

Abstract
Polysialic acids are bioactive carbohydrates found in eukaryotes and some bacterial pathogens. The bacterial polysialyltransferases (PSTs), which catalyze the synthesis of polysialic acid capsules, have previously been identified in select strains of Escherichia coli and Neisseria meningitidis and are classified in the Carbohydrate-Active enZYmes Database as glycosyltransferase family GT-38. In this study using DNA sequence analysis and functional characterization we have identified a novel polysialyltransferase from the bovine/ovine pathogen Mannheimia haemolytica A2 (PSTMh). The enzyme was expressed in recombinant form as a soluble maltose-binding-protein fusion in parallel with the related PSTs from E. coli K1 and N. meningitidis group B in order to perform a side-by-side comparison. Biochemical properties including solubility, acceptor preference, reaction pH optima, thermostability, kinetics, and product chain length for the enzymes were compared using a synthetic fluorescent acceptor molecule. PSTMh exhibited biochemical properties that make it an attractive candidate for chemi-enzymatic synthesis applications of polysialic acid. The activity of PSTMh was examined on a model glycoprotein and the surface of a neuroprogenitor cell line where the results supported its development for use in applications to therapeutic protein modification and cell surface glycan remodelling to enable cell migration at implantation sites to promote wound healing. The three PSTs examined here demonstrated different properties that would each be useful to therapeutic applications.
Introduction
Polysialic acid (PSA) plays a crucial role on the surface of eukaryotic neuronal cells and neuro-invasive pathogens like Neisseria meningitidis. In mammals PSA is presented as a homopolymer of α2,8-linked N-acetyl-neuraminic acid (Neu5Ac) residues on a small number of proteins of which the major carrier is the neural cell adhesion molecule (NCAM). It has been shown that the PSA on NCAM modulate cell-cell interactions through anti-adhesive properties during development (reviewed in [1]). The NCAM molecule has been implicated in numerous normal and pathological processes, including mammalian development, neuronal plasticity, and tumour metastasis [2]. PSA is also found on CD36 [3], as well as on the recently described SynCAM [4]. Recently the PSA portion of NCAM was shown to have a very important role in the brain since removal of the polysialylation resulted in severe brain defects and precocious death [5], [6]. In addition, changes to the amount and chain length of PSA may influence psychiatric disorders through impaired binding to brain-derived neurotrophic factor and dopamine, which are linked to schizophrenia and other psychiatric disorders, such as depression and bipolar disorder [7].
In bacteria three PSA variants are produced as capsular polysaccharides (for a review of sialic acid biosynthesis see [8]). Neuro-invasive human pathogens Escherichia coli K1 and N. meningitidis group B as well as the bovine/ovine pathogen Mannheimia haemolytica A2 produce a capsule composed of homopolymer of α2,8-linked Neu5Ac residues [9], [10]. Additionally, N. meningitidis group C produces a capsule with a homopolymeric α2,9-linked structure [11], whereas E. coli K92 produces a capsule with alternating linkages of α2,8/α2,9-linked Neu5Ac [12]. It has been established that the mimicry of PSA by these meningitis-causing strains of E. coli and N. meningitidis allows them to avoid detection by the host immune system, thereby contributing to their pathogenesis [13].
Polysialyltransferases (PSTs) are membrane-associated enzymes that catalyze the transfer of Neu5Ac from the activated sugar donor CMP-Neu5Ac to the non-reducing end of the growing polymer. In mammals the PSA chain is synthesized by two Golgi-resident enzymes, ST8Sia2 and ST8Sia4, which have a strong specificity for their protein target(s) [14], [15]. These enzymes are found in the Carbohydrate-Active enZYmes database (CAZy) family GT-29 [16], along with all other mammalian sialyltransferases. In bacteria, genes encoding PSTs in E. coli K1 and K92 as well as from N. meningitidis group B and C are located in the capsule biosynthesis locus of the respective organisms [17], [18]. These enzymes have been characterized in recombinant form [19], [20], [21], [22], [23] and grouped into CAZy family GT-38, which consists exclusively of bacterial PSTs. Bacterial PSTs function at the cytoplasmic side of the inner membrane next to the capsule export machinery, which has posed a challenge for expression and analysis of recombinant forms of these peripheral membrane enzymes.
Recently the PSA polysaccharide has been discovered to have a function in biotechnology where it is used as a chemical means of improving therapeutic protein half-life [24], [25]. This chemical conjugation is not the only way of attaching PSA on therapeutic proteins; we have also shown it can be added enzymatically to existing N-linked glycans using the bacterial PST enzyme from N. meningitidis group B [26]. Since the site-specific PSA addition could be applied to many recombinant therapeutic glycoproteins it would be advantageous to improve the PST enzyme's profile, in particular the specific activity and purity.
The animal pathogen M. haemolytica A2 has a PSA capsule and has been previously tested for polysialyltransferase activity [27], although to our knowledge the enzyme has not been purified or characterized. Our primary objective in this study was to identify the gene encoding this enzyme and characterize the enzyme in recombinant form. We further expanded this analysis to include a side-by-side comparison with two other recombinantly-expressed PSTs from N. meningitidis group B and E. coli K1. Although the latter two enzymes have been previously characterized by us and others, there is significant variation in their performance depending on the expression construct, purification method, and activity assay used. Furthermore, an improved purification method has resulted in significantly improved enzyme performance [26]. Characterizing the three enzymes in parallel allowed us to evaluate their utility as tools for glycoprotein modification, and for other applications of PSA synthesis.
Materials and Methods
Analysis of the M. haemolytica A2 genome sequence
All molecular biology methods were performed according to manufacturer's instructions, where appropriate. Genomic DNA from M. haemolytica A2 (ATCC 29694) was isolated using the DNeasy Tissue kit (Qiagen Inc., Toronto, Canada). Genomic DNA was amplified using Phusion polymerase (New England Biolabs, Pickering, Canada) with primers based on the sequences of M. haemolytica A1 capsule biosynthetic genes wzf and phyB, which were expected to be conserved (Forward 5′-CAGTTGGAATAATTACAGTTAGCCA and Reverse 5′- CTTTTTGCTCACTTTCTAACCAACG). The ∼8.2 kb PCR amplicon was sequenced using primer walking followed by submission of the sequence to GenBank (FJ217347). The sequence of pstMh was confirmed following the subsequent publication of the M. haemolytica A2 genome sequence (NZ_ACZY010000016.1, COK_0618) [28].
Cloning of bacterial polysialyltransferases
PSTMh was amplified as a full-length and a Δ20 N-terminal truncation using forward primers 5′-GTAGGTTAACATATGATTAAGACAATAAAGAAATTATTAGTATC and 5′-CTTTCAGGATCATATGTTTTTAAAATTTCATTTAGCAGAAG, respectively, with the following reverse primer 5′-CGTTGGGTCGACTTACTATATTTCATTTGTATTATTTAATATTC. A Δ15PSTEc amplicon was generated by amplification of the PSTEc DNA sequence with primers 5′-GGCTACGCATATGAATCCAATTGGGTTTTTCCGT and 5′-CCGAGCCGTCGACCTATTACTCCCCCAAGAAAATCCTT. PCR amplicons were then digested NdeI and SalI restriction enzymes (underlined in primer sequence) (New England Biolabs, Pickering, Canada) followed by ligation with T4 DNA ligase (Roche Diagnostics, Laval, Canada) into the expression vectors pCW or pCWMalE-thrombin, where appropriate. MalE-PSTEc, MalE-PSTNm, and MalE-Δ19PSTNm were expressed from constructs previously generated by our laboratory [22], [26]. PST constructs were then transformed into E. coli AD202 (E. coli Genetic Stock Center, CGSC 7297) for expression.
Expression and Purification of Recombinant Polysialyltransferases
The various PST constructs were expressed and purified using the conditions previously described for MalE-glycosyltransferase fusion proteins [26], [29]. Briefly, cells were grown in 2YT media at 37°C until the culture reached OD600∼0.5 followed by induction with 0.5 M IPTG and overnight growth at 25°C. Cells were harvested by centrifugation and stored at −80°C. To extract the enzyme the cells were thawed and re-suspended in PBS buffer followed by lysis with an Emulsiflex-C5 (Avestin) and centrifugation (27 000× g). Soluble protein was purified using a 5 mL HiTrap Heparin HP column (GE Healthcare, Baie d'Urf'é, Canada) with a linear gradient of 0–60% B using the following buffers: A, PBS pH 7.4; B, PBS pH 7.4+2 M NaCl. Fractions containing the purest enzyme were pooled together and stored in the column elution buffer. Attempts to desalt the protein resulted in partial loss of activity, so the preparations were used without desalting and therefore contained approximately 500 mM NaCl.
Polysialyltransferase Activity Assays on Small Molecule Acceptors
The activity of the PSTs was determined by using synthetic monosialyl-lactosyl (GM3), disialyl-lactosyl (GD3), and trisialyl-lactosyl (GT3) ganglioside analogues (glycan portion only) coupled to the fluorophore 6-(5-fluoresceincarboxamido)-hexanoic acid succinimidyl ester (FCHASE) (Invitrogen, Burlington, Canada), which were synthesized as previously described [22]. The standard assay conditions were: 0.5 mM GD3-FCHASE, 50 mM HEPES pH 7.5, 10 mM MgCl2, and 10 mM CMP-Neu5Ac at 30°C. Where indicated in the text some of these parameters were changed including the acceptor molecule (GT3-FCHASE), buffer and pH (MES pH 5.5, HEPES pH 7.0, Tris-HCl pH 8.0, Tris-HCl pH 9.5), temperature (25°C, 37°C), and the concentration of CMP-Neu5Ac for kinetic analysis (0.25 mM to 20 mM). The assays were analysed by capillary electrophoresis as previously described [22].
Polysialyltransferases Activity Assays on Fetuin
The activity of the PSTs was determined on the bovine serum glycoprotein fetuin which had been enzymatically modified so that the N-glycans terminated in two Neu5Ac residues (disialyl-fetuin, diSA-Fet) as previously described [26]. The PSTs were incubated with either 1 mg/mL or 3 mg/mL diSA-Fet in the presence of 50 mM sodium phosphate buffer pH 8.0, 10 mM MgCl2, and 10 mM CMP-Neu5Ac at 30°C for a period of time indicated in the text. The amount of enzyme used in the reactions (1 mU) was standardized according to the specific activity determined from reactions with GD3-FCHASE. The reactions were stopped using 1× protein sample buffer and analyzed by SDS-PAGE. The presence of PSA was confirmed by performing Western blots probed with anti-PSA antibodies (mAb735, kind gift of Dr. Rita Gerardy-Schan, Hanover Medical School).
Production of Polysialic Acid on the Surface of PC12 Cells
PC12 cells (CRL-1721, ATCC) were plated at a density of 1×106 cells/well on 12 mm coverslips (Bellco), coated with 25 µg/ml poly-l-lysine, in 24 well sterile culture plates. PC12 cells were grown in DMEM media (containing 2.5% heat inactivated fetal calf serum, 15% heat inactivated horse serum, 100 U/ml penicillin, 100 µg/ml streptomycin) for 24–48 hours prior to treatment. The cells were washed once with serum-free media immediately prior to treatment with the PSTs. The PST reaction was conducted for 1 hour at 37°C in the presence of 5% CO2 in a humidified incubator in a 200 µl volume of DMEM containing 5 mM CMP-Neu5Ac and 4 mU PST. Subsequent to the PST reaction, the cells were washed twice with PBS and fixed with 200 µl formalin for 10 min at room temperature. The cells were then washed twice with PBS and stored in PBS at 4°C until use in immunocytochemistry.
Immunocytochemical Detection of PSA on PC12 Cells
Polysialylation of PC12 cells was determined using α-PSA-NCAM immunocytochemistry. Briefly, fixed cells were blocked in PBS containing 1% BSA and 0.05% Triton X-100 for 30 min at room temperature. The cells were then incubated for 60 min at room temperature with α-PSA-NCAM (1∶200, mouse IgM clone 2-2B, Millipore, MAB5324) in PBS buffer (with BSA and Triton as above). The cells were then washed three times with PBS prior to incubation with goat anti-mouse IgM Alexa488 conjugate (1∶600, Molecular Probes, A-21042) in PBS (with BSA and Triton as above). Thereafter, the cells were washed 3 times with PBS and once with dH2O prior to mounting with Vectashield (Vector Labs, Burlingame, CA) containing 5 µg/ml Hoechst 33258. Confocal fluorescent images were obtained using an Olympus Fluoview FV1000 upright microscope with a 40× objective (UPLSAPO, 0.95 NA).
Results
Identification of a putative polysialyltransferase from the Mannheimia haemolytica A2 genome sequence
The DNA coding sequence for a putative PST was determined by sequence analysis of a region of the M. haemolytica A2 genome. Using primers based on the sequences of M. haemolytica A1 capsule biosynthesis genes wzf and phyB, a region of the M. haemolytica A2 genome was amplified and the sequence was analyzed using primer walking. A 1.2 kb DNA sequence was identified that encoded a putative protein (henceforth referred to as PSTMh) of 401 amino acids long and a predicted molecular weight of 47.7 kDa. An examination of the corresponding amino acid sequence using a BLAST alignment showed that it shared 35% and 31% identity, respectively, with the previously described PSTNm and PSTEc, which is at a similar level to the identity reported for the latter two enzymes (33%) [21], [22]. Based on the observed sequence similarity, this gene was cloned and expressed to detect for PST activity.
Evaluation of Solubility and Activity of Polysialyltransferase Constructs
PSTMh was cloned and expressed as three variants: full length (PSTMh), full length with an N-terminal maltose binding protein fusion (MalE-PSTMh), and with a 20 amino acid N-terminal truncation fused with MalE (MalE-Δ20PSTMh). The truncation of amino acids at the N-terminus to remove a putative trans-membrane domain as well as the addition of MalE has previously been shown to improve the solubility and specific activity of PST [22], [23], [30].
In order to measure the solubility of the various PST constructs, the specific activities of the soluble and insoluble fractions of the cell lysates were determined using the fluorescent synthetic acceptor molecule GD3-FCHASE followed by detection using capillary electrophoresis (CE). PST activity was detected in both the soluble and insoluble fractions of all construct expressions. The fusion to MalE resulted in approximately a five-fold increase in activity compared to PSTMh (Table 1). Furthermore, the truncation of PSTMh (MalE-Δ20PSTMh) resulted in an increase in the proportion of activity in the soluble fraction (86% compared to ∼45–50% for the full-length constructs). In support of these results, qualitative SDS-PAGE analysis indicated that MalE-Δ20PSTMh demonstrated the best combination of expression and solubility of the three constructs (Figure 1). The activity of the soluble and insoluble fractions for the corresponding PSTNm and PSTEc constructs was also examined. Similarly, both MalE-Δ19PSTNm and MalE-Δ15PSTEc demonstrated an increased ratio of soluble activity from 1∶1 to 3∶1 compared to MalE-PSTNm and MalE-PSTEc (Table 1 and Figure 1). Interestingly, the specific activity of the soluble material from the cell lysate was detectably higher for the full-length MalE fusions compared to their truncated counterparts. However, once purified, the activity of the truncated proteins was restored to similar levels as the full-length purified proteins (data not shown). Since the truncated MalE-fusion proteins resulted in the best solubility, expression, and activity they were chosen for further biochemical analysis.
Table 1. Comparison of Soluble Activity of PST Constructs1.
| Enzyme | Supernatant (mU/mg) | Pellet (mU/mg) | % Sup | % Pel |
| PSTMh | 76.4±15.7 | 101.1±9.4 | 43.1% | 56.9% |
| MalE-PSTMh | 439.7±10.4 | 454.9±80.4 | 49.2% | 50.8% |
| MalE-Δ20PSTMh | 341.0±4.8 | 55.3±7.9 | 86.0% | 14.0% |
| MalE-PSTNm | 351.1±185.9 | 290.3±135.6 | 54.7% | 45.3% |
| MalE-Δ20PSTNm | 88.0±42.9 | 29.9±17.7 | 74.6% | 25.4% |
| MalE-PSTEc | 169.6±15.2 | 201.3±1.0 | 45.7% | 54.3% |
| MalE-Δ20PSTEc | 68.8±9.3 | 24.2±1.9 | 74.0% | 26.0% |
Experiments were performed in triplicate (n = 3) with mean values and standard error reported.
Figure 1. Expression and solubility of PSTMh constructs.

The PST constructs were expressed in E. coli AD202 followed by cell disruption and centrifugation. The soluble (Sup) and insoluble (Pel) fractions were analyzed by SDS-PAGE and Coomassie Blue staining. Bands corresponding to the predicted sizes of the protein variants were observed (indicated with an asterisk). The increased intensity of the bands of the MalE-fusions indicated increased expression compared to the non-fusion protein. Furthermore, the truncated protein (MalE-Δ20PSTMh) variant demonstrated the best solubility.
Acceptor Molecule and pH Conditions for Optimal Activity
Having established the optimal construct (MalE-Δ20PSTMh, hereafter referred to simply as PSTMh) we next wanted to evaluate several biochemical properties of the enzyme and compare them to the equivalent constructs of the other two bacterial PSTs (PSTNm and PSTEc). We first examined if PSTMh had a preference for a disialyl (GD3-FCHASE) or trisialyl (GT3-FCHASE) acceptor molecule. In the case of both acceptors the specific activities of PSTMh and PSTNm were significantly greater than that of PSTEc (Table 2). PSTMh and PSTNm demonstrated a clear preference (nearly two-fold greater) for GT3-FCHASE, in contrast to PSTEc, which has a slight preference for GD3-FCHASE. None of the PSTs demonstrated activity on the monosialyl acceptor molecule GM3-FCHASE (data not shown), which is consistent with published evidence that at least two Neu5Ac residues on the acceptor molecule are required [22], [26]. We next wanted to determine the optimal reaction pH for PSTMh in the range between pH 7.0–pH 8.0, since these pH values would likely be most suitable for downstream applications as they are near physiological pH and also since previous studies have shown that PSTs from E. coli and N. meningitidis have a broad range of pH tolerance with an optimum at pH 8.0 [22], [23]. We also analyzed reactions at outlying pH values, including pH 5.5, which was expected to be unsuitable for large-scale reactions because of its acidity, as well as a very basic pH at 9.5. PSTMh demonstrated optimal activity at pH 7.0, while still retaining relatively high levels of activity at pH 7.5 and pH 8.0 (Figure 2). Similarly, PSTNm and PSTEc demonstrated activity across a range of pH values, although these enzymes demonstrated higher activity at a slightly more basic pH range from pH 7.5–pH 9.5.
Table 2. Biochemical Properties of Bacterial Polysialyltransferases1.
| Specific activity2 | CMP-Neu5Ac Kinetics2 | ||||
| Enzyme | GD3-FCHASE (mU/mg) | GT3-FCHASE (mU/mg) | Km (mM) | Vmax (µmol/min/mg) | Vmax/Km |
| PSTMh | 1433.4±83.8 | 2670.8±92.1 | 1.1±0.05 | 1519.0±96.2 | 1380.9 |
| PSTNm | 939.6±222.2 | 1885.7±173.1 | 5.7±0.7 | 1318.5±40.3 | 251.9 |
| PSTEc | 322.1±56.1 | 225.5±50.3 | 4.9±0.7 | 800.4±112.4 | 162.4 |
Experiments were performed in triplicate (n = 3) with mean values and standard error reported.
Experiments were performed with enzymes from three separate purification batches in duplicate (n = 6) with mean values and standard error reported.
Figure 2. pH optima of PSTNm, PSTEc, and PSTMh.
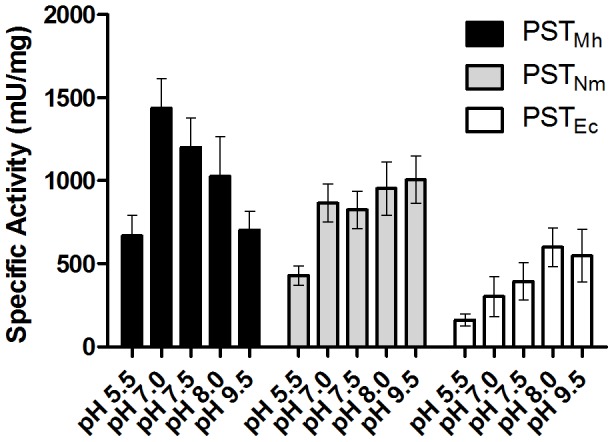
The specific activities of the three bacterial PSTs were compared at different reaction pHs. . Enzymes from three separate purification batches were tested in duplicate (n = 6). All enzymes demonstrated activity across the range of pHs tested. PSTMh demonstrated a reaction pH optimum of 7.0. PSTNm and PSTEc demonstrated optimal activity at slightly higher pH (7.5–8.0).
Stability of the Enzymes During a Timecourse
We wanted to examine the performance of the three enzymes over a two hour time course at different incubation temperatures (25°C, 30°C, and 37°C) that may be required in downstream applications. We observed that all of the enzymes demonstrated a decrease in activity when measured throughout the time course as a result of the intrinsic labile nature of these enzymes, as well as the decrease in donor substrate and the changing nature of the acceptor molecule. Interestingly, higher incubation temperatures had different effects on the performance of the enzymes. At early time points (1–2 min) both PSTNm and PSTEc demonstrated optimal activity at 37°C, with decreased amounts of activity at lower temperatures (Figure 3). By 10 min (which was chosen because the majority of the drop in activity occurs by this time) the advantage imparted by the higher temperature was lost and the different temperatures gave similar activity values. In contrast, incubation of the reaction at 37°C was inhibitory to PSTMh, giving the lowest amount of activity compared to the other two temperatures. This trend continued throughout the course of the reaction. In addition to examining the activity at different temperatures we also looked at the percentage of activity remaining compared to the level at 1 min of incubation. In addition to having the highest activity of the three enzymes, PSTNm had the highest percentage of activity remaining at all time points . For example at 10 min, the activity remaining for PSTNm was 50.7% (25°C), 44.4% (30°C), and 34.7% (37°C). PSTMh was intermediate in its stability with 33.8% (25°C), 30.3% (30°C), and 15.8% (37°C) of the activity was remaining at 10 min. PSTEc was the most thermo-labile with only 26.9% (25°C), 20.4% (30°C), and 12.9% (37°C) of activity at 10 min.
Figure 3. Stability and thermolability of PSTNm, PSTEc, and PSTMh.
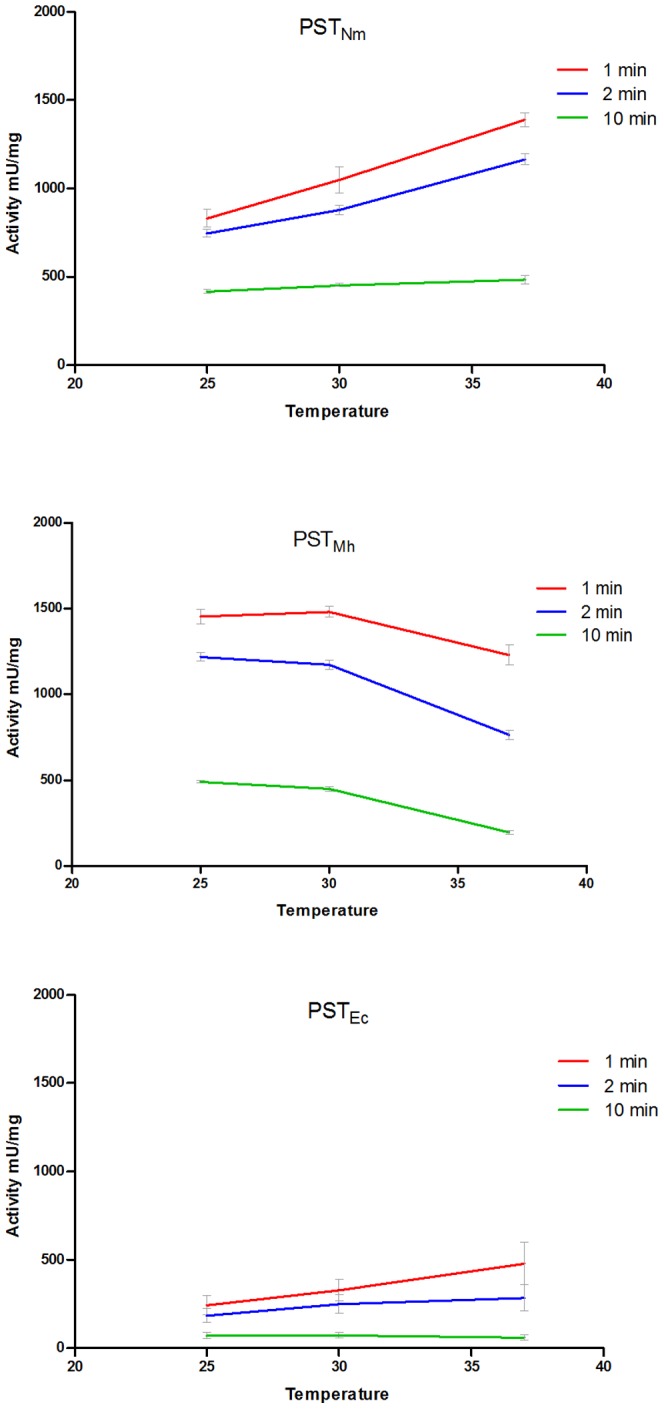
The specific activities of the three bacterial PSTs were monitored during a two-hour time course at three different reaction temperatures (25°C, 30°C, and 37°C). Both PSTNm and PSTEc demonstrated optimal temperature at 37°C in constrast to PSTMh, which demonstrated lower activity at this higher temperature compared to other incubation temperatures. After 10 min incubation there is no longer an advantage for the activity at a higher temperature for these two enzymes. . Enzymes from three separate purification batches were tested in duplicate (n = 6).
Donor Kinetics for CMP-Neu5Ac
In the development of glycosyltransfereases for synthetic purposes, one must consider the ability of an enzyme to use substrates efficiently. In the case of PSTs, this is a particularly relevant consideration since the donor molecular, CMP-Neu5Ac, is relatively expensive to produce and therefore could be a limiting reagent for synthesis.
Hence, we wanted to compare the kinetic parameters of the three bacterial PSTs for CMP-Neu5Ac rather than the acceptor molecule, since we did not anticipate the endogenous acceptor or our fluorescent synthetic acceptors to be used in downstream applications. PSTMh was able to efficiently use CMP-Neu5Ac at lower concentrations than PSTEc or PSTNm when modifying the acceptor molecule GD3-FCHASE. Using concentrations of CMP-Neu5Ac ranging from 0.25 mM to 10–20 mM, PSTMh was determined have a Km of 1.10 mM compared to 5.67 mM and 4.93 mM for PSTNm and PSTEc, respectively (Table 2). It is important to note that there is significant variation in the published Km values for PSTNm and PSTEc depending on the assay, acceptor molecule, expression construct, and purification methods [21], [22], which is why testing the different enzymes in parallel is important for establishing their utility in downstream applications.
PSA Chain Length on Synthetic Acceptors
The ability to form different chain lengths may be needed for applications of these enzymes, such as therapeutic glycoprotein modification [26]. To determine product chain length heterogeneity we performed an analysis of the chain length distribution of the products generated from GD3-FCHASE at three different points during product formation: ∼15%, ∼30%, and ∼65% conversion. At all the stages of the reactions tested with the three enzymes, the predominant product is the acceptor plus one Neu5Ac residue. Each successive product is present in smaller amounts under these reaction conditions. Both PSTMh and PSTNm are capable of making PSA chains longer than twenty Neu5Ac residues in length, whereas PSTEc makes short oligomers with a maximum length of ∼10 Neu5Ac. As can be seen in Figure 4, at 15% conversion PSTNm and PSTMh formed chains twice the length of that formed by PSTEc (a total of 18, 16, and 7 Neu5Ac residues, respectively). This same trend was seen at higher percent conversions as well. The chain lengths of products formed by PSTNm and PSTMh increased to 22 and 18 at 30% conversion and then to chains longer than 20 Neu5Ac residues at 65% conversion. (It is difficult to determine the actual chain length of products longer than 20 Neu5Ac due to a limitation in the resolution of the products when analyzed by CE.) Unlike PSTNm and PSTMh, the products formed by PSTEc remained as short Neu5Ac oligomers at all percent conversions: 8 and 11 Neu5Ac, respectively, at 30% and 65% conversion. Interestingly at high product conversion (65%), the product with two Neu5Ac residues added represents a considerably higher proportion of the products formed by PSTEc than the other two enzymes (∼30% of the total product compared to ∼15% for PSTMh and less than 10% for PSTNm). In the case of PSTMh and PSTNm at the same stage of the reaction, the products longer than 6 Neu5Ac residues represent the majority of the products (∼40% for PSTMh and ∼70% for PSTNm). This observation is interesting because it may be possible to use different enzymes in order to produce products with varying chain lengths and heterogeneity.
Figure 4. Analysis of PSA Chain Length by Capillary Electrophoresis.

Capillary electrophoresis was used to analyze the reactions of GD3-FCHASE and CMP-Neu5Ac catalyzed by the three bacterial PSTs. Shown here are the reactions stopped at approximately 15% conversion. The large peak at ∼9.4 min represents the starting material and subsequent peaks are the products with additional Neu5Ac residues added. Acceptor plus 2 and 10 Neu5Ac products are indicated with arrows. The chain length of the products generated by PSTNm (18 Neu5Ac) and PSTMh (16 Neu5Ac) were significantly longer than that of PSTEc (7 Neu5Ac). The electropherograms of the reactions have been overlaid on the same axis to aid in the comparison.
Activity on Disialyated-Fetuin and PC12 Cells
Recently we demonstrated the ability of PSTNm to polysialylate therapeutic glycoproteins, which in turn resulted in an improved pharmacokinetic profile for the therapeutics [26]. Here we wanted to determine if PSTMh is able to modify the model serum glycoprotein fetuin, which has been previously modified such that its glycans terminate in a disialyl moiety (diSA-Fet), in order to be a substrate for bacterial PSTs. When the three PSTs were incubated with diSA-Fet and CMP-Neu5Ac, all three enzymes were able to synthesize polysialylated material, as was evidenced by the change in migration of the product protein on an SDS-PAGE gel and was confirmed by a Western blot with anti-PSA antibodies (Figure 5). An examination of the intensity (based on gel densitometry) and molecular mass of the converted material on an SDS-PAGE gel showed that PSTNm demonstrated the highest activity on this protein (69% conversion in 1 h). Under standard reaction conditions, both PSTMh and PSTEc were also able to modify diSA-Fet, however to a lesser degree (21% and 25.5% conversion, respectively). With all three enzymes, doubling the reaction time resulted in an increase of product formation. Next, the amount of PSTEc and PSTMh in the reaction was increased to ∼13 µg (chosen based on the maximal volume of enzyme that could be added to the reaction) in order to determine if it was possible to increase the amount of product formed. When additional enzyme was added to the reaction the amount of polysialylated product increased dramatically (39% conversion for PSTEc and 63% for PSTMh).
Figure 5. SDS-PAGE Analysis of PST Reactions on diSA-fetuin.
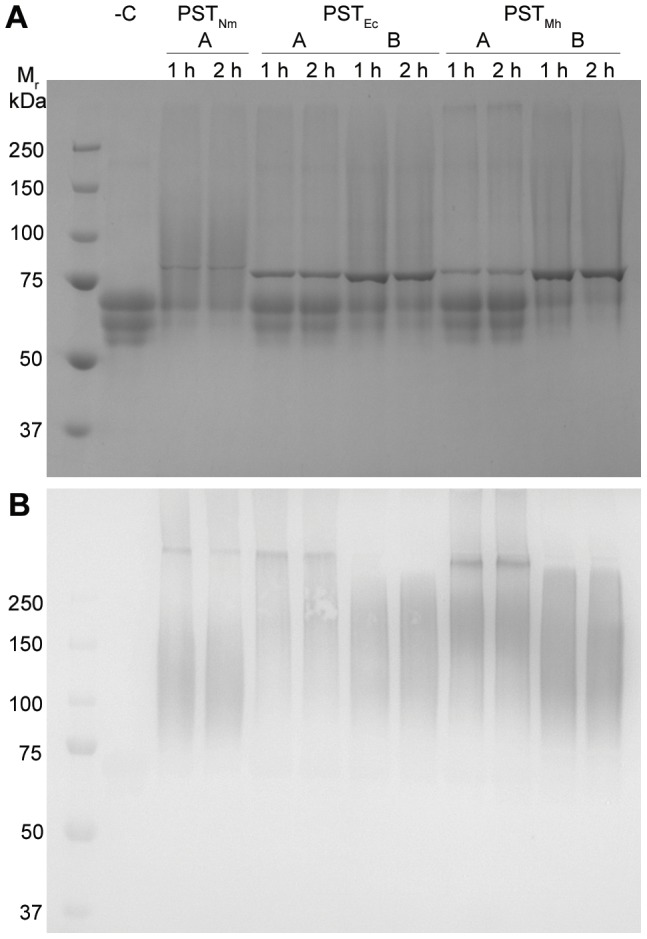
A.The three PSTs were incubated with the acceptor molecule disialyl-fetuin using standard reaction conditions (lanes denoted “A”) as well as a reaction condition to improve the formation of the product (13 µg enzyme, lanes denoted “B”) for 1 or 2 h. A negative control reaction (containing no enzyme) was included (“-C”). The small band at ∼75 kDa indicates the PST added to the reaction. B. Following SDS-PAGE analysis, the proteins were transferred to a membrane and probed with α-PSA antibodies. All reactions were positive for PSA except the negative control lane containing unmodified disialyl-fetuin.
In addition to the utility of bacterial PSTs in the modification of therapeutic proteins to improve pharmacokinetics, a recent application in tissue repair has been developed [31], whereby PSTNm was used to synthesize PSA on the surface of cells in culture and in brain tissue. We therefore wanted to establish if PSTMh was also capable of forming PSA on the surface of cells in culture. The three enzymes were incubated with CMP-Neu5Ac and the neuroprogenitor cell line PC12, which are known to over-express the native target of PST in mammalian systems, NCAM [32]. PSA production was detected by anti-PSA immunostaining and microscopy. In a live cell assay, all three enzymes were able to synthesize PSA on the surface of PC12 cells in the presence of CMP-Neu5Ac (Figure 6). Labeling in the absence of CMP-Neu5Ac was limited to levels comparable to non-specific binding of the secondary antibody (data not shown). Confocal microscopy confirmed that the PSA staining was restricted to the cell surface, consistent with the enzymes being excluded from entering live cells. Quantitation of PSA staining indicated that the three enzymes produced comparable levels of polysialylation under the experimental conditions tested (Figure 6 inset). As the bacterial PST enzymes require acceptor glycans terminating in a disialyl moiety, the present experiment indicated that PC12 cell surface proteins must contain this moiety. As was expected for a homogenous cell line culture, all cells were uniformly labelled by the PSTs. Interestingly, it was consistently observed that fewer cells were present on the coverslips treated with the PST enzymes, which is likely due to reduced adhesion of the PSA labelled cells. This observation is in line with the known effect of PSA on NCAM adhesion [1].
Figure 6. Polysialylation of cell-surface proteins on live PC12 cells.

PC12 cells were incubated with the three PSTs in the absence and presence of 5 mM CMP-Neu5Ac for 1 hour at 37°C. Cell-surface PSA was detected using PSA-NCAM immunocytochemistry and nuclei were detected with Hoechst 33258 and confocal microscopy imaging. Following treatment of PC12 cells with all three PSTs in the presence of CMP-Neu5Ac specific cell-surface PSA labeling was detected (as indicated in the panels denoted “+CMP-Neu5Ac). Control reactions were also performed with the CMP-Neu5Ac absent; in these reactions PSA labeling was not detected. Quantitation of cellular polysialylation was performed using ImageJ (v 10.2) on the immunocytochemical images from two independent experiments by measuring the fluorescent intensity of 15 randomly selected cells per image. Images were pre-processed by performing background subtraction (50 pixel rolling ball radius) prior to measuring pixel intensity. Relative fluorescence values were calculated by dividing the PSA intensity (green fluorescence) by DAPI intensity (blue fluorescence). Data were normalized to average+CMP fluorescence for each experiment (n = 2).
Discussion
We have characterized the PST from the bacterium M. haemolytica A2 and compared several biochemical parameters to those of other glycosyltransferases of family GT-38. Based on the identification of this sequence and the following demonstration of PST activity, we propose that PSTMh be classified as a member of family GT-38. This is the first GT-38 member that is not from either E. coli or N. meningitidis species.
The interest in using such PSTs for potential therapeutic applications requires that the enzyme of choice have qualities that are compatible with modification of high value therapeutic proteins, such as high purity and easy removal after modification. The production of high purity (recombinant) enzymes requires good over-expression, simple reproducible purification and good storage properties. The application of the enzyme requires that it be very efficient in conversion of the substrates that are used, so that the kinetic parameters should be such that large excesses of expensive substrates are not required for production. Because PSTs do not produce a single homogeneous product, it is also important to be able to consistently produce a product of desired length by changing the reaction conditions or by the generation of mutant enzymes with altered properties. With these parameters in mind as a starting point we sought to examine the performance of the three GT-38 polysialyltransferases.
We began the development of PST's for in vitro modification of glycoproteins and application to tissue engineering with the characterization of PSTNm (expressed as the full-length version MalE-PSTNm) [22]. Our preliminary work with this enzyme showed that while the expression was adequate, the purification lead to material that was not as pure as needed for these applications, and that the enzyme had some stability issues that made it a good candidate for enzyme improvement. Next, we were able to show that truncation of the N-terminus of the protein greatly improved solubility and that an improved purification method could lead to higher purity, yields, and activity [26]. We also performed proof-of-concept experiments demonstrating that therapeutic proteins were modifiable by PSTNm in vitro. The other well studied GT-38 enzyme from E. coli serotype K1 has also been applied to small scale glycolipid synthesis applications by others, but not as a pure protein or on protein or cell based acceptors [33]. While we are confident that glycolipids could be modified based on that work, we did not pursue gangliosides as acceptors as there is currently no demonstrated therapeutic need for these types of molecules. We have also shown that the application of PSTs to cell surface modification does not involve significant modification of glycolipids as the PSA formed on cell surfaces is completely removed by a trypsin treatment [31].
Having now made improvements to the issues of expression, solubility, purity, and utility of PSTNm, we wanted to address the same issues with PSTEc. While we were working on PSTNm and PSTEc we identified PSTMh through genome mining, so that we were now able to test all three enzymes for the applications mentioned above, which is the basis for this study. It was readily apparent that all three enzymes could become more soluble when an N-terminal deletion was made between the enzyme and its fusion partner. The increase in expression was the greatest for the PSTMh and in fact it yielded the highest level of active PST both in the soluble fraction and in terms of specific activity on the small synthetic acceptors. More specifically when PSTMh was purified, over 21 mg of enzyme was recovered, in contrast to ∼4 mg of PSTEc and ∼6 mg of PSTNm from the same amount of cell extract (data not shown). Furthermore, the increase in yield came specifically from the protein of interest (and not any co-purified contaminant proteins) and therefore the degree of purity was also the highest for PSTMh. Despite the increased yield and purity, tests using diSA-Fet showed that it required larger amounts of PSTMh to achieve the same level of protein modification with PSTNm, but unlike PSTEc, it could produce the same level of modification seen with PSTNm. Testing on the PC-12 neuroprogenitor cells showed that all three enzymes were capable of modifying cell surface proteins, with no overt differences in the ability to label proteins on the surface of live cells in situ, despite any differences in biochemical properties including (thermo)stability, product chain-length formation, and kinetics. Together the results of this study indicate that each of the three enzymes has specific qualities that may be useful in future therapeutic applications.
Conclusions
As we have demonstrated that the GT-38 enzymes can be used to modify glycoproteins and mammalian cell surfaces, we are now tasked with improving them further for application in the production of therapeutics. We are planning to improve the specific activity, stability, and substrate range through directed evolution. It would be of interest to try to maintain the feature of the enzyme that enables it to form short chains of limited size distribution on glycoprotein acceptors, as that may be advantageous to applications requiring a more homogeneous polysialylation. The lower degree of stability at 37°C presents an ideal parameter to improve through directed evolution or site-directed mutagenesis, since any in vivo applications would require an enzyme optimally active at this temperature. In order to fully exploit these enzymes for in vitro synthesis we will improve the protein acceptor range of the enzyme as well so that more proteins/cells can be modified. In conclusion we have shown through a side-by-side comparison of three GT-38 polysialyltransferases that they have very different properties as recombinant proteins, and that the newest member from M. haemolytica A2 shows promise as a catalyst for the modification of therapeutic proteins and cell surfaces.
Acknowledgments
We wish to thank Lisa M. Willis and Marie-France Goneau for technical assistance.
Funding Statement
Canadian Institutes for Health Research grant MOP-84272. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rutishauser U (2008) Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nature reviews Neuroscience 9: 26–35. [DOI] [PubMed] [Google Scholar]
- 2. Kiss JZ, Muller D (2001) Contribution of the neural cell adhesion molecule to neuronal and synaptic plasticity. RevNeurosci 12: 297–310. [DOI] [PubMed] [Google Scholar]
- 3. Yabe U, Sato C, Matsuda T, Kitajima K (2003) Polysialic acid in human milk. CD36 is a new member of mammalian polysialic acid-containing glycoprotein. The Journal of biological chemistry 278: 13875–13880. [DOI] [PubMed] [Google Scholar]
- 4. Galuska SP, Rollenhagen M, Kaup M, Eggers K, Oltmann-Norden I, et al. (2010) Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proceedings of the National Academy of Sciences of the United States of America 107: 10250–10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinhold B, Seidenfaden R, Rockle I, Muhlenhoff M, Schertzinger F, et al. (2005) Genetic Ablation of Polysialic Acid Causes Severe Neurodevelopmental Defects Rescued by Deletion of the Neural Cell Adhesion Molecule. Journal of Biological Chemistry 280: 42971–42977. [DOI] [PubMed] [Google Scholar]
- 6. Muhlenhoff M, Oltmann-Norden I, Weinhold B, Hildebrandt H, Gerardy-Schahn R (2009) Brain development needs sugar: the role of polysialic acid in controlling NCAM functions. Biological chemistry 390: 567–574. [DOI] [PubMed] [Google Scholar]
- 7. Isomura R, Kitajima K, Sato C (2011) Structural and Functional Impairments of Polysialic Acid by a Mutated Polysialyltransferase Found in Schizophrenia. Journal of Biological Chemistry 286: 21535–21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferrero MA, Aparicio LR (2010) Biosynthesis and production of polysialic acids in bacteria. Applied Microbiology and Biotechnology 86: 1621–1635. [DOI] [PubMed] [Google Scholar]
- 9. Adlam Knight J. M MAWJM, Lindon J. C C (1987) Production of colominic acid by Pasteurella haemolytica serotype A2 organisms. FEBS Microbiology Letters 42: 23–25. [Google Scholar]
- 10. Bhattacharjee AK, Jennings HJ, Kenny CP, Martin A, Smith IC (1975) Structural determination of the sialic acid polysaccharide antigens of Neisseria meningitidis serogroups B and C with carbon 13 nuclear magnetic resonance. Journal of Biological Chemistry 250: 1926–1932. [PubMed] [Google Scholar]
- 11. Jennings HJ, Bhattacharjee AK, Bundle DR, Kenny CP, Martin A, et al. (1977) Strucutres of the capsular polysaccharides of Neisseria meningitidis as determined by 13C-nuclear magnetic resonance spectroscopy. Journal of Infectious Diseases 136 Suppliment: S78–S83. [DOI] [PubMed] [Google Scholar]
- 12. Whitfield C, Roberts IS (1999) Structure, assembly and regulation of expression of capsules in Escherichia coli . Molecular Microbiology 31: 1307–1319. [DOI] [PubMed] [Google Scholar]
- 13.Silver RP, Vimr ER (1990) Polysialic acid capsule of Escherichia coli K1. In: Iglewski BH, Clark VL, editors. The Bacteria 11, Molecular Basis of Bacterial Pathogenesis. New York: Academic Press. pp. 39–60.
- 14. Foley DA, Swartzentruber KG, Colley KJ (2009) Identification of Sequences in the Polysialyltransferases ST8Sia II and ST8Sia IV That Are Required for the Protein-specific Polysialylation of the Neural Cell Adhesion Molecule, NCAM. Journal of Biological Chemistry 284: 15505–15516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zapater JL, Colley KJ (2012) Sequences prior to conserved catalytic motifs of polysialyltransferase ST8Sia IV are required for substrate recognition. The Journal of biological chemistry 287: 6441–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, et al. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Research 37: D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vimr ER (1991) Map position and genomic organization of the kps cluster for polysialic acid synthesis in Escherichia coli K1. Journal of Bacteriology 173: 1335–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frosch M, Weisgerber C, Meyer TF (1989) Molecular characterization and expression in Escherichia coli of the gene complex encoding the polysaccharide capsule of Neisseria meningitidis group B. Proceedings of the National Academy of Sciences of the United States of America 86: 1669–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andreishcheva EN, Vann WF (2006) Gene products required for de novo synthesis of polysialic acid in Escherichia coli K1. Journal of Bacteriology 188: 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen GJ, Datta AKAK, Izumi M, Koeller KM, Wong CH, et al. (1999) Expression of alpha-2,8/2,9-polysialyltransferase from Escherichia coli K92. Characterization of the enzyme and its reaction products. Journal of Biological Chemistry 274: 35139–35146. [DOI] [PubMed] [Google Scholar]
- 21. Freiberger F, Claus H, Gunzel A, Oltmann-Norden I, Vionnet J, et al. (2007) Biochemical characterization of a Neisseria meningitidis polysialyltransferase reveals novel functional motifs in bacterial sialyltransferases. Molecular Microbiology 65: 1258–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willis LM, Gilbert M, Karwaski MF, Blanchard MC, Wakarchuk WW (2008) Characterization of the alpha-2,8-polysialyltransferase from Neisseria meningitidis with synthetic acceptors, and the development of a self-priming polysialyltransferase fusion enzyme. Glycobiology 18: 177–186. [DOI] [PubMed] [Google Scholar]
- 23. Peterson DC, Arakere G, Vionnet J, McCarthy PC, Vann WF (2011) Characterization and acceptor preference of a soluble meningococcal group C polysialyltransferase. Journal of bacteriology 193: 1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gregoriadis G, Jain S, Papaioannou I, Laing P (2005) Improving the therapeutic efficacy of peptides and proteins: a role for polysialic acids. International Journal of Pharmaceutics 300: 125–130. [DOI] [PubMed] [Google Scholar]
- 25. Wu JR, Lin Y, Zheng ZY, Lin CC, Zhan XB, et al. (2010) Improvement of the CuZn-superoxide dismutase enzyme activity and stability as a therapeutic agent by modification with polysialic acids. Biotechnology Letters 32: 1939–1945. [DOI] [PubMed] [Google Scholar]
- 26. Lindhout T, Iqbal U, Willis LM, Reid AN, Li J, et al. (2011) Site-specific enzymatic polysialylation of therapeutic proteins using bacterial enzymes. Proceedings of the National Academy of Sciences of the United States of America 108: 7397–7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barrallo S, Reglero A, Revilla-Nuin B, Martinez-Blanco H, Rodriguez-Aparicio LB, et al. (1999) Regulation of capsular polysialic acid biosynthesis by temperature in Pasteurella haemolytica A2. FEBS Letters 445: 325–328. [DOI] [PubMed] [Google Scholar]
- 28. Lawrence PK, Kittichotirat W, Bumgarner RE, McDermott JE, Herndon DR, et al. (2010) Genome sequences of Mannheimia haemolytica serotype A2: ovine and bovine isolates. Journal of bacteriology 192: 1167–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernatchez S, Gilbert M, Blanchard MC, Karwaski MF, Li J, et al. (2007) Variants of the beta-1,3-galactosyltransferase CgtB from the bacterium Campylobacter jejuni have distinct acceptor specificities. Glycobiology 17: 1333–1343. [DOI] [PubMed] [Google Scholar]
- 30. Schwarzer D, Stummeyer K, Haselhorst T, Freiberger F, Rode B, et al. (2009) Proteolytic release of the intramolecular chaperone domain confers processivity to endosialidase F. The Journal of biological chemistry 284: 9465–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Maarouf A, Moyo-Lee Yaw D, Lindhout T, Pearse DD, Wakarchuk W, et al. (2012) Enzymatic engineering of polysialic acid on cells in vitro and in vivo using a purified bacterial polysialyltransferase. The Journal of biological chemistry 287: 32770–32779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prentice HM, Moore SE, Dickson JG, Doherty P, Walsh FS (1987) Nerve growth factor-induced changes in neural cell adhesion molecule (N-CAM) in PC12 cells. The EMBO Journal 6: 1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cho J, Troy FaII (1994) Polysialic Acid Engineering: Synthesis of Polysialylated Neoglycosphingolipids by Using the Polysialyltransferase from Neuroinvasive Escherichia coli K1. Proceedings of the National Academy of Sciences of the United States of America 91: 11427–11431. [DOI] [PMC free article] [PubMed] [Google Scholar]