

Abstract
Introduction
The homeostatic intracellular repair response (HIR2) is an endogenous beneficial pathway that eliminates damaged mitochondria and dysfunctional proteins in response to stress. The underlying mechanism is adaptive autophagy. The purpose of this study was to determine whether the HIR2 response is activated in the heart in patients undergoing cardiac surgery and to assess whether it is associated with the duration of ischemic arrest and predicted surgical outcome.
Methods
Autophagy was assessed in 19 patients undergoing coronary artery bypass or valve surgery requiring cardiopulmonary bypass (CPB). Biopsies of the right atrial appendage obtained before initiation of CPB and after weaning from CPB were analyzed for autophagy by immunoblotting for LC3, Beclin-1, Atg5-12, and p62. Changes in p62, a marker of autophagic flux, were correlated with duration of ischemia and with the mortality/morbidity risk scores obtained from the Society of Thoracic Surgeons Adult Cardiac Surgery Database (v2.73).
Results
Heart surgery was associated with a robust increase in autophagic flux indicated by depletion of LC3-I, LC3-II, Beclin-1, and Atg5-12; the magnitude of change for each of these factors correlated significantly with changes in the flux marker p62. Moreover, changes in p62 correlated directly with cross clamp time and inversely with the mortality/morbidity risk scores.
Conclusion
These findings are consistent with preclinical studies indicating that HIR2 is cardioprotective, and reveal that it is activated in patients in response to myocardial ischemic stress. Strategies designed to amplify HIR2 during conditions of cardiac stress may have therapeutic utility and represent an entirely new approach to myocardial protection in patients undergoing heart surgery.
Introduction
Heart surgery patients continue to experience significant post-operative complications such as myocardial stunning and infarction. This is due in part to inadequate intraoperative myocardial protection and ischemia/reperfusion injury [1]. Myocardial stunning (reversible post ischemic myocardial dysfunction) manifests as low cardiac output and lasts hours to days after surgery. In patients with limited functional reserve, stunning can lead to multi-organ failure and death. Similarly, some degree of myocardial necrosis occurs in up to 20% of patients undergoing heart surgery with subsequent adverse short and long term adverse consequences [2-5]. Because irreversible tissue injury involves cell death, previous efforts focused on preventing apoptosis and necrosis [6]. However, attention is now focused on mechanisms to salvage cells by amplifying their endogenous protective mechanisms. One such mechanism is the homeostatic intracellular repair response (HIR2). The underlying process is adaptive autophagy, a lysosomal salvage pathway that eliminates damaged mitochondria and misfolded aggregated proteins in response to stress.
Adaptive autophagy is characterized by the formation of a cup-shaped pre-autophagosomal double-membrane structure which surrounds cytoplasmic material and closes to form the autophagosome [Fig. 1]. The outer membrane of the autophagosome fuses with a lysosome to create a single membrane structure, the autophagolysosome. Here, the cargo is degraded; amino acids, free fatty acids, and sugars are exported to the cytosol for use as metabolic substrates [7-9]. This process involves multiple autophagy (Atg) proteins. For example, activation of the class III PI3K/Vps34 and Beclin1 (Atg6) complex results in the formation of an isolation membrane to which other Atg proteins are recruited. The conjugate of Atg12-Atg5 is involved in the expansion of the isolation membrane. Another protein, p62, functions as an adaptor protein to link ubiquitin-rich mitochondria and protein aggregates to the forming autophagosome [10-12]. Autophagic activity is reflected in changes in the abundance of proteins such as Beclin-1, Atg5-12, and p62. Since p62 levels increase when autophagy is inhibited chronically, and decrease when autophagy is induced, p62 can be used as a marker to assess autophagic flux [13]. For the purpose of this study, we selected Beclin-1 as a catalytic factor that also participates in regulating lysosomal fusion, Atg5-12 as a consumable structural component, and p62 as a dynamically regulated adaptor protein for ubiquitinated cargo.
Figure 1.
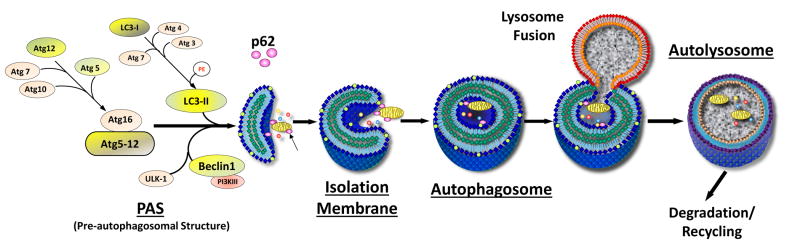
Autophagy Machinery Involved in HIR2. Initiation of autophagy involves the participation of autophagy (Atg) proteins involved in formation of the pre-autophagosomal structure and elongation of the isolation membrane. Atg12 is covalently linked to Atg5 by a ubiquitin ligase system (Atg7, Atg10). Damaged mitochondria, protein aggregates, and other cytosolic components are flagged for engulfment by the adaptor protein p62. The autophagosome fuses with a lysosome and the cargo is degraded along with many of the Atg proteins. Under conditions of rapid autophagic flux, Atg proteins may be consumed before they can be replaced by new protein synthesis.
Autophagy has been extensively investigated in cell culture and animal models subjected to stresses such as starvation or ischemia/reperfusion [9, 14-16]. To measure autophagy it is necessary to obtain tissue for Western blot analysis, immunofluorescence microscopy, or electron microscopy. For that reason, few studies have examined autophagy in the human heart [17, 18], but there is evidence that HIR2 is active in animal models [10, 19-21]. Given that inadequate myocardial protection remains a major clinical problem, the purpose of this study was to determine if HIR2 is activated in patients undergoing surgery, and whether the duration of ischemic arrest or patient risk factors affect the pathway.
Methods
Study Design
The study protocol was fully approved by the Institutional Review Boards of Wayne State University School of Medicine and Oakwood Hospital Medical Center. All tissue procurements were done in compliance with the IRB approved protocol. The transfer of human tissue between Oakwood Healthcare Inc. and the San Diego State University (SDSU) was governed by a Universal Biological Materials Transfer Agreement (UBMTA) between the two institutions. Nineteen patients undergoing conventional heart surgery with cardiopulmonary bypass (CPB) and intraoperative cardioplegia were studied. Inclusion criteria were limited to adult patients undergoing first time non-emergent cardiac surgery on CPB with aortic cross clamping. The patient population included those undergoing coronary artery bypass grafting (CABG) alone, valve repair or replacement alone, or CABG combined with valve repair or replacement. Right atrial tissue (100-200mg) was excised during atrial cannulation before initiation of CPB and aortic cross clamping (“a” samples), and again after removal of cross clamp and weaning from CPB (“b” samples). Atrial biopsies were frozen in liquid nitrogen and stored at -80° C until they were analyzed.
Biochemical Analysis
Samples were shipped on dry ice to San Diego State University, where they were processed and analyzed in three lots (“a” and “b” samples from patients 1-5, 6-12, and 13-19). For all samples, proteins were extracted by Dounce homogenization in ice-cold RIPA buffer with freshly added Protease Inhibitor Cocktail Complete (Roche) followed by centrifugation (1000g × 5min, 4°C) and recovery of supernatants and determination of protein concentrations (Bio-Rad DC Protein assay). Equal amounts of protein (20 μg) were resolved on 10-20% Tris-Glycine SDS-PAGE gels (Invitrogen) and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat dry milk for 1h and then incubated overnight with 1:1000 diluted primary antibodies against actin (Sigma), Beclin-1 (Cell Signaling), Atg5 (Santa Cruz), and p62/SQSTM1 (MBL International). Membranes were washed (3 × 10min) in 150mM NaCl buffered with 50mM Tris (pH 7.6) with 0.1% Tween-20 (TBST) at room temperature and incubated with the appropriate peroxidase-conjugated secondary antibodies (KPL, 1:2500) for 1 hour. Following washing in TBST, blots were developed with SuperSignal West Dura Extended Duration Substrate (Thermo-Pierce), and immuno-reactive bands were visualized with the Bio-Rad ChemiDoc XRS system. Captured images were quantified by densitometry using NIH Image J. Relative density values were calculated for samples run on the same blot, and to normalize between blots, index samples were included on each blot and signal intensity was normalized to those samples.
Prior work has established p62 as a surrogate marker of autophagic flux [22]. Changes in p62 protein abundance before and after CPB were calculated as the difference in the normalized densitometry values (before minus after) and recorded as the Δp62 value for each patient. Changes in Δp62 were correlated retrospectively with cross clamp time and with the predicted risk of operative mortality and morbidity obtained from the Society of Thoracic Surgeons (STS) Adult Cardiac Surgery Database (v2.73).
STS Database
The Society of Thoracic Surgeons (STS) Adult Cardiac Surgery Database (v2.73)predicted risk is based on patient demographics and clinical variables, and employs different models for different surgical procedures. Database elements were obtained by an independent data analyst who abstracted the required elements from the patient's electronic medical record or paper record and manually inserted into the database software. The software assigns a predicted risk of various events based on different STS risk models. Predicted morbidity and mortality is defined as the composite end point of death or permanent stroke, renal failure, prolonged ventilation > 24hr, deep sternal wound infection, and reoperation for any reason. Mortality is defined as occurring within the same hospitalization after the index operation even after 30d of surgery or within 30d of surgery even if discharged home, unless it was clearly unrelated to surgery.
Statistical Analysis
Paired two-tailed Student's t-test was used to analyze the changes in autophagy proteins before cross clamping and after unclamping. Linear regression was used to examine the relationship between the HIR2 response and each patient's predicted risk score obtained from the STS database. Multiple linear regression was used to adjust for potential confounding variables. Pearson correlation coefficients were calculated between autophagy proteins and patient variables. Continuous variables are expressed as mean ± standard deviation; categorical variables as percentages. A p-value of 0.05 was considered significant.
Results
Patient Characteristics
Patient demographics and clinical characteristics are shown in Table 1. Among the 19 patients, 13 underwent CABG, 5 underwent a valve procedure and 1 patient had a combined procedure (aortic valve replacement and CABG). Predicted risk for morbidity/mortality ranged from 3.6 to 26.4%. All patients were discharged from the hospital in less than 30d; one patient developed appendicitis postoperatively and required operative intervention. In this group of patients the pre-operative ejection fraction ranged from 40-65%; no patients were NYHA Class III or IV.
Table 1.
Patient Characteristics. Pertinent characteristics of the 19 patients are summarized. Values represent mean ± SD, or the number of patients with that characteristic and percentage in parentheses.
| Patient Characteristics | |
|---|---|
| Age (years) | 60 ± 12 |
| Male gender | 17 (89%) |
| Weight (kg) | 103.2 ± 22.3 |
| Smoker | 5 (26%) |
| Previous MI | 8 (42%) |
| BSA (m2) | 2.22 ± 0.25 |
| BMI (kg/m2) | 31.5 ± 5.6 |
| Diabetes mellitus | 7 (37%) |
| Dyslipidemia | 16 (84%) |
| Hypertension | 18 (95%) |
| Preoperative ejection fraction (%) | 58 ± 7 |
| CABG only | 13 (68%) |
| Valve only | 5 (26%) |
| CABG + valve | 1 (6%) |
| Cross clamp time (min) | 118 ± 30 |
| Cardiopulmonary bypass time (min) | 145 ± 35 |
| # vessels bypassed | 3.35 |
| Predicted morbidity and mortality | 11% ± 6% |
Analysis of Intraoperative Autophagy
Western blotting was performed to determine the protein abundance of actin at the two sampling times. There was no change in actin levels before and after cross clamp (data not shown), and actin was used as a loading control for all samples. Many autophagy proteins are consumed during brisk autophagic flux, including Atg5-12 and p62. We have previously observed depletion of Beclin-1 and LC3-II in the porcine heart under conditions of accelerated autophagic flux (unpublished data). Under conditions of ischemic and nutritional stress, the consumption of these factors exceeds the capacity of the cell to replace them. Thus the rate of disappearance of these factors is a reflection of the magnitude of autophagic flux. As shown in Fig. 2, Beclin-1 protein levels were higher before CPB than after (0.422±0.017 vs. 0.296±0.026 relative density units, p=0.001), indicating an increase in autophagic flux during the surgical procedure. Similar changes were observed for Atg5-12 (0.420±0.062 vs. 0.343±0.062, p=0.002) (Fig. 3). The adaptor protein p62 is important for clearance of damaged mitochondria in the heart [10] and is similarly subject to lysosomal degradation. Levels of p62 also dropped during surgery (0.233±0.022 vs. 0.128±0.011, p=0.001) (Fig. 4). Changes before and after CPB for Beclin-1, Atg5-12, LC3-I and LC3-II all showed a statistically significant correlation with Δp62 (Fig. 5), suggesting that this represented coordinated engagement of the autophagy machinery during surgery.
Figure 2.
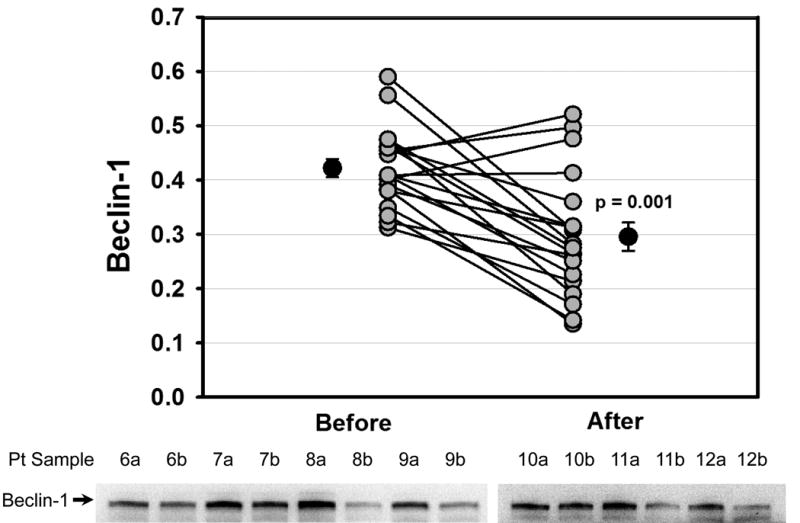
Reduced Levels of Beclin-1 during Cardiac Stress. Graph shows densitometry values for Beclin-1 from right atrial appendage biopsies taken before and after CPB for each patient. Lower panel shows representative Western blots from 7 of the 19 patients. Arrow indicates Beclin-1.
Figure 3.
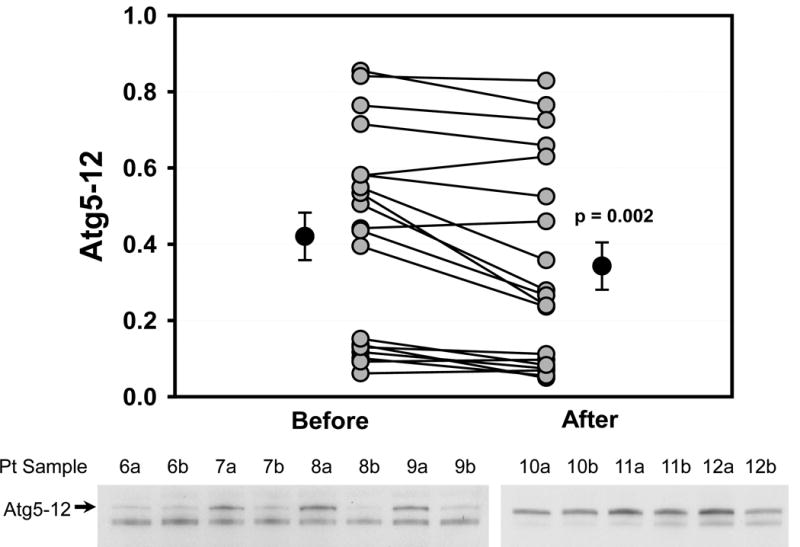
Reduced Levels of Atg5-12 during Cardiac Stress. Graph shows densitometry values for Atg5-12 from right atrial appendage biopsies taken before and after CPB for each patient. Lower panel shows representative Western blots from 7 of the 19 patients. Arrow indicates Atg5-12.
Figure 4.
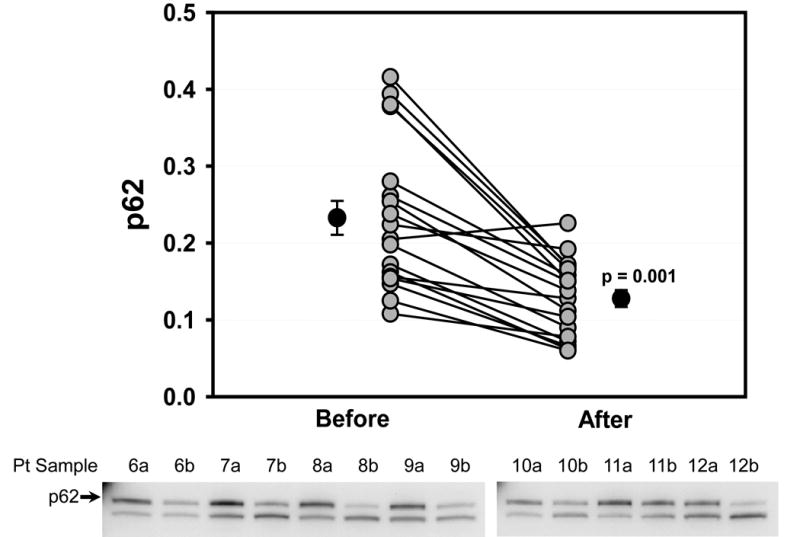
Reduced Levels of p62 during Cardiac Stress. Graph shows densitometry values for p62 from right atrial appendage biopsies taken before and after CPB for each patient. Lower panel shows representative Western blots from 7 of the 19 patients. Arrow indicates p62.
Figure 5.
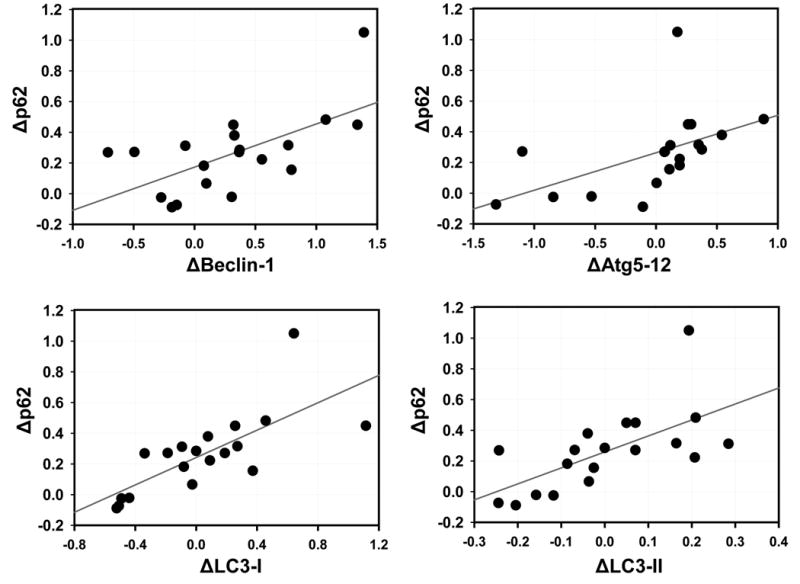
Autophagy Protein Depletion Is Coordinated. Each graph shows the change in the indicated autophagy protein (e.g. ΔBeclin-1) plotted against the change in p62 (Δp62) for each patient.
To assess whether the decrease in p62 was time-dependent, we performed a linear regression analysis of cross clamp time versus Δp62 (Fig. 6) (model r2=0.1808, p=0.0696). Because the Δp62 value is calculated from the baseline and post-operative p62 values, we then performed a multivariate analysis of post-operative p62 versus cross clamp time, after adjusting for the baseline p62. In this multivariate analysis (model r2=0.3630), post-operative p62 was independently associated with both cross clamp time (p=0.0310) and the baseline p62 value (p=0.0396). Taken together, these results suggest that autophagic flux is upregulated in the heart in response to the stress of intraoperative ischemia.
Figure 6.

Depletion of p62 as a Function of Cross Clamp Time. Plot shows linear regression analysis of Δp62 vs. cross clamp time (p=0.0696). Each point corresponds to an individual patient. With multivariate analysis to take the starting level of p62 into account, there was a statistically significant association between the second p62 level and cross clamp time (r2 =0.3785; p=0.0384).
Preclinical studies have shown that autophagy plays a protective role in the setting of myocardial ischemia and reperfusion [10, 21, 23-25]. As the patients in this study exhibited variable degrees of autophagic flux, we wished to determine whether this flux might have a bearing on the patient's ability to tolerate the stress. In the absence of significant morbidity in this small patient series, we focused on the on the predicted risk of operative morbidity and mortality based on the STS Database models. As shown in Fig. 7, there is an inverse correlation between the magnitude of autophagic flux (based on Δp62) and the predicted risk (model r2=0.2572, p=0.0267). Because cross clamp time predicts Δp62 it is a potential confounding variable. Therefore, we performed a multivariate analysis to adjust for cross clamp time in the Δp62 versus risk score relationship. In this multivariate analysis (model r2=0.3785), Δp62 remained independently associated with predicted risk (p=0.0384).
Figure 7.
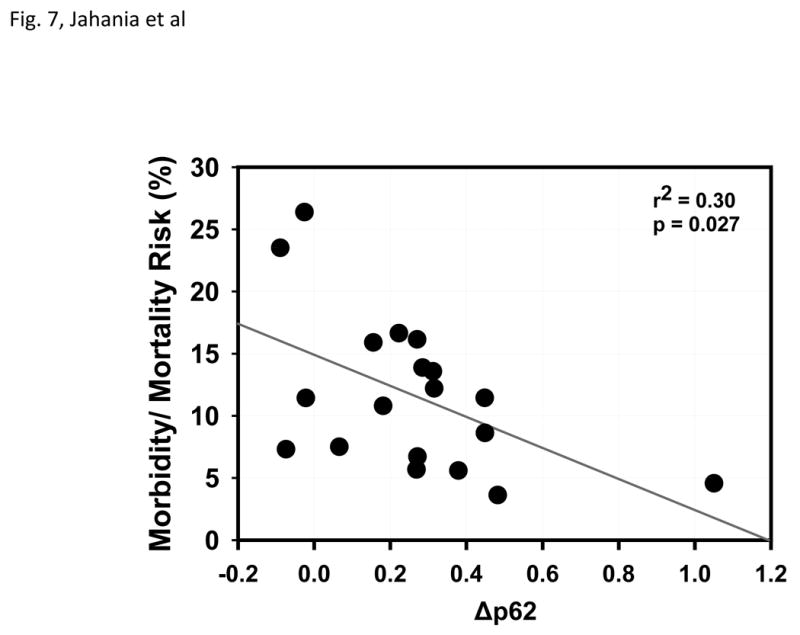
Operative Risk Correlates with Changes in Autophagy. Plot shows linear regression of morbidity and mortality risk score versus Δp62. Each point represents an individual patient.
Discussion
The results show that autophagy protein levels diminish, reflecting an increase in autophagic flux during cardiac surgery. All factors measured in this study (LC3-I, LC3-II, Beclin-1, Atg5-12, and p62) decreased during CPB, and the magnitude of change for each of these factors correlated significantly with Δp62. Moreover, the change in p62 correlated significantly with cross clamp time, suggesting that this pathway is ongoing during ischemic arrest and cold cardioplegia. Interestingly, Δp62 correlated significantly with the STS Database risk score, even after accounting for cross clamp time. This suggests that the patient characteristics that contribute to statistical risk (including features of metabolic syndrome and age) may also affect autophagy.
While the clearance of p62 has become widely accepted as an index of autophagic flux [11, 26, 27], p62 can be upregulated within minutes in response to a stimulus (e.g., ischemic preconditioning) [23]. Levels then decline as a result of lysosomal degradation of p62 and its cargo of damaged mitochondria and ubiquitinated protein aggregates [10]. In these heart samples, autophagy proteins including Beclin-1, Atg5-12, and LC3-II, were degraded in parallel with p62, consistent with accelerated flux. An increase in LC3-II is often interpreted as increased autophagy, but a low LC3-II level may be due to minimal induction of autophagy or accelerated flux and inadequate replacement. Interpretation of dynamic protein changes in the heart undergoing ischemic arrest and cold cardioplegia is complex. To simplify interpretation, we have made two assumptions: (1) the decrease in autophagy proteins during surgery is solely due to lysosomal degradation (not proteasomal), and (2) the magnitude of the decrease is not confounded by new protein synthesis. Proteasome function is impaired during myocardial ischemia and reperfusion [28]. We did not measure mRNA and cannot comment on new protein synthesis. With these caveats, we conclude that autophagy is activated and that the magnitude of change is a reflection of autophagic flux.
There is little information about autophagy in the human heart. Kassiotis et al. showed that in biopsies from left ventricles of failing hearts, mRNA and protein levels of autophagy were downregulated after LVAD support [17]. In biopsies of right atrial appendages obtained at the time of CABG surgery, Garcia et al. reported evidence of impaired autophagic flux in patients who developed postoperative atrial fibrillation [18]. Both groups concluded that autophagy was an adaptive response in the stressed heart. In our study, autophagy was measured in the right atrial appendage. Whether this parallels autophagy in the left ventricle is unknown.
We suggest that adaptive autophagy is the underlying mechanism of the homeostatic intracellular repair response, which is elicited by cellular challenges such as nutrient limitation, transient ischemia, and endoplasmic reticulum stress. Removal of damaged organelles and misfolded proteins via autophagy is required for preservation of ATP production and myocardial contractility [30-33], and to prevent apoptotic or necrotic cell death. In a study of pigs with hibernating myocardium induced by chronic ischemia, autophagosomes were absent from apoptotic cells whereas viable cells appeared to upregulate autophagy [34].
Autophagy is suppressed by insulin signaling, hyperlipidemia, hypercholesterolemia, and amino acids, and has been reported to be impaired in animal models of obesity and metabolic syndrome [35-38]. Impairment of HIR2 may account for the increased vulnerability to ischemic injury in patients with obesity and metabolic syndrome. Similarly, autophagy diminishes with age [39-41]. Presence of these comorbid conditions may limit the patient's ability to upregulate HIR2 in response to an ischemic challenge [42]. This leads us to consider whether it is possible to amplify HIR2 in the most vulnerable patients. A growing number of cardioprotective drugs have been reported to induce autophagy in the heart, including adenosine, diazoxide, ranolazine, rapamycin, sildenafil, chloramphenicol, and statins, but have not been tested in settings that mimic the comorbidities of cardiac surgery patients [23][19, 24, 43, 44]. In this study, all 19 patients were on statin therapy, which is known to induce autophagy [45] and which could have enhanced their ability to tolerate ischemia [46, 47].
Our findings show that the human heart responds to ischemic stress by activating HIR2, a pathway that may be compromised by comorbid conditions. Amplifying HIR2 during cardiac surgery could mitigate the operative risk in vulnerable patients and would represent an entirely new approach to intraoperative myocardial protection.
Footnotes
Presented at the Southern Surgical Association, 124th Annual Meeting, Palm Beach, FL, 2012
The research was funded by NHLBI 2R01 HL034579-25 (RA Gottlieb and RM Mentzer, Jr.) and NHLBI 2R01 HL060590-15 (RA Gottlieb).
References
- 1.Bayat H, Swaney JS, Ander AN, Dalton N, Kennedy BP, Hammond HK, et al. Progressive heart failure after myocardial infarction in mice. Basic Res Cardiol. 2002;97:206–13. doi: 10.1007/s003950200013. [DOI] [PubMed] [Google Scholar]
- 2.Werner ME, Meredith AL, Aldrich RW, Nelson MT. Hypercontractility and impaired sildenafil relaxations in the BKCa channel deletion model of erectile dysfunction. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2008;295:R181–R8. doi: 10.1152/ajpregu.00173.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao CM, Xia Q, Gao Q, Chen M, Wong TM. Calcium-Activated Potassium Channel Triggers Cardioprotection of Ischemic Preconditioning. Journal of Pharmacology and Experimental Therapeutics. 2005;312:644–50. doi: 10.1124/jpet.104.074476. [DOI] [PubMed] [Google Scholar]
- 4.Cushman M, McClure LA, Lakoski SG, Jenny NS. Eligibility for statin therapy by the JUPITER trial criteria and subsequent mortality. Am J Cardiol. 2010;105:77–81. doi: 10.1016/j.amjcard.2009.08.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salloum FN, Takenoshita Y, Ockaili RA, Daoud VP, Chou E, Yoshida Ki, et al. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial KATP channels when administered at reperfusion following ischemia in rabbits. Journal of Molecular and Cellular Cardiology. 2007;42:453–8. doi: 10.1016/j.yjmcc.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Botha P, MacGowan GA, Dark JH. Sildenafil citrate augments myocardial protection in heart transplantation. Transplantation. 2010;89:169–77. doi: 10.1097/TP.0b013e3181c42b22. [DOI] [PubMed] [Google Scholar]
- 7.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore–dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. The Journal of Clinical Investigation. 2010;120:3680–7. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–61. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravingerova T, Adameova A, Kelly T, Antonopoulou E, Pancza D, Ondrejcakova M, et al. Changes in PPAR gene expression and myocardial tolerance to ischaemia: relevance to pleiotropic effects of statins. Can J Physiol Pharmacol. 2009;87:1028–36. doi: 10.1139/Y09-071. [DOI] [PubMed] [Google Scholar]
- 10.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 12.Chen YP, Xiang XJ, Min S. Effect of ranolazine postconditioning on the reperfusion injury salvage kinase and mitochondrial permeability transition pore against ischemia-reperfusion injury in isolated rat heart. Chinese Pharmacological Bulletin. 2010 [Google Scholar]
- 13.Vilahur G, Casani L, Pena E, Duran X, Juan-Babot O, Badimon L. Induction of RISK by HMG-CoA reductase inhibition affords cardioprotection after myocardial infarction. Atherosclerosis. 2009;206:95–101. doi: 10.1016/j.atherosclerosis.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Hamacher-Brady A, Brady NR, Gottlieb RA, Gustafsson AB. Autophagy as a protective response to Bnip3-mediated apoptotic signaling in the heart. Autophagy. 2006;2:307–9. doi: 10.4161/auto.2947. [DOI] [PubMed] [Google Scholar]
- 15.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–87. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 16.Przyklenk K, Whittaker P, Wider J, Undyala VV, Gottlieb RA, Mentzer RM. Metabolic Support of Mitochondrial Complex I with Yeast Ndi1 Evokes Significant Cardioprotection in the in vivo Rat. Model of Acute Myocardial Infarction Circulations. 2011;124:A17970. [Google Scholar]
- 17.Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, et al. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. The Journal of Clinical Investigation. 2004;114:837–45. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pateliya BB, Singh N, Jaggi AS. Possible Role of Opioids and KATP Channels in Neuroprotective Effect of Postconditioning in Mice. Biological and Pharmaceutical Bulletin. 2008;31:1755–60. doi: 10.1248/bpb.31.1755. [DOI] [PubMed] [Google Scholar]
- 19.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Jr, et al. Autophagy Induced by Ischemic Preconditioning is Essential for Cardioprotection. J Cardiovasc Transl Res. 2010 doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr, et al. Profound cardioprotection with chloramphenicol succinate in the swine model of ischemia-reperfusion injury. Circulation. 2010;122 doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy. 2009;5:709–12. doi: 10.4161/auto.5.5.8510. [DOI] [PubMed] [Google Scholar]
- 22.Kim SH, Lu HF, Alano CC. Neuronal Sirt3 Protects against Excitotoxic Injury in Mouse Cortical Neuron Culture. PLoS One. 2011;6:e14731. doi: 10.1371/journal.pone.0014731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Jr, et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010;3:365–73. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RM, Jr, et al. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol. 2009;104:157–67. doi: 10.1007/s00395-009-0006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, et al. Impaired Autophagosome Clearance Contributes to Cardiomyocyte Death in Ischemia-Reperfusion Injury. Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy. 2011;7:572–83. doi: 10.4161/auto.7.6.14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Kem DC. Proteasome inhibition during myocardial infarction. Cardiovasc Res. 2010;85:312–20. doi: 10.1093/cvr/cvp309. [DOI] [PubMed] [Google Scholar]
- 29.Ludman A, Venugopal V, Yellon DM, Hausenloy DJ. Statins and cardioprotection--more than just lipid lowering? Pharmacol Ther. 2009;122:30–43. doi: 10.1016/j.pharmthera.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochimica et Biophysica Acta (BBA) - Bioenergetics. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, et al. Autophagy and Protein Kinase C Are Required for Cardioprotection by Sulfaphenazole. Am J Physiol Heart Circ Physiol. 2009 doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. The Journal of Clinical Investigation. 2007;117:2825–33. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, et al. Functional Significance and Morphological Characterization of Starvation-Induced Autophagy in the Adult Heart. Am J Pathol. 2009;174:1705–14. doi: 10.2353/ajpath.2009.080875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–12. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javadov S, Purdham DM, Zeidan A, Karmazyn M. NHE-1 inhibition improves cardiac mitochondrial function through regulation of mitochondrial biogenesis during postinfarction remodeling. American Journal of Physiology - Heart and Circulatory Physiology. 2006;291:H1722–H30. doi: 10.1152/ajpheart.00159.2006. [DOI] [PubMed] [Google Scholar]
- 36.Fretwell L, Dickenson JM. Role of large-conductance Ca2+-activated K+ channels in adenosine A1 receptor-mediated pharmacological postconditioning in H9c2 cells. Canadian Journal of Physiology and Pharmacology. 2011;89:24–30. doi: 10.1139/y10-106. [DOI] [PubMed] [Google Scholar]
- 37.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Headrick JP, Lasley RD. Adenosine receptors and reperfusion injury of the heart. Handb Exp Pharmacol. 2009:189–214. doi: 10.1007/978-3-540-89615-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nair S, Ren J. Autophagy and cardiovascular aging: lesson learned from rapamycin. Cell Cycle. 2012;11:2092–9. doi: 10.4161/cc.20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prasad H, Ryan DA, Celzo MF, Stapleton D. Metabolic syndrome: definition and therapeutic implications. Postgrad Med. 2012;124:21–30. doi: 10.3810/pgm.2012.01.2514. [DOI] [PubMed] [Google Scholar]
- 41.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiological Reviews. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 42.Ferdinandy P, Schulz R, Baxter GF. Interaction of Cardiovascular Risk Factors with Myocardial Ischemia/Reperfusion Injury, Preconditioning, and Postconditioning. Pharmacological Reviews. 2007;59:418–58. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 43.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–64. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 44.D'Annunzio V, Donato M, Erni L, Miksztowicz V, Buchholz B, Carrion CL, et al. Rosuvastatin given during reperfusion decreases infarct size and inhibits matrix metalloproteinase-2 activity in normocholesterolemic and hypercholesterolemic rabbits. J Cardiovasc Pharmacol. 2009;53:137–44. doi: 10.1097/FJC.0b013e318197c5e9. [DOI] [PubMed] [Google Scholar]
- 45.Araki M, Motojima K. Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem Biophys Res Commun. 2008;367:462–7. doi: 10.1016/j.bbrc.2007.12.166. [DOI] [PubMed] [Google Scholar]
- 46.Kocsis GF, Pipis J, Fekete V, Kovacs-Simon A, Odendaal L, Molnar E, et al. Lovastatin interferes with the infarct size-limiting effect of ischemic preconditioning and postconditioning in rat hearts. Am J Physiol Heart Circ Physiol. 2008;294:H2406–9. doi: 10.1152/ajpheart.00862.2007. [DOI] [PubMed] [Google Scholar]
- 47.Ludman AJ, Hausenloy DJ, Babu G, Hasleton J, Venugopal V, Boston-Griffiths E, et al. Failure to recapture cardioprotection with high-dose atorvastatin in coronary artery bypass surgery: a randomised controlled trial. Basic Res Cardiol. 2011;106:1387–95. doi: 10.1007/s00395-011-0209-5. [DOI] [PubMed] [Google Scholar]