

Abstract
As a non-ruminant herbivore, the white rhinoceros has the ability to utilize fibrous plant matter through microbial fermentation in the hindgut. So far, there has been no report using molecular techniques to study the gut microbiota of the white rhinoceros. We used barcoded pyrosequencing to characterize 105,651 sequences of 16S rRNA genes obtained from fecal samples from five white rhinoceroses. Results showed that Firmicutes and Bacteroidetes were the predominant phyla in the samples, which were comprised largely of unclassified bacteria. The microbiota of one animal treated with drug therapy differed from those in other healthy animals, and was dominated by Aerococcus -related bacteria. The core microbiota in the healthy rhinoceros were dominated by phyla Firmicutes and Bacteroidetes, represented by the Ruminococcaceae, Lachnospiraceae, Rikenellaceae and Prevotellaceae families. The present work provides a phylogenetic framework for understanding the complex microbial community of the rhinoceros; however, further studies are required to link the distinctive microbiota with their digestive role in the hindgut of the white rhinoceros.
Introduction
The rhinoceros is one of five surviving species of odd-toed ungulates in the Rhinocerotidae family. The five different species of rhinoceros include two African species, the white rhinoceros (Ceratotherium simum) and the black rhinoceros (Diceros bicornis), and three Asian species, Indian rhinoceros (Rhinoceros unicornis), Sumatran rhinoceros (Dicerorhinus sumatrensis), and Javan rhinoceros (Rhinoceros sondaicus). Relative to the four other species which are in the list of endangered wild animals, the white rhinoceros is classed as vulnerable, with roughly 16,000 remaining in the wild in 2007 (IUCN 2008).
The white rhinoceros is, after the elephant, the largest extant mammalian herbivore [1]. As a hindgut fermenter, the white rhinoceros has the ability to utilize fibrous plant matter through microbial fermentation in the hindgut. Comparative studies among non-ruminant herbivores showed that the rhinoceros had a similar digestive system to horses and elephants [2], [3]. Costa et al. found that Firmicutes predominated (68%) in the feces of healthy horses, followed by Bacteroidetes (14%) and Proteobacteria (10%) [4]. At the genus level, previous studies showed that cellulose-digesting microflora (e.g., Ruminococcus and Fibrobacter species) were important members of the microbial community in the rumen or the hindgut of non-ruminant herbivores [5], [6], which enabled the host to gain nutrients from fibrous plant materials. However, information on microbial diversity in the hindgut of the white rhinoceros remains limited. To our knowledge, there has been no report using molecular techniques to study microbial flora in the feces of white rhinoceros.
As a specialized grazing species (focusing on leaves and grass), the white rhinoceros is able to eat plants that are toxic to other animals. To understand whether this animal has distinctive gut microbiota, and whether the tolerance of the white rhinoceros to toxicants is related to gut microbiota, comprehensive analysis of the bacterial community is required. The development of high throughput sequencing has led to a revolution in the characterization of complex microbial populations [4], [7], [8]. Thus, the aim of this study was to investigate the microbial community in the feces of white rhinoceroses using the high throughput pyrosequencing analysis.
Results
Across all five samples, 105,651 quality sequences from 116,208 reads were classified as bacteria. The average length of quality sequences was 482 bp. The total number of sequences, coverage, the number of OTUs, and statistical estimates of species richness for 16,929-sequence subsets from each sample at a genetic distance of 3% are presented in Table 1. The rarefaction curves generated by MOTHUR plotting the number of reads by the number of OTUs tended to approach the saturation plateau (Figure 1). Libshuff analysis indicated that differences in the bacterial community structure between the library of X1 and libraries of other animals were significant (P<0.001).
Table 1. Phylotype coverage and diversity estimation of the 16S rRNA gene libraries of the feces of rhinoceroses from the pyrosequencing analysis1.
| Rhinoceros | Reads | OTUs | ACE | Chao | Shannon | Simpson | coverage |
| X1 | 16,929 | 686 | 861.4 | 903.4 | 4.649 | 0.0355 | 0.9890 |
| X2 | 16,929 | 636 | 760.5 | 786.0 | 4.762 | 0.0284 | 0.9912 |
| X3 | 16,929 | 765 | 946.4 | 1033.7 | 5.254 | 0.0130 | 0.9885 |
| X4 | 16,929 | 762 | 890.5 | 946.7 | 5.300 | 0.0131 | 0.9904 |
| X5 | 16,929 | 757 | 901.9 | 917.6 | 5.282 | 0.0122 | 0.9900 |
The operational taxonomic units (OTUs) were defined with 3% dissimilarity. The coverage percentages, richness estimators (ACE and Chao), and diversity indices (Shannon and Simpson) were calculated.
Figure 1. Rarefaction curves.
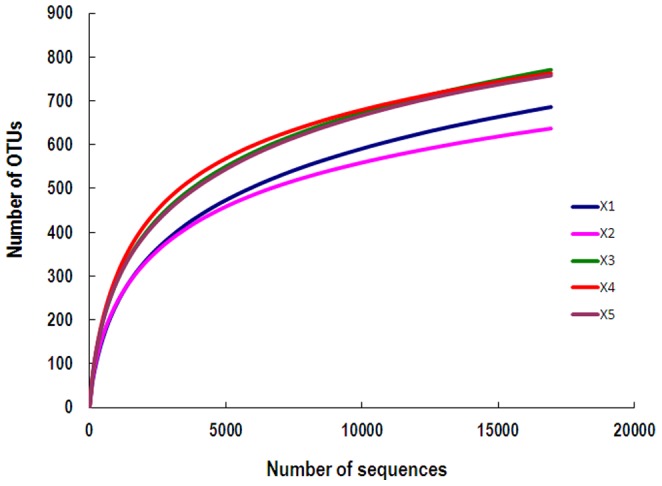
Rarefaction curves comparing the number of reads with the number of phylotypes found in the DNA in the feces of five rhinoceroses.
Taxonomic Composition
A total of 16 prokaryotic phyla were identified from the 16S rRNA gene sequences (Figure 2). In the feces of five rhinoceroses, Firmicutes were predominant, represented by 49.48%-72.52% of 16S rRNA gene sequences. Bacteroidetes was the second most abundant phylum at 18.18%-43.83%. These two phyla were more than 90% of the total sequences in all five animals. A high relative abundance of Actinobacteria (4.10%) was found in rhinoceros X1 compared to the other four animals (lower than 0.7%). In contrast, rhinoceros X1 had a lower abundance of Bacteroidetes (18.18%) in its feces than the other four animals (33.05%-43.83%).
Figure 2. Fecal bacterial community at the phylum level.

Relative abundance of bacterial groups (phylum level) in the feces of five white rhinoceroses.
At the family level, the abundances of unclassified bacteria in the samples from X1, X2, X3, X4 and X5 were 42.23%, 14.56%, 21.66%, 21.01% and 18.10%, respectively (Figure 3). In the feces of X1, Aerococcaceae was predominant with the abundance of 17.10%, followed by Lachnospiraceae and Ruminococcaceae and unclassified Lactobacillales. In the feces of other four healthy animals, the most abundant families were Ruminococcaceae, Lachnospiraceae, Rikenellaceae, Prevotellaceae and unclassified Bacteroidales, which made up approximantely 65% (59.70% to 72.22%) of total sequences.
Figure 3. Fecal bacterial community at the family level.

Relative abundance of bacterial groups (family level) in the feces of five white rhimoceroses.
At the genus level, 57.94% of total reads classified as bacteria in the feces of rhinoceros X1 were unclassified, while the abundances of unclassified bacteria in the samples from X2, X3, X4, and X5 were higher, approximantely 80% (75.40% to 84.78%) (Figure 4). In the feces of X1, genus Aerococcus was predominant with the abundance higher than 17%, followed by unclassified Lachnospiraceae, unclassified Ruminococcaceae, and unclassified Lactobacillales. In the feces of X2, the most abundant genera were unclassified Ruminococcaceae, unclassified Lachnospiraceae, RC9 gut group, unclassified Prevotellaceae, unclassified Bacteroidales, and unclassified Rikenellaceae, which made up 68.9% of total sequences. Unclassified Ruminococcaceae, unclassified Lachnospiraceae, and unclassified Bacteroidales were the three most predominant groups in the feces of X3, X4, and X5. Genera Corynebacterium and Ruminobacter were only observed in samples of X1 with abundances of 1.04% and 0.93% of total bacteria, respectively. The abundances of genera Aerococcus, Corynebacterium, and Weissella in the feces of X1 were more than 200 times higher than those of other animals. Clustered heatmap analysis based on the bacterial community profiles at the genus level disclosed that samples from animals X3, X4 and X5 were grouped together with a similarity higher than 70%, while X1 was outlier from the other four animals (Figure 5).
Figure 4. Fecal bacterial community at the genus level.

Relative abundance of bacterial groups (genus level) in the feces of five white rhinoceroses.
Figure 5. Bacterial distribution among the five samples.

Double dendrogram showing the bacterial distribution among the fecal samples of five rhinoceroses. The bacterial phylogenetic tree was calculated using the neighbor-joining method, and the relationship among samples was determined using Bray distance and the complete clustering method. Total 50 genera with the abundance higher than 0.1% within total bacteria were sorted for the analysis. The heatmap plot depicts the relative percentage of each bacterial genus (variables clustering on the Y-axis) within each sample (X-axis clustering). The relative values for the bacterial genus are depicted by color intensity in the legend indicated at the top of the figure. Clusters based on the distance of the five samples along the X-axis and the bacterial genera along the Y-axis are indicated at the top and bottom of the figure, respectively.
As shown in Table 2, Aerococcus- viridans related OTU dominated in the X1 library with the abundance of 15.24%. Lactobacillales and Bacillales-related OTUs, which were only found in the X1 sample, represented 8.08% and 3.30% of sequences in the X1 library. Prevotellaceae, Ruminococcaceae, Lachnospiraceae, Bacteroidales and Clostridium-related OTUs predominated in all five libraries.
Table 2. Relative abundance of predominant OTUs (percentage) in the feces of five African white rhinoceroses1.
| OTUs | Rhinoceros | Annotation2 | ||||
| X1 | X2 | X3 | X4 | X5 | ||
| OTU1 | 1.365 | 7.591 | 4.495 | 4.141 | 5.718 | f: Prevotellaceae |
| OTU3 | 1.819 | 11.944 | 1.932 | 0.898 | 0.904 | g: RC9_gut_group |
| OTU4 | 3.763 | 1.979 | 3.544 | 4.519 | 2.168 | g: Clostridium |
| OTU2 | 15.240 | 0.012 | 0.000 | 0.000 | 0.012 | s: Aerococcus viridans |
| OTU8 | 0.662 | 0.189 | 5.464 | 2.416 | 2.936 | g: Bacteroides |
| OTU6 | 1.500 | 0.703 | 2.197 | 5.216 | 1.069 | o: Bacteroidales |
| OTU12 | 2.339 | 2.688 | 1.802 | 0.768 | 2.268 | o: Bacteroidales |
| OTU9 | 0.000 | 3.704 | 1.170 | 0.012 | 3.905 | f: Lachnospiraceae |
| OTU13 | 0.721 | 0.106 | 1.223 | 2.889 | 2.776 | f: Christensenellaceae |
| OTU5 | 0.112 | 1.465 | 2.127 | 0.490 | 2.570 | f: Ruminococcaceae |
| OTU10 | 0.219 | 1.181 | 1.022 | 2.505 | 1.506 | f: Rikenellaceae |
| OTU23 | 0.000 | 0.295 | 1.459 | 2.989 | 1.394 | o: Bacteroidales |
| OTU19 | 0.006 | 1.802 | 1.559 | 2.132 | 0.419 | f: Ruminococcaceae |
| OTU11 | 0.000 | 0.461 | 2.865 | 0.000 | 2.056 | p: Bacteroidetes |
| OTU14 | 5.381 | 0.000 | 0.000 | 0.000 | 0.000 | o: Lactobacillales |
| OTU15 | 0.254 | 3.237 | 0.916 | 0.059 | 0.862 | f: Lachnospiraceae |
| OTU49 | 2.262 | 0.502 | 0.089 | 1.175 | 1.235 | p: Bacteroidetes |
| OTU18 | 0.106 | 0.892 | 2.121 | 0.603 | 1.081 | f: Ruminococcaceae |
| OTU55 | 0.425 | 0.656 | 1.412 | 0.939 | 1.270 | f: Ruminococcaceae |
| OTU7 | 0.366 | 0.431 | 0.951 | 1.967 | 0.981 | f: Prevotellaceae |
| OTU20 | 0.000 | 3.343 | 0.821 | 0.000 | 0.484 | p: Bacteroidetes |
| OTU16 | 0.354 | 0.679 | 0.715 | 1.388 | 1.370 | f: Ruminococcaceae |
| OTU25 | 1.760 | 0.242 | 0.443 | 1.240 | 0.443 | f: Ruminococcaceae |
| OTU91 | 0.969 | 0.478 | 0.650 | 1.370 | 0.626 | f: Lachnospiraceae |
| OTU17 | 0.035 | 0.041 | 2.446 | 0.006 | 1.441 | f: Ruminococcaceae |
| OTU30 | 0.437 | 1.743 | 0.154 | 0.396 | 0.927 | o: Bacteroidales |
| OTU21 | 3.219 | 0.012 | 0.071 | 0.089 | 0.106 | g: Streptococcus |
| OTU22 | 2.097 | 0.089 | 0.083 | 0.673 | 0.461 | f: Lachnospiraceae |
| OTU62 | 3.302 | 0.000 | 0.000 | 0.000 | 0.000 | o: Bacillales |
| OTU24 | 1.311 | 0.750 | 0.230 | 0.744 | 0.089 | f: Prevotellaceae |
| OTU85 | 0.012 | 1.270 | 0.809 | 0.071 | 0.727 | g: Acidaminococcus |
| OTU42 | 2.694 | 0.000 | 0.000 | 0.000 | 0.000 | o: Lactobacillales |
| OTU31 | 0.289 | 0.413 | 0.650 | 0.727 | 0.555 | f: Christensenellaceae |
| OTU44 | 0.000 | 1.382 | 0.508 | 0.006 | 0.738 | o: Clostridiales |
| OTU67 | 2.629 | 0.000 | 0.000 | 0.000 | 0.006 | s: Weissella salipiscis |
| OTU28 | 0.030 | 0.691 | 0.496 | 0.951 | 0.431 | f: Lachnospiraceae |
| OTU27 | 0.313 | 0.419 | 0.449 | 0.591 | 0.792 | g: Prevotella |
Thirty-seven OTUs with abundances higher than 0.5% in the microbial community were sorted from a total of 1409 OTUs, and defined as predominant OTUs.
The consensus sequence of each OTU was annotated to the closest lineage using the MOTHUR program against the SILVA 16S rRNA reference database. s: = species; g: = genus; f: = family; o: = order; c: = class; p: = phylum.
Core Fecal Microbiota
The bacterial species in the feces of five rhinoceroses were further investigated for the presence of core gut microbiota (Table 3). The five libraries had 266 OTUs in common, which comprised 46.61%, 72.19%, 67.60%, 73.85%, and 68.16% of reads in X1, X2, X3, X4, and X5 libraries, respectively. Firmicutes and Bacteroidetes dominated in the shared OTUs, as well as the reads of shared OTUs. Of the total 1409 OTUs, the four libraries from healthy animals X2, X3, X4, and X5 had 350 OTUs in common, which comprised 85.60%, 78.40%, 83.55% and 82.34% of reads in each library, respectively (Table 4). The core microbiota were dominated by phyla Firmicutes and Bacteroidetes, represented by the Ruminococcaceae, Lachnospiraceae, Rikenellaceae and Prevotellaceae families (Table 5). In addition, 176 OTUs, which were only observed in the X1 library, were further analyzed. These X1-specific OTUs comprised 21.32% of total reads in the X1 library, and were dominated by Firmicutes and Actinobacteria phyla.
Table 3. Shared phyla among the 16S rRNA gene libraries from five rhinoceroses.
| Phylum | Shared OTUs | Reads of shared OTUs | Reads of shared OTUs/Total reads (%) | ||||||||
| X1 | X2 | X3 | X4 | X5 | X1 | X2 | X3 | X4 | X5 | ||
| Actinobacteria | 4 | 50 | 36 | 30 | 29 | 31 | 0.30 | 0.21 | 0.18 | 0.17 | 0.18 |
| Bacteroidetes | 55 | 2,742 | 5,776 | 4,230 | 5,024 | 4,386 | 16.20 | 34.12 | 24.99 | 29.68 | 5.91 |
| Chloroflexi | 2 | 134 | 110 | 93 | 121 | 84 | 0.79 | 0.65 | 0.55 | 0.71 | 0.50 |
| Firmicutes | 189 | 4,745 | 5,986 | 6,899 | 6,918 | 6,884 | 28.03 | 5.36 | 40.75 | 40.86 | 0.66 |
| Lentisphaerae | 4 | 86 | 57 | 85 | 89 | 50 | 0.51 | 0.34 | 0.50 | 0.53 | 0.30 |
| Planctomycetes | 1 | 49 | 11 | 32 | 19 | 21 | 0.29 | 0.06 | 0.19 | 0.11 | 0.12 |
| Proteobacteria | 1 | 14 | 132 | 7 | 158 | 14 | 0.08 | 0.78 | 0.04 | 0.93 | 0.08 |
| Spirochaetes | 2 | 3 | 3 | 7 | 20 | 10 | 0.02 | 0.02 | 0.04 | 0.12 | 0.06 |
| Tenericutes | 1 | 8 | 50 | 11 | 8 | 15 | 0.05 | 0.30 | 0.06 | 0.05 | 0.09 |
| Unclassified | 7 | 60 | 60 | 50 | 116 | 43 | 0.35 | 0.35 | 0.30 | 0.69 | 0.25 |
| Total shared sequences | 266 | 7,891 | 12,221 | 11,444 | 12,502 | 11,538 | 46.61 | 72.19 | 67.60 | 73.85 | 68.16 |
Table 4. Shared phyla among the 16S rRNA gene libraries from four healthy rhinoceroses1.
| Phylum | Shared OTUs | Reads of shared OTUs | Reads of shared OTUs/Total reads | ||||||
| X2 | X3 | X4 | X5 | X2 | X3 | X4 | X5 | ||
| Actinobacteria | 7 | 59 | 44 | 48 | 57 | 0.35 | 0.26 | 0.28 | 0.34 |
| Bacteroidetes | 66 | 6,062 | 4,709 | 5,798 | 4,890 | 35.81 | 27.82 | 34.25 | 28.89 |
| Candidate_division_TM7 | 1 | 10 | 3 | 2 | 5 | 0.06 | 0.02 | 0.01 | 0.03 |
| Chloroflexi | 2 | 110 | 93 | 121 | 84 | 0.65 | 0.55 | 0.71 | 0.50 |
| Firmicutes | 241 | 7,633 | 8,033 | 7,483 | 8,562 | 45.09 | 47.45 | 44.20 | 50.58 |
| Lentisphaerae | 10 | 294 | 206 | 249 | 128 | 1.74 | 1.22 | 1.47 | 0.76 |
| Planctomycetes | 1 | 11 | 32 | 19 | 21 | 0.06 | 0.19 | 0.11 | 0.12 |
| Proteobacteria | 4 | 142 | 20 | 176 | 18 | 0.84 | 0.12 | 1.04 | 0.11 |
| Spirochaetes | 4 | 16 | 10 | 31 | 14 | 0.09 | 0.06 | 0.18 | 0.08 |
| Tenericutes | 3 | 75 | 52 | 30 | 102 | 0.44 | 0.31 | 0.18 | 0.60 |
| Unclassified | 11 | 79 | 71 | 188 | 58 | 0.47 | 0.42 | 1.11 | 0.34 |
| Total shared sequences | 350 | 14,491 | 13,273 | 14,145 | 13,939 | 85.60 | 78.40 | 83.55 | 82.34 |
The phyla in bold letters represent core fecal microbiota.
Table 5. The predominant core microbiota (family level) in the samples from four healthy rhinoceroses1.
| Family | Shared OTUs | Reads of shared OTUs | Reads of shared OTUs/Total reads | ||||||
| X2 | X3 | X4 | X5 | X2 | X3 | X4 | X5 | ||
| Lachnospiraceae | 55 | 2,548 | 1,466 | 1,495 | 2,294 | 15.05 | 8.66 | 8.83 | 13.55 |
| Prevotellaceae | 7 | 1,626 | 1,128 | 1,348 | 1,391 | 9.06 | 6.66 | 7.96 | 8.22 |
| Rikenellaceae | 36 | 3,180 | 1,311 | 1,584 | 1,312 | 18.78 | 7.74 | 9.36 | 7.75 |
| Ruminococcaceae | 90 | 2,667 | 3,739 | 2,918 | 3,271 | 15.75 | 22.09 | 17.24 | 19.32 |
| Total shared sequences | 188 | 10,021 | 7,644 | 7,345 | 8,268 | 59.19 | 45.15 | 43.39 | 48.84 |
The core microbiota were generated from Table 4.
Discussion
The microbial population in the hindgut plays a key role in the health and welfare of the herbivore [9]. An active and functional fibrolytic bacterial population in the hindgut converts fibrous feeds into volatile fatty acids which make a significant contribution to the energy requirements of the host [6]. So far, studies regarding the intestinal microbial flora of the white rhinoceros are relatively limited [10]. In the current study, the fecal bacterial community of the white rhinoceros has been determined comprehensively for the first time using high throughput sequencing technology. In the present study, it was not unexpected to find that a large number of bacteria in the feces of the white rhinoceros belonged to the unclassified genera based on the current database of 16S RNA gene sequences, since little work on this kind of wild herbivorous animal has been done before. However, to some extent, the result also reflects the weakness of high throughput sequencing that it is not precise for lower levels taxonomic classification because of the short-read lengths and background ‘noise' introduced by PCR and sequencing [11]. Nevertheless, the results might suggest that the white rhinoceros might possess specific intestinal microbiota for its special feeding habits. However, to fully understand these unknown bacteria and their special role to the hosts, further studies are still needed.
The white rhinoceros is an ungulate animal, with hoofs that have three toes on each foot. They are more closely related to horses (who are also ungulates) than hippos [12]. In addition, also like horses, rhinoceroses are hindgut fermenters with the ability to eat less nutritious vegetation than ruminants due to their faster digestion. In previous studies, the fecal bacterial communities of horses have been intensively investigated [4], [6], [13], [14]. Costa et al. compared the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing, and found Firmicutes predominated among healthy horses, followed by Bacteroidetes and Proteobacteria [4]. Similar to the healthy horses, Firmicutes were also found to be the most predominant phylum in the feces of the five white rhinoceroses. This phylum has also been reported to be the most abundant in the hindgut of healthy humans and most of mammals [10], [15], [16]. Within the Firmicutes phylum, we found that the Lachnospiraceae and Ruminococcaceae families dominated in the feces of the five white rhinoceroses, which is consistent with previous studies on hindgut microbiota of humans and other mammals [17], [18]. Although most of these families were not classified at the genus and species levels, numerous bacteria such as Ruminococcus spp., Butyrivibrio spp., and Clostridium spp. are regarded as fiber-degraders in the rumen and the hindgut of herbivores [5], [6]. However, as another main fiber-degrader in the rumen, Fibrobacter was not detected in the feces of the white rhinoceros, likely due to our methodology. Considering that the bacterial communities in the hindgut of animals were believed to similar to those in feces [18], this result indicate that the fiber degrading bacteria in the hindgut of herbivores are dissimilar to those in the rumen. Nevertheless, the influence of the DNA extraction method used in this study could not be ignored.
Bacteroidetes is also one of the most abundant phyla in the gut of humans and herbivores [6], [15]. In the present study, this phylum was the second most abundant in the fecal bacterial community of all five white rhinoceroses, which is consistent with the finding in healthy horses and other mammals [4], [10]. However, in the rumen of dairy cows, Bacteroidetes was regarded as the most abundant phylum, which represented around 40–70% of abundance within the total bacterial community [5], [19]. The results indicate that unlike the ruminant, Bacteroidetes might play a lesser role in hindgut fermentation compared to the dominant Firmicutes phylum in non-ruminant herbivores. Nevertheless, we found that families Rikenellaceae and Prevotellaceae dominated in this phylum, which is also similar to the findings on other mammals [20], [21]. Interestingly, Bacteroidetes became predominant in the feces of horses affected by colitis. In contrast, the abundance of this phylum in the sample from X1 was much lower than in the samples from the four healthy animals. The possible reason for this variation may be the drug therapy for X1.
Noticeably, we found that the genus Aerococcus was predominant in the X1 library; in particular, Aerococcus viridans-related OTU was only found in the feces of rhinoceros X1 with the relative abundance of 4.62% within the total bacteria. A. viridans has been associated with different human infections, such as endocarditis, urinary tract infections, and meningitis [22]–[24]. In addition, this species has also been isolated from the milk of cows with subclinical mastitis [25], and from different clinical specimens of normally sterile body sites of pigs [26]. Moreover, A. viridans was found to be resistant to many antimicrobial drugs, including penicillins, cefotaxime, amikacin, trimethoprim/sulfamethoxazole, and glycopeptides [27]–[29], which is consistent with the fact that rhinoceros X1 had been treated with cefotaxime and amikacin due to constipation.
In this study, we found three female rhinoceroses had relatively high similar microbiota, although X3 was much older than X4 and X5, which suggests that the fecal microbiota of the rhinoceros might be influenced by the animal’s sex. This is consistent with the findings of a previous study where female and male macaques possessed distinctive microbiota in fecal and colonic contents [7]. Similarly, partitioning of the gut microbiota by sex has also been noted in mice [30]; however, the physiological mechanism such as circulating levels of hormones for the observed sexual dimorphism is unknown.
In the wider area of gut microbiology, there is active debate concerning the existence of a core stable microbiota. It is estimated that there are perhaps 5000 unique bacterial OTUs in the human gut when considered over a range of individuals under different spatial and temporal conditions [31]. However, it is speculated that there are perhaps 300 OTUs that make up the core stable microbial population in a healthy individual [32]. In the present study, 350 OTUs representing more than 75% of abundance within the total microbiota were regarded as core bacteria in four healthy rhinoceroses. Even when considering the outlier microbiota, the five rhinoceroses still had 266 core OTUs in the fecal microbiota. In addition, we found that the core bacteria in four healthy rhinoceroses were dominated by phyla Firmicutes and Bacteroidets including Prevotellaceae, Rikenellaceae, Ruminococcaceae, and Lachnospiraceae families, which is different from those reported by Ley et al for 106 mammals [10]. Costa et al found that only family Lachnospiraceae dominated the core bacterial population in the feces of healthy horses [4]. In the rumen of cows, the predominant core bacteria belonged to the Prevotella genus, Lachnospiraceae family, and the Butyrivibrio genus [5]. Possible reasons for the high percentage of core bacteria in the rhinoceros are that we evaluated only a few animals and the animals had the same diet in the same conditions. In addition, we removed the bacteria from fiber materials in the feces before DNA extraction, which can also affect the subsequent bacterial community using pyrosequencing analysis [33]. A higher diversity of predominant core bacteria in rhinoceroses compared with horses and cows might be responsible for its strong ability to adapt to the diet (e.g. toxic phytochemicals in the diet).
In summary, the work presented here describes the composition of the overall bacterial communities in the feces of five white rhinoceros living in a zoo. Our data reveals the presence of a complex bacterial community in the feces of the white rhinoceros. The rhinoceros possesses distinctive microbiota and core bacteria in the feces compared to horses. These observations increase our understanding of the bacterial ecosystem of this endangered animal, however, further study is still needed to know whether rhimoceroses in the wild have specific gut microbiota compared to other non-ruminant herbivores.
Materials and Methods
Collection of Fecal Samples
Five African white rhinoceroses were housed in the same room with a 3000-square meters outdoor playgroud at the Shanghai Wild Animal Park (see Table 6). X1 was treated with neostigmine bromide (0.6 g), cisapride (0.4 g), and rhubarb-soda tablet (50 g) through oral administration accompanying with intramuscular injection of cefotaxime (20 g) and amikacin (4 g) twice per day for six days because of the bad appetite and constipation, and had recovered one month prior to the study. X2, X3, X4, and X5 were healthy animals. The twice-daily diet for each animal consisted of 100–125 kg fresh, local grass (mainly gramineous pasture including Digitaria spp., Eleusine indica and Setaria viridis), 7.5 kg hay pellets (Leymus chinensis), and 2 kg carrot. In September 2011, fresh fecal sample (approximate 100 g) were immediately collected by the animal raiser when each animal was upon defecation in the morning, sent to the laboratory in foamed plastic containers with dry ice, and processed immediately after arrival. The samples were pretreated according to Wang et al. [34] and Wei et al. [35] as follows: 50 g of feces was suspended in a sterile plastic beaker containing 250 ml of sterile phosphate-buffered saline (PBS) (0.05 mol/l, pH 7.4). The sample was stirred with a sterile plastic rod for about 30 min to remove the bacteria from the plant residue. The suspension then was divided into 60-ml aliquots and transferred to eight sterile 80-ml centrifuge tubes and vortexed vigorously for 15 min. The samples were centrifuged at 200 g for 5 min three times (each time the supernatant was transferred to a new tube) to remove coarse particles. The cells in the supernatant were collected and washed three times by centrifuging at 9000 g for 3 min with 30 ml fresh PBS. Finally, the washed cell pellets were re-suspended in one tube in 10 ml of sterile PBS, divided into 1-ml aliquots, and stored at −20°C for DNA extractions within one week.
Table 6. Information on the rhinoceroses used in this study.
| Rhinoceros | Sex | Age (years) | Health condition |
| X1 | male | 26 | treated with neostigmine bromide, cisapride and rhubarb-soda tablet through oral administration accompanying with intramuscular injection of antibiotic therapy one month prior to the study |
| X2 | male | 26 | healthy |
| X3 | female | 25 | healthy |
| X4 | female | 17 | healthy |
| X5 | female | 17 | healthy |
Ethics Statement
The study was approved by Nanjing Agricultural University Animal Care and Use Committee. Fecal samples of the rhinoceros were collected with the permission of Chunzhong Xu, the director of Shanghai Wild Animal Park. The study did not involve endangered or protected species.
DNA Extraction
The total genomic DNA was isolated from the pretreated fecal samples using the commercially available stool DNA extraction kit according to the instructions of the manufacturer (QIAamp DNA Stool Mini Kit, Qiagen, Hilden, Germany). The concentration of extracted DNA was determined using a Nano-Drop 1000 spectrophotometer (Thermo Scientific Inc., Wilmington, DE, USA).
PCR Amplification, Amplicon Quantitation, and Pyrosequencing
To analyze the taxonomic composition of the bacterial community, universal primers (8F 5′-AGA GTT TGA TCC TGG CTC AG-3′ and 533R 5′-TTA CCG CGG CTG CTG GCA C-3′) targeting the V1–V3 region of 16S rRNA gene were chosen for the amplification and subsequent pyrosequencing of the PCR products [36]. The PCR were carried out in triplicate with: 50 ml reactions with 10 µl 5-fold reaction buffer, 50 ng of DNA, 0.4 mM each primer, 0.5 U Pfu polymerase (TransStart-FastPfu DNA Polymerase, TransGen Biotech), and 2.5 mM dNTPs. The amplification program consisted of an initial denaturation step at 94°C for 4 min. This was followed by 25 cycles, where 1 cycle consisted of 94°C for 30 s (denaturation), 55°C for 30 s (annealing), 72°C for 30 s (extension), and a final extension of 72°C for 10 min. All PCR products were visualized on agarose gels (2% in TBE buffer) containing ethidium bromide, and purified with a DNA gel extraction kit (Axygen, China).
Prior to sequencing, the DNA concentration of each PCR product was determined using a Quant-iT PicoGreen double-stranded DNA assay (Invitrogen, Germany), and was quality controlled on an Agilent 2100 bioanalyzer (Agilent, USA). Amplicon pyrosequencing was performed from the A-end using a 454/Roche A sequencing primer kit on a Roche Genome Sequencer GS-FLX Titanium platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.
Bioinformatics Analysis
The end fragments were blunted and tagged on both ends with ligation adaptors that contained a unique 10-bp sequence (sample specific barcode sequence) and a short 4-nucleotide sequence (TCAG) called sequencing key, which were recognized by the system software and the priming sequences. All pyrosequencing reads were filtered according to barcode and primer sequences. The resulting sequences were further screened and filtered for quality. Sequences that were shorter than 200 bp in length, contained ambiguous characters, contained over two mismatches to the primers, or contained mononucleotide repeats of over six nt were removed. To assess bacterial diversity among samples in a comparable manner, a randomly selected, 16929-sequence (the lowest number of sequences in the five samples) subset from each sample was aligned using the ‘align.seqs’ command and compared with the Bacterial SILVA database (SILVA version 108; http://www.arb-silva.de/documentation/background/release-108/). The aligned sequences were clustered into operational taxonomic units (OTUs) defined by 97% similarity [37] using CD-HIT-OUT program [38]. We also calculated the coverage percentage using Good’s method [39], abundance based coverage estimator (ACE), bias-corrected Chao richness estimator, and the Shannon and Simpson diversity indices using the MOTHUR program (http://www.mothur.org) [40]. Libshuff analysis was used to compare population structure between different aminals. The heatmap figure was generated using custom Perl scripts. The raw pyrosequencing reads were submitted to Sequencing Read Archive (SRA) database under the accession id: SRA073469.
Acknowledgments
We thank Chunzhong Xu in Shanghai Wild Animal Park for his assistance in collecting the fecal samples.
Funding Statement
This study was supported by a grant from the Fundamental Research Funds for the Central Universities (KYZ201153). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. McNaughton SJ, Georgiadis NJ (1986) Ecology of African grazing and browsing mammals. Ann Rev Ecol Syst 17: 39–65. [Google Scholar]
- 2. Clemens ET, Maloiy GMO (1982) The digestive physiology of three East African herbivores: the elephant, rhinoceros and hippopotamus. J Zool 198: 141–156. [Google Scholar]
- 3. Kienzle E, Schramel P, Dierenfeld ES, Flach EJ, Behlert O, et al. (2006) Macromineral absorption in the black rhinoceros (Diceros bicornis) compared with the domestic horse. J Nutr 136: 2017S–2020S. [DOI] [PubMed] [Google Scholar]
- 4. Costa MC, Arroyo LG, Allen-Vercoe E, Stampfli HR, Kim PT, et al. (2012) Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3–V5 region of the 16S rRNA gene. PLoS ONE 7(7): e41484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jami E, Mizrahi I (2012) Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 7(3): e33306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Daly K, Proudman CJ, Duncan SH, Flint HJ, Dyer J, et al. (2012) Alterations in microbiota and fermentation products in equine large intestine in response to dietary variation and intestinal disease. J Bri Nutr 107: 989–995. [DOI] [PubMed] [Google Scholar]
- 7. McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, et al. (2008) The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog 4(2): e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Su Y, Li B, Zhu W (2013) Fecal microbiota of piglets prefer utilizing dl-lactate mixture as compared to d-lactate and l-lactate in vitro . Anaerobe 19: 27–33. [DOI] [PubMed] [Google Scholar]
- 9. Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA (2008) Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol 6: 121–131. [DOI] [PubMed] [Google Scholar]
- 10. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, et al. (2008) Evolution of mammals and their gut microbes. Science 320: 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lynch MDJ, Bartram AK, Neufeld JD (2012) Targeted recovery of novel phylogenetic diversity from next-generation sequence data. ISME J 6: 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Henry JS, Lance VA, Conlon JM (1991) Primary structure of pancreatic polypeptide from four species of perissodactyla (Przewalski’s horse, zebra, rhino, tapir). Gern Comp Endo 84: 440–446. [DOI] [PubMed] [Google Scholar]
- 13. Garrett LA, Brown R, Poxton IR (2002) A comparative study of the intestinal microbiota of healthy horses and those suffering from equine grass sickness. Vet Microbiol 87: 81–88. [DOI] [PubMed] [Google Scholar]
- 14. Daly K, Stewart CS, Flint HJ, Shirazi-Beechey SP (2001) Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiol Ecol 38: 141–151. [Google Scholar]
- 15. Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444: 1022–1023. [DOI] [PubMed] [Google Scholar]
- 16. Guo X, Xia X, Tang R, Zhou J, Zhao H, et al. (2008) Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett Appl Microbiol 47: 367–373. [DOI] [PubMed] [Google Scholar]
- 17. Hooda S, Boler BMV, Serao MCR, Brulc JM, et al. (2012) 454 pyrosequencing reveals a shift in fecal microbiota of healthy adult men consuming polydextrose or soluble corn fiber. J Nutr 142: 1259–1265. [DOI] [PubMed] [Google Scholar]
- 18. Steelman SM, chowdhary BP, Dowd S, Suchodolski J, Janecka JE (2012) Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet Res 27: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li RW, Wu S, Baldwin RL, Li W, Li C (2012) Perturbation dynamics of the rumen microbiota in response to exogenous butyrate. PLoS One 7: e29392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zened A, Combes S, Cauquil L, Mariette J, Klopp C (2013) Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol Ecol 83: 504–514. [DOI] [PubMed] [Google Scholar]
- 21.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, et al.. (2009) High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137: 1716–1724, e1711–1712. [DOI] [PMC free article] [PubMed]
- 22. Facklam RR, Elliot JA (1995) Identification, classification, and clinical relevance of catalase-negative, gram-positive cocci, excluding the streptococci and enterococci. Clin Microbiol Rev 8: 470–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gopalachar A, Akins RL, Davis WR, Siddiqui AA (2004) Urinary tract infection caused by Aerococcus viridans, a case report. Med Sci Monit 10: CS73–CS75. [PubMed] [Google Scholar]
- 24. Popescu GA, Benea E, Mitache E, Piper C, Horstkotte D (2005) Anunusual bacterium, Aerococcus viridans, and four cases of infective endocarditis. J Heart Valve Dis 14: 317–319. [PubMed] [Google Scholar]
- 25. Devriese LA, Hommez J, Laevens H, Pot B, Vandamme P, et al. (1999) Identification of aesculin-hydrolyzing streptococci, lactococci, aerococci, and enterococci from subclinical intramammary infections in dairy cows. Vet Microbiol 70: 87–94. [DOI] [PubMed] [Google Scholar]
- 26. Martin V, Vela AI, Gilbert M, Cebolla J, Goyache J, et al. (2007) Characterization of Aerococcus viridans isolates from swine clinical specimens. J Clin Microbiol 45: 3053–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Facklam R, Lovgren M, Shewmaker PL, Tyrrell G (2003) Phenotypic description and antimicrobial susceptibilities of Aerococcus sanguinicola isolates from human clinical samples. J Clin Microbiol 41: 2587–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen L, Yu W, Huang S, Lin M, Chen T, et al. (2012) Successful treatment of Aerococcus viridans endocarditis in a patient allergic to penicillin. J Microbiol Immunol Infect 45: 158–160. [DOI] [PubMed] [Google Scholar]
- 29. Uh Y, Son JS, Jang IH, Yoon KJ, Hong SK (2002) Penicillin-resistant Aerococcus viridans bacteremia associated with granulocytopenia. J Korean Med Sci 17: 113–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schloss PD, Handelsman J (2006) Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72: 6773–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, et al. (2007) Molecular-phylogenetic characterization of microbial community imbalances in human inammatory bowel diseases. Proc Natl Acad Sci USA 104: 13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manichanh C, Varela E, Martinez C, Antolin M, Llopis M, et al. (2008) The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J Gastroenterol 103: 1754–1761. [DOI] [PubMed] [Google Scholar]
- 33. Yu ZT, Morrison M (2004) Improved extraction of PCR-quality community DNA from digesta and fecal samples. BioTchniques 36: 808–812. [DOI] [PubMed] [Google Scholar]
- 34. Wang RF, Cao WW, Cerniglia CE (1996) PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Appl Environ Microbiol 62: 1242–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wei G, Lu H, Zhou Z, Xie H, Wang A, et al. (2007) The microbial community in the feces of the giant panda (Ailuropoda melanoleuca) as determined by PCR-TGGE profiling and clone library analysis. Microbiol Ecol 54: 194–202. [DOI] [PubMed] [Google Scholar]
- 36. Baker G, Smith JJ, Cowan DA (2003) Review and re-analysis of domain-specific 16S primers. J Microbiol Meth 55: 541–555. [DOI] [PubMed] [Google Scholar]
- 37. Stackebrandt E, Goebel BM (1994) Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44: 846–849. [Google Scholar]
- 38. Wu S, Zhu Z, Fu L. Niu B, Li W (2011) WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12: 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Good IL (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40: 237–264. [Google Scholar]
- 40. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]