, mandibular process; Max, maxillary process; MT, mesencephalic tegmentum; FP, frontonasal prominence; Ctrl, control; cKO, Fgfr1 conditional knock-out; WT, Fgfr1 wild type.
The structures constituted by fused midline nasal prominence were missing in mutant embryos at the E14.5 and neonatal stage (Fig. 1C). Completely cleft primary and secondary palates as well as cleft lip were readily seen. Partially formed palate rugae were observed in mutant mice at the neonatal stage. The palatal shelves were widely separated in mutant mice, allowing direct view of the underlying presphenoid bone. In addition, the mutant embryos also exhibited other severe orofacial dysformation, including tooth bud defect and micrognathia. Embryos bearing one conditional null Fgfr1 allele, one or two Fgfr1flox alleles without the Cre allele, or one Cre allele were indistinguishable from wild type embryos, and hereafter were defined as controls.
Ablation of Fgfr1 in NCCs Does Not Disrupt Participation of CNC Cells in Palate Shelves
To characterize the palate defects in Fgfr1cKO embryos, coronal sections of E13.5, E14.5, and E15.5 embryos were H&E-stained for histological analyses. At the stage of E14.5, the anterior portion of the secondary palate in Fgfr1flox embryos was undergoing fusing process, and the MEE was formed, whereas the posterior portion had finished the elevation but was still undergoing expansion processes. The mutant palate shelves, however, were still growing downward at both anterior and posterior positions (Fig. 2, A and B). The palate shelves failed to elevate and finish the fusion processes even at E15.5 stages. Although mesoderm-derived mesenchymal cells also contribute to palate shelves as a minor cell population, the majority of palate shelf mesenchymal cells were derived from the CNC (30, 31). Lineage tracing with the ROSA26lacZ reporter allele revealed that similar to the Fgfr1flox palate shelves, the majority of mesenchymal cells in mutant palate shelves were derived from Wnt1Cre-expressing CNC cells (Fig. 3, A and B). However, there were a fraction of cells adjacent to the epithelial cells that were not X-Gal-labeled. The origin of these cells remains to be determined. In situ hybridization revealed that Fgfr1 expression in the palate shelf mesenchyme was diminished (Fig. 3C). In addition, all cells in the mutant palate shelf mesenchyme were vimentin-positive (Fig. 3D), indicating mesenchymal cells.
FIGURE 2.

Failure of palate shelf elevation in Fgfr1cKO embryos during palatogenesis. A, coronal sections of anterior and posterior portions of control or Fgfr1cKO embryos collected from the indicated stages were H&E-stained for tissue histological analyses. Unlike control embryos, Fgfr1cKO palate shelves failed to elevate and fuse. B, enlarged pictures of the boxed areas in A. Ctrl, control; MES, medline epithelial seam; Oc, oral cavity; Sp, secondary palate shelf; T, tongue.
FIGURE 3.

Ablation of Fgfr1 in NCC did not affect participation of CNC cells in the palate shelf mesenchyme. A, whole mount lacZ staining showing CNC cells in the palate shelves in E12.5 embryos. B, same tissue in A was paraffin-embedded and sectioned for demonstrating X-Gal-stained cells in the mesenchyme. C, in situ hybridization showing Fgfr1 expression in palate shelf mesenchymal cells was diminished in E12.5 Fgfr1cKO embryos. D, coronal sections of E14.5 embryos were immunostained with anti-vimentin antibody. To-Pro3 was used for nuclear counterstaining. Insets were two-channel images of the areas indicated by arrows, showing both the FITC and To-Pro3 signal to demonstrate that the epithelial cells were vimentin-negative.
The size of mutant palate shelves appeared to be smaller than the control at E13.5 (Fig. 2). To determine whether cell proliferation was a causal factor for small palate shelves, BrdU incorporation assays were employed to assess the proliferating cell numbers in palate shelves at the stages of E12.5 to E14.5 (Fig. 4). The results showed that cell proliferation in the epithelium of anterior palate shelves and the mesenchyme at the posterior shelves were reduced at E12.5–13.5, although no differences were found in the anterior mesenchyme and the posterior epithelium in palate shelves (Fig. 4, A and B). However, both the epithelium and mesenchyme of anterior and the mesenchyme of posterior Fgfr1cKO palate shelves had a higher proliferation than control at E14.5, although the posterior epithelium of Fgfr1cKO palate shelves had a similar proliferation activity as the control (Fig. 4C). Thus, it appeared that the cell proliferation in Fgfr1cKO palate shelves was delayed, rather than simply reduced. The data suggest that lack of FGFR1 signals in CNC-derived mesenchymal cells in palate shelves results in reduced epithelium proliferation at a non-cell autonomous manner.
FIGURE 4.

Delayed cellular proliferation in Fgfr1cKO palate shelves. BrdU incorporation assays demonstrate that Fgfr1cKO palate shelves had compromised proliferation at the E12.5–13.5 (A and B) stages and enhanced proliferation at E14.5 (C) stage. Bottom panels, numbers of labeled cells were scored from three independent samples and expressed as mean ± S.D. *, p < 0.05; dashed lines indicate the areas where the proliferative cells were counted; Epi, epithelium; Mes, mesenchyme.
Lacking FGFR1 Signals in CNC-derived Mesenchymal Cells Affects Degeneration of MEE in Palate Shelves
During the fusion process of palate shelves, cells in the MEE undergo degeneration and the midline epithelial seam disappears at the end. Fgfr1cKO embryos exhibited multiple craniofacial defects, including micrognathia and heightened tongue, which had been shown to provide a steric hindrance for palate shelf elevation without the fusion process (32–39). To determine whether lacking mesenchymal FGFR1 affected the fusion directly by compromising epithelial degeneration or indirectly by providing a steric hindrance, E13.5 embryonic heads without the tongue and mandibles were dissected for ex vivo cultures for 48 h. Although the palate shelves of both control and Fgfr1cKO embryos did not complete the fusion process, the gap between the two shelves was significantly reduced (Fig. 5A). However, when the two palate shelves were dissected and placed in a proximal apposition, the fusion took place in both control and mutant palate shelves in 2 days (Fig. 5B). H&E staining of the ex vivo cultured E13.5 palate shelves showed that both control and Fgfr1cKO palate shelves fused after being cultured for 2 days. However, the gap between the two mutant shelves was not fully filled by mesenchymal cells as the control. In addition, there was still a fraction of midline epithelial seam remained (Fig. 5C). Immunostaining further revealed that expression of epithelial characteristic cytokeratins in the mutant MEE was not eliminated as completely as in controls (Fig. 5D). The results indicate that although it did not abolish the process, ablation of Fgfr1 in CNC-derived mesenchymal cells compromised the deterioration of MEE cells during the fusion.
FIGURE 5.
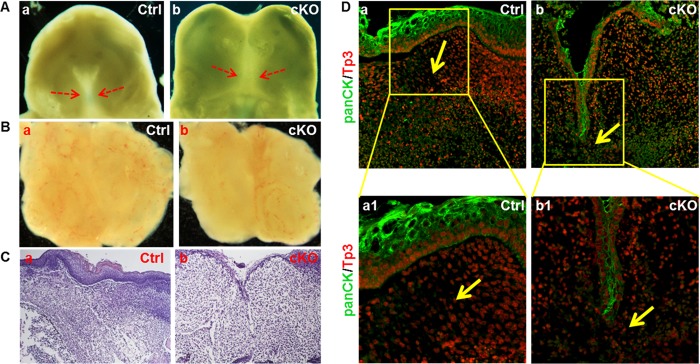
Ablation of Fgfr1 in NCCs impairs palate shelf fusion. A and B, heads without the tongue and mandible or palate shelves dissected from E13.5 embryos were cultured at 37 °C for 48 h showing fusion of the two palate shelves. C and D, palate tissues in B were sectioned and H&E-stained or immunostained with anti-pan-cytokeratin antibodies showing the loss of MEE in fused palate shelves.
p63 is expressed in palate epithelial cells and is often used as an indicator for palate epithelial cell integrity. Immunostaining showed that the MEEs in control palate shelves had a fuzzy E-cadherin staining, which was a sign of deteriorating epithelial cells. The MEE cells in mutant anterior palate shelves also were not intact even though they did not undergo elevation and fusion processes. However, the severity of degeneration was apparently compromised (Fig. 6A). Similarly, E-cadherin staining also showed that, compared with the control, Fgfr1cKO palate shelves had less defused E-cadherin staining in MEE cells at this stage (Fig. 6B). In addition, TUNEL analyses showed that although MEE cells in mutant palate shelves still had apoptotic cells, the numbers were significantly lower than those in control palate shelves (Fig. 6C). Consistently, expression of Tgfβ3 in MEE cells was reduced in Fgfr1cKO embryos (Fig. 6D), which plays a critical role in promoting fusion of the palatal shelves (40). Together, the results showed that these characteristic changes in MEE were apparently less obvious in E14.5 Fgfr1cKO embryos than those in the control. Therefore, although not essential, FGFR1 signaling in CNC-derived mesenchymal cells is involved in regulating degeneration of the MEE during palate shelf fusion through non-cell autonomous mechanisms. However, the molecular mechanism by which FGFR1 signaling in CNC-derived mesenchymal cells affects MEE cell degeneration and proliferation remains to be characterized.
FIGURE 6.
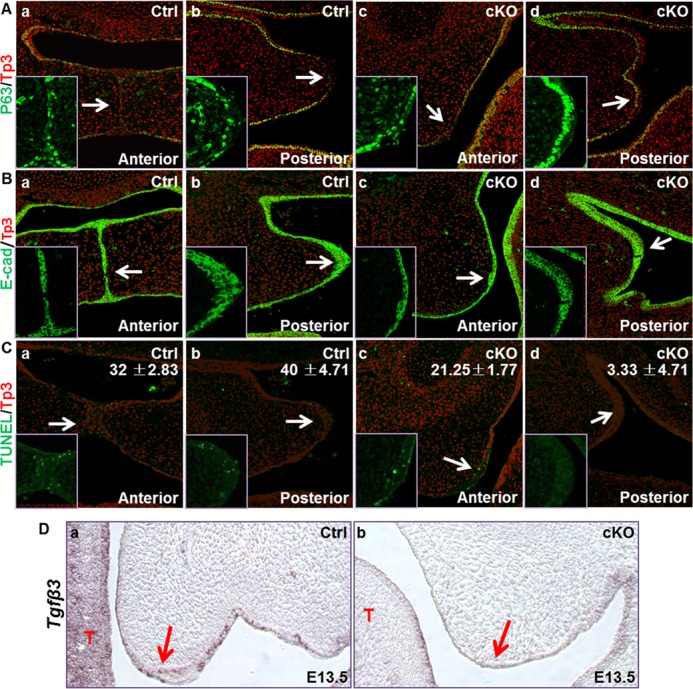
Compromised deterioration of the MEE cells in Fgfr1cKO palate shelves. A–C, tissue sections from E14.5 embryos were subjected to immunostaining with anti-p63, E-cadherin (E-Cad) antibody, or TUNEL analyses as indicated showing compromised deterioration of epithelial cells in control and Fgfr1cKO palate shelves. Numbers in C are average apoptotic epithelial cell numbers in three replicated samples. D, in situ hybridization showing Tgfβ3 expression in MEE cells in E13.5 embryos.
Ablation of Fgfr1 in NCCs Affects Cell Signaling in Frontofacial Tissues
To further characterize how ablation of Fgfr1 in CNC cells affected cell signaling during craniofacial development, real time RT-PCR analyses were carried out to assess expression of key regulatory molecules in craniofacial development at E10.5 embryos and E14.5 frontofacial tissues. The results clearly demonstrated that expression of Notch1 and Bmp4 was increased, and the expression of Wnt1 was reduced in the E10.5 Fgfr1cKO embryos (Fig. 7A). To better assess the FGFR1-regulated gene expression in frontofacial development, the frontofacial part of E14.5 embryos was dissected for real time RT-PCR analyses. The results confirmed that both Wnt and BMP signalings were changed by NCC-specific deletion of Fgfr1 in E14.5 frontofacial tissues (Fig. 7B). In addition, expression of Msx1 and Ptx1 was also reduced in mutant frontofacial tissues. Furthermore, immunostaining showed that phosphorylated Smad1/5/8-positive cells were significantly increased in Fgfr1cKO palate shelves, especially in the anterior part at E14.5 stage (Fig. 7C). This further demonstrated increased BMP signaling in Fgfr1cKO palate shelves.
FIGURE 7.

Ablation of Fgfr1 in NCCs affects cell signaling in the embryos. A and B, total RNAs were extracted from E10.5 embryos (A) or E14.5 frontal facial areas defined in panels a and b (B) for real time RT-PCR analyses of the indicated gene expression. Data are means ± S.D. of three independent analyses. C, immunostaining of coronal sections of E14.5 embryos with anti-phosphorylated Smad1/5/8 showing increased BMP signaling in Fgfr1cKO palate shelves.
DISCUSSION
The FGF signaling axis is critical for palate development through directional and reciprocal communications between the mesenchyme and epithelium of palate shelves. To date, the majority of studies has been focused on the FGF7/10-FGFR2 signaling axis that directionally mediates the mesenchyme to epithelium communication. How FGF signaling, especially that mediated by FGFR1, in the CNC cells regulates palate formation is not understood. Here, we report that tissue-specific ablation of Fgfr1 in NCCs with Wnt1Cre caused primary and secondary cleft palate, cleft lip, and other craniofacial detects. Detailed analyses revealed that ablation of Fgfr1 in NCCs did not abrogate patterning of CNC-derived mesenchymal cells in the palate shelves. However, it delayed cell proliferation in both the mesenchyme and epithelium, and impeded development of medial nasal processes and the lift of palate shelves during palatogenesis. In addition, although it did not fully prevent the fusion process of ex vivo cultured palate shelves once they were placed in a close contact position, the deterioration of the MEE in mutant palatal shelves was compromised. The detailed mechanism regarding how ablation of Fgfr1 in the mesenchyme affects cell proliferation in Fgfr1-intact epithelial cells remains to be determined, although quantitative RT-PCR analyses showed altered expression of Bmp, Wnt, and other signaling molecules in the frontofacial tissue of E14.5 Fgfr1 mutant embryos. In addition, ablation of Fgfr1 in NCCs affected BMP and Wnt signaling in E10.5 embryos. This further demonstrates the cross-talk between FGF and other signaling pathways during embryonic development.
Palate development is a multifactorial event and is regulated by multiple signaling pathways. The cross-talk of these intertwining pathways is important in regulating palatogenesis (1), and the FGF signaling pathway is no exception. Balance in FGF signaling appears to be important for palate formation. Constitutively activating mutations in FGFR1 and FGFR2 as well as loss of function mutation of FGFR1 lead to cleft palate. Suppression of FGFR signaling via FGFR kinase inhibitor causes palate defects (41). Many sporadic mutations of FGFR1 and FGFR2, as well as FGF3, FGF7, FGF8, FGF10, and FGF18, cause palate malformations in human. These include Kallmann, Pfeiffer, Apert, and Crouzon syndromes where cleft palate is part of a broad craniofacial phenotype (22, 42, 43). In addition, FGFR1 is widely expressed in the myofibroblasts of injured palate, suggesting that FGFR1 signaling is also important for palate repair during injury (44). Sprys are decoys for FGFR substrates functioning as negative regulators of FGFR signaling, which are expressed in the FGF signaling domains during mouse craniofacial development (45). On the one hand, deletion of Spry2 intensifies FGF signaling intensity, which leads to increased cellular proliferation in the palate shelves and cleft palate (46), without affecting palate shelf fusion in the in vitro organ culture system (17). On the other hand, insufficient FGFR1 signaling due to loss-of-function mutations or haploid insufficiency of Fgfr1 also leads to defects in palatogenesis. However, how FGFR1 regulates palatogenesis has not been completely clear. Interestingly, our data here showed that although ablation of Fgfr1 in NCCs did not prevent patterning of CNC cells in palate shelves, it affected cell proliferation and abrogated palate shelf elevation. It did not fully disrupt palate shelf fusion once they were adjacent. Together with the literature, these results suggest that a precise balance of FGFR1 signaling is required for CNC-derived mesenchymal cell proliferation and change in palate shelf conformation. Both excess and deficiency of FGFR1 signaling led to defects in palatogenesis.
As a key step in palatogenesis, the palate shelves changed their conformation and elevated prior to the fusion stage. Ablation of Fgfr1 in NCCs led to failure in changing palate shelf conformation during palatogenesis (Fig. 2). To date, the mechanism underlying conformational change of palate shelves is not well understood. Several mechanisms have been proposed, which include alteration in cellular proliferation, apoptosis, cell adhesion, or glycosaminoglycan (GAG) content in the extracellular matrix (3). Our data showed that mesenchymal proliferation was decreased at first and then increased in Fgfr1 mutant palate shelves, indicating that proliferation was delayed but not disrupted. Therefore, failure of changing palate shelf conformation in Fgfr1 mutants is not likely due to compromised cell proliferation in the palate shelves. Emerging data show that the composition of GAG in the extracellular matrix varies in different cell types and that the GAG also participates in cell signaling (11). FGF2 has been shown to modulate GAG expression and stimulate hyaluronan synthase genes in vitro (47). Embryos bearing the Fgfr2C342Y mutant show delayed palate elevation and reduced levels of mesenchymal GAGs. Reduced levels of feedback regulators of FGF signaling suggest that this gain-of-function mutation in FGFR2 ultimately resembles loss of FGF function in the palate mesenchyme (15). Future work to characterize GAG compositions in Fgfr1cKO palate shelves is needed to assess the possibility that loss of Fgfr1 in CNC-derived mesenchymal cells changes GAG compositions and impairs palate shelf elevation required for palate shelf fusion.
In vitro experiments with control and Fgfr1 mutant palate shelves showed that although mutant palate shelves also fused and formed the MEE when they were placed in a proximal position, the degeneration of MEE cells was compromised (Fig. 5). This indicates that although not essential, FGFR1 signals in CNC-derived mesenchymal cells play an important role in controlling MEE degeneration during palate fusion. The results are different from previous findings that palatal fusion is not affected by the Crouzon gain-of-function FGFR mutation or Spry2 deficiency (15, 17, 45). Interestingly, it has been shown that FGF18 from the mesenchyme induces Runx1 that is expressed in the epithelium, which regulates epithelial-mesenchymal transition and morphological changes in the MEE cells during palate fusion (48).
The expression patterns and roles of Msx1, Bmp2, and Bmp4 in palate formation have been extensively studied (49, 50). Msx1 is expressed in palatal mesenchyme and is required for expression of Bmp4 in the mesenchyme but not the epithelium. The expression of Bmp2 can also be indirectly regulated by Msx1 (49). Msx1-BMP signaling is required for the proliferation of palatal mesenchyme as well as apoptosis of palatal epithelium, (8, 49). Gain-of-function of BMP signaling due to loss of Noggin leads to deregulated cell proliferation, cell death, and changes in gene expression in palate shelves (50). Wnt signaling also plays critical roles in palate formation (51). Enhanced cell apoptosis in the palatal epithelium and deregulation of cell proliferation in palatal shelves are associated with increased Bmp signaling intensity (50). In line with these reports, our RT-PCR analyses showed that ablation of Fgfr1 in CNC cells led to aberrant expression of Msx1, Bmp2, Bmp4, and Wnt signaling components in Fgfr1cKO E10.5 embryos and frontofacial tissues of E14.5 embryos (Fig. 7). However, whether changes in expression of the Msx1-Bmp axis during E10.5–E14.5 are causal factors of deregulated cell proliferation and apoptosis in Fgfr1cKO palatal shelves remains unanswered. Further ex vivo or in vivo studies are needed to reveal the molecular mechanism by which mesenchymal FGFR1 signals regulate expression of these signaling molecules during palate formation.
In summary, ablation of Fgfr1 in the NCCs delayed cell proliferation in both mesenchymal and epithelial compartments of palate shelves and abrogated palate shelf elevation; it did not fully disrupt the palate shelf fusion but compromised deterioration of the MEE during the process. This is the first report demonstrating the mechanism by which the FGFR1 signaling in CNC cells regulates palatogenesis.
Acknowledgments
We thank Dr. Juha Patanen, for generously providing the Fgfr1-floxed mice and Dr. Stefan Siwko for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant K08DE020883 (to J. Y. C.) and Grants CA96824 and CA140388 from USPHS (to F. W.). This work was also supported by the Natural Science Foundation of Zhejiang Province of China Grant Y2110492 (to C. W.), the J. S. Dunn Research Foundation (to W. L. M.), and Grant CPRIT110555 from the Cancer Prevention and Research Institution of Texas (to F. W. and W. L. M.), and the National Natural Science Foundation of China Grants 81101712 and 81270761 (to X. L.).
- MEE
- medial edge epithelium
- CNC
- cranial neural crest
- NCC
- neural crest cell
- FGFR
- FGF receptor
- GAG
- glycosaminoglycan.
REFERENCES
- 1. Jiang R., Bush J. O., Lidral A. C. (2006) Development of the upper lip: morphogenetic and molecular mechanisms. Dev Dyn. 235, 1152–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alappat S., Zhang Z. Y., Chen Y. P. (2003) Msx homeobox gene family and craniofacial development. Cell Res. 13, 429–442 [DOI] [PubMed] [Google Scholar]
- 3. Snyder-Warwick A. K., Perlyn C. A. (2012) Coordinated events: FGF signaling and other related pathways in palatogenesis. J. Craniofac. Surg. 23, 397–400 [DOI] [PubMed] [Google Scholar]
- 4. Khrapunov S. M., Zima V. L., Tiuleniev V. I., Berdishev H. D. (1975) Change in conformation of histones F2a and F2b in solutions of different ionic strength. Ukrains'kyi Biokhimichnyi Zhurnal 47, 284–289 [PubMed] [Google Scholar]
- 5. Ferguson M. W. (1988) Palate development. Development 103, 41–60 [DOI] [PubMed] [Google Scholar]
- 6. Shuler C. F. (1995) Programmed cell death and cell transformation in craniofacial development. Crit. Rev. Oral Biol. Med. 6, 202–217 [DOI] [PubMed] [Google Scholar]
- 7. Wilkie A. O., Morriss-Kay G. M. (2001) Genetics of craniofacial development and malformation. Nat. Rev. Genet. 2, 458–468 [DOI] [PubMed] [Google Scholar]
- 8. Liu W., Sun X., Braut A., Mishina Y., Behringer R. R., Mina M., Martin J. F. (2005) Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132, 1453–1461 [DOI] [PubMed] [Google Scholar]
- 9. Cobourne M. T., Xavier G. M., Depew M., Hagan L., Sealby J., Webster Z., Sharpe P. T. (2009) Sonic hedgehog signalling inhibits palatogenesis and arrests tooth development in a mouse model of the nevoid basal cell carcinoma syndrome. Dev Biol. 331, 38–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin Y. R., Han X. H., Taketo M. M., Yoon J. K. (2012) Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development 139, 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKeehan W. L., Wang F., Luo Y. (2009) Handbook of Cell Signaling, 2nd Ed., pp. 253–259, Academic/Elsevier Press, New York [Google Scholar]
- 12. Nie X., Luukko K., Kettunen P. (2006) FGF signalling in craniofacial development and developmental disorders. Oral Dis. 12, 102–111 [DOI] [PubMed] [Google Scholar]
- 13. Rice R., Spencer-Dene B., Connor E. C., Gritli-Linde A., McMahon A. P., Dickson C., Thesleff I., Rice D. P. (2004) Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest. 113, 1692–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hosokawa R., Deng X., Takamori K., Xu X., Urata M., Bringas P., Jr., Chai Y. (2009) Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J. Exp. Zool. B Mol. Dev. Evol. 312B, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Snyder-Warwick A. K., Perlyn C. A., Pan J., Yu K., Zhang L., Ornitz D. M. (2010) Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate. Proc. Natl. Acad. Sci. U.S.A. 107, 2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martínez-Abadías N., Holmes G., Pankratz T., Wang Y., Zhou X., Jabs E. W., Richtsmeier J. T. (2013) From shape to cells: mouse models reveal mechanisms altering palate development in Apert syndrome. Dis. Model Mech. 6, 768–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsumura K., Taketomi T., Yoshizaki K., Arai S., Sanui T., Yoshiga D., Yoshimura A., Nakamura S. (2011) Sprouty2 controls proliferation of palate mesenchymal cells via fibroblast growth factor signaling. Biochem. Biophys. Res. Commun. 404, 1076–1082 [DOI] [PubMed] [Google Scholar]
- 18. Takamori K., Hosokawa R., Xu X., Deng X., Bringas P., Jr., Chai Y. (2008) Epithelial fibroblast growth factor receptor 1 regulates enamel formation. J. Dental Res. 87, 238–243 [DOI] [PubMed] [Google Scholar]
- 19. Lee S., Crisera C. A., Erfani S., Maldonado T. S., Lee J. J., Alkasab S. L., Longaker M. T. (2001) Immunolocalization of fibroblast growth factor receptors 1 and 2 in mouse palate development. Plast. Reconstr. Surg. 107, 1776–1784 [DOI] [PubMed] [Google Scholar]
- 20. Trokovic N., Trokovic R., Mai P., Partanen J. (2003) Fgfr1 regulates patterning of the pharyngeal region. Genes Dev. 17, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim H. G., Herrick S. R., Lemyre E., Kishikawa S., Salisz J. A., Seminara S., MacDonald M. E., Bruns G. A., Morton C. C., Quade B. J., Gusella J. F. (2005) Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J. Med. Genet. 42, 666–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pitteloud N., Acierno J. S., Jr., Meysing A., Eliseenkova A. V., Ma J., Ibrahimi O. A., Metzger D. L., Hayes F. J., Dwyer A. A., Hughes V. A., Yialamas M., Hall J. E., Grant E., Mohammadi M., Crowley W. F., Jr. (2006) Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U.S.A. 103, 6281–6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sato N., Katsumata N., Kagami M., Hasegawa T., Hori N., Kawakita S., Minowada S., Shimotsuka A., Shishiba Y., Yokozawa M., Yasuda T., Nagasaki K., Hasegawa D., Hasegawa Y., Tachibana K., Naiki Y., Horikawa R., Tanaka T., Ogata T. (2004) Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1 or KAL2) in five families and 18 sporadic patients. J. Clin. Endocrinol. Metab. 89, 1079–1088 [DOI] [PubMed] [Google Scholar]
- 24. Danielian P. S., Muccino D., Rowitch D. H., Michael S. K., McMahon A. P. (1998) Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326 [DOI] [PubMed] [Google Scholar]
- 25. Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 [DOI] [PubMed] [Google Scholar]
- 26. Trokovic R., Trokovic N., Hernesniemi S., Pirvola U., Vogt Weisenhorn D. M., Rossant J., McMahon A. P., Wurst W., Partanen J. (2003) FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 22, 1811–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chai Y., Bringas P., Jr., Shuler C., Devaney E., Grosschedl R., Slavkin H. C. (1998) A mouse mandibular culture model permits the study of neural crest cell migration and tooth development. Int. J. Dev. Biol. 42, 87–94 [PubMed] [Google Scholar]
- 28. Shiota K., Kosazuma T., Klug S., Neubert D. (1990) Development of the fetal mouse palate in suspension organ culture. Acta Anatomica 137, 59–64 [DOI] [PubMed] [Google Scholar]
- 29. Casey L. M., Lan Y., Cho E. S., Maltby K. M., Gridley T., Jiang R. (2006) Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev. Dyn. 235, 1830–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trainor P. A., Tan S. S., Tam P. P. (1994) Cranial paraxial mesoderm: regionalisation of cell fate and impact on craniofacial development in mouse embryos. Development 120, 2397–2408 [DOI] [PubMed] [Google Scholar]
- 31. Ito Y., Yeo J. Y., Chytil A., Han J., Bringas P., Jr., Nakajima A., Shuler C. F., Moses H. L., Chai Y. (2003) Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 130, 5269–5280 [DOI] [PubMed] [Google Scholar]
- 32. Song Z., Liu C., Iwata J., Gu S., Suzuki A., Sun C., He W., Shu R., Li L., Chai Y., Chen Y. (2013) Mice with Tak1 deficiency in neural crest lineage exhibit cleft palate associated with abnormal tongue development. J. Biol. Chem. 288, 10440–10450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bjork B. C., Turbe-Doan A., Prysak M., Herron B. J., Beier D. R. (2010) Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 19, 774–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang X., Goudy S. L., Ketova T., Litingtung Y., Chiang C. (2008) Gli3-deficient mice exhibit cleft palate associated with abnormal tongue development. Dev. Dyn. 237, 3079–3087 [DOI] [PubMed] [Google Scholar]
- 35. Barrow J. R., Capecchi M. R. (1999) Compensatory defects associated with mutations in Hoxa1 restore normal palatogenesis to Hoxa2 mutants. Development 126, 5011–5026 [DOI] [PubMed] [Google Scholar]
- 36. Murray S. A., Oram K. F., Gridley T. (2007) Multiple functions of Snail family genes during palate development in mice. Development 134, 1789–1797 [DOI] [PubMed] [Google Scholar]
- 37. Miettinen P. J., Chin J. R., Shum L., Slavkin H. C., Shuler C. F., Derynck R., Werb Z. (1999) Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat. Genet. 22, 69–73 [DOI] [PubMed] [Google Scholar]
- 38. Gendron-Maguire M., Mallo M., Zhang M., Gridley T. (1993) Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell 75, 1317–1331 [DOI] [PubMed] [Google Scholar]
- 39. Ricks J. E., Ryder V. M., Bridgewater L. C., Schaalje B., Seegmiller R. E. (2002) Altered mandibular development precedes the time of palate closure in mice homozygous for disproportionate micromelia: an oral clefting model supporting the Pierre-Robin sequence. Teratology 65, 116–120 [DOI] [PubMed] [Google Scholar]
- 40. Kaartinen V., Cui X. M., Heisterkamp N., Groffen J., Shuler C. F. (1997) Transforming growth factor-β3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev. Dyn. 209, 255–260 [DOI] [PubMed] [Google Scholar]
- 41. Lee J. M., Kim J. Y., Cho K. W., Lee M. J., Cho S. W., Kwak S., Cai J., Jung H. S. (2008) Wnt11/Fgfr1b cross-talk modulates the fate of cells in palate development. Dev. Biol. 314, 341–350 [DOI] [PubMed] [Google Scholar]
- 42. Alappat S. R., Zhang Z., Suzuki K., Zhang X., Liu H., Jiang R., Yamada G., Chen Y. (2005) The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev. Biol. 277, 102–113 [DOI] [PubMed] [Google Scholar]
- 43. Riley B. M., Mansilla M. A., Ma J., Daack-Hirsch S., Maher B. S., Raffensperger L. M., Russo E. T., Vieira A. R., Dodé C., Mohammadi M., Marazita M. L., Murray J. C. (2007) Impaired FGF signaling contributes to cleft lip and palate. Proc. Natl. Acad. Sci. U.S.A. 104, 4512–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanda T., Funato N., Baba Y., Kuroda T. (2003) Evidence for fibroblast growth factor receptors in myofibroblasts during palatal mucoperiosteal repair. Arch. Oral Biol. 48, 213–221 [DOI] [PubMed] [Google Scholar]
- 45. Minowada G., Jarvis L. A., Chi C. L., Neubüser A., Sun X., Hacohen N., Krasnow M. A., Martin G. R. (1999) Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 126, 4465–4475 [DOI] [PubMed] [Google Scholar]
- 46. Welsh I. C., Hagge-Greenberg A., O'Brien T. P. (2007) A dosage-dependent role for Spry2 in growth and patterning during palate development. Mech. Dev. 124, 746–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shimabukuro Y., Ichikawa T., Terashima Y., Iwayama T., Oohara H., Kajikawa T., Kobayashi R., Terashima H., Takedachi M., Terakura M., Hashikawa T., Yamada S., Murakami S. (2008) Basic fibroblast growth factor regulates expression of heparan sulfate in human periodontal ligament cells. Matrix Biol. 27, 232–241 [DOI] [PubMed] [Google Scholar]
- 48. Charoenchaikorn K., Yokomizo T., Rice D. P., Honjo T., Matsuzaki K., Shintaku Y., Imai Y., Wakamatsu A., Takahashi S., Ito Y., Takano-Yamamoto T., Thesleff I., Yamamoto M., Yamashiro T. (2009) Runx1 is involved in the fusion of the primary and the secondary palatal shelves. Dev. Biol. 326, 392–402 [DOI] [PubMed] [Google Scholar]
- 49. Zhang Z., Song Y., Zhao X., Zhang X., Fermin C., Chen Y. (2002) Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129, 4135–4146 [DOI] [PubMed] [Google Scholar]
- 50. He F., Xiong W., Wang Y., Matsui M., Yu X., Chai Y., Klingensmith J., Chen Y. (2010) Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev. Biol. 347, 109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. He F., Xiong W., Wang Y., Li L., Liu C., Yamagami T., Taketo M. M., Zhou C., Chen Y. (2011) Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev. Biol. 350, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]