

Abstract
MicroRNA miR-376c was expressed in normal intrahepatic biliary epithelial cells (HIBEpiC), but was significantly suppressed in the HuCCT1 intrahepatic cholangiocarcinoma (ICC) cell line. The biological significance of the down-regulation of miR-376c in HuCCT1 cells is unknown. We hypothesized that miR-376c could function as a tumor suppressor in these cells. To test this hypothesis, we sought the targets of miR-376c, and characterized the effect of its down-regulation on HuCCT1 cells. We performed proteomic analysis of miR-376c-overexpressing HuCCT1 cells to identify candidate targets of miR-376c, and validated these targets by 3′-UTR reporter assay. Transwell migration assays were performed to study the migratory response of HuCCT1 cells to miR-376c overexpression. Furthermore, microarrays were used to identify the signaling that were potentially involved in the miR-376c-modulated migration of HuCCT1. Finally, we assessed epigenetic changes within the potential promoter region of the miR-376c gene in these cells. Proteomic analysis and subsequent validation assays showed that growth factor receptor-bound protein 2 (GRB2) was a direct target of miR-376c. The transwell migration assay revealed that miR-376c significantly reduced epidermal growth factor (EGF)-dependent cell migration in HuCCT1 cells. DNA microarray and subsequent pathway analysis showed that interleukin 1 beta and matrix metallopeptidase 9 were possible participants in EGF-dependent migration of HuCCT1 cells. Bisulfite sequencing showed higher methylation levels of CpG sites upstream of the miR-376c gene in HuCCT1 relative to HIBEpiC cells. Combined treatment with the DNA-demethylating agent 5-aza-2′-deoxycytidine and the histone deacetylase inhibitor trichostatin A significantly upregulated the expression of miR-376c in HuCCT1 cells. We revealed that epigenetic repression of miR-376c accelerated EGF-dependent cell migration through its target GRB2 in HuCCT1 cells. These findings suggest that miR-376c functions as a tumor suppressor. Since metastasis is the major cause of death in ICC, microRNA manipulation could lead to the development of novel anti-cancer therapy strategies for ICC.
Introduction
Deep sequencing and transcriptome analysis revealed the existence of non-coding RNAs (ncRNAs) in mammalian cells [1]–[3]. MicroRNAs (miRNAs) are single-stranded 19- to 25-nucleotide ncRNAs that play a critical role in posttranscriptional gene regulation. The miRNA-mediated gene silencing is regulated by complementarity between nucleotides at positions 2–8 of the miRNAs (i.e., the seed sequences) and the 3′-untranslated regions (3′-UTR) of their target genes [4], [5]. Dysregulated expression of miRNA is involved in a variety of human diseases, including cancer and liver disease [6], [7]. Recent studies show that expression levels of various miRNAs are downregulated in cancer, indicating that they may function as tumor suppressors [8]–[10]. For example, downregulation of miR-15 and miR-16 in most chronic lymphocytic leukemia cells leads to upregulation of anti-apoptotic B cell lymphoma 2 (Bcl-2) protein [8]. The upregulated Bcl-2 averts apoptotic cell death of leukemia cells and thereby promotes their survival. Let-7, which is downregulated in lung cancers relative to normal lung tissue, targets ras oncogenes [10]. The increased levels of Ras protein in lung cancer cells leads to upregulated cell growth. The miR-200 family and miR-205 target zinc finger homeodomain enhancer-binding protein (ZEB) transcription factors, which are known to be inducers of the epithelial-mesenchymal transition in breast cancer [9]. Downregulation of these miRNAs are likely to be an essential early step in breast cancer metastasis.
Cholangiocarcinoma (CC) is a bile duct cancer, and is classified as intrahepatic or extrahepatic [11]–[13]. Intrahepatic CC (ICC) is derived from epithelial cells of the bile ducts. Although ICCs comprise only 5–10% of all cases of liver cancer, they are the second most common liver malignancy [14]. The incidence and mortality rate of ICC are increasing worldwide. Despite advances in surgical techniques, chemotherapies and radiotherapies, long-term survival remains low because of the late presentation of the disease [14], [15]. Even after resection, the prognosis for patients with advanced ICC is extremely poor [14], [16], [17]. Some researchers have examined the miRNA expression profiles in ICC, to understand the molecular and clinical basis of carcinogenesis and the progression of this disease [18], [19].
We reported previously that miR-376c (formerly designated as miR-368 in miRBase Release 12; currently designated as miR-376c-3p in miRBase Release 19) was expressed in a normal intrahepatic biliary epithelial cell line (HIBEpiC), but was significantly suppressed in an ICC cell line (HuCCT1) [18]. However, the biological significance of the downregulation of miR-376c in HuCCT1 cells was unknown. We hypothesized that miR-376c could function as a tumor suppressor in these cells. To test this hypothesis, we sought the targets of this miRNA, and characterized the effect of miR-376c down-regulation in HuCCT1 cells. We identified a direct target mRNA of miR-376c by proteomic analysis. Enforced expression of miR-376c significantly impaired migration of HuCCT1 cells by reducing levels of the targeted mRNA, indicating that downregulation of miR-376c is critical for this response. Finally, we investigated the DNA methylation and histone modification status of the putative promoter regions of miR-376c gene in HuCCT1 cells.
Materials and Methods
Cell culture and RNA isolation
HIBEpiC were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA). HuCCT1 was obtained from the American Type Culture Collection (Manassas, VA, USA). The ICC cell lines TKKK, Huh28, and IHGGK, and the extrahepatic CC cell line TFK-1 were from RIKEN BioResource Center (Tsukuba, Japan). All cell lines were maintained in the media recommended by the suppliers, at 37°C in a humidified incubator with a 5% CO2 atmosphere. Total RNA was extracted from each sample using RNAiso plus (Takara Bio, Ohtsu, Japan), according to the manufacturer's instructions.
Proteomic analysis
Proteomic analysis was performed based on our previous method [20]. HuCCT1 cells grown to semi-confluence were transfected with Pre-miR-376c (mature miR-376c-3p mimic) or Pre-miR negative control #1 (Applied Biosystems, Foster City, CA, USA) at a final concentration of 30 nM using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. Pre-miR-transfected HuCCT1 cells were harvested 72 h after transfection and subjected to the following proteomic analysis. Cells were lysed with a thiourea lysis buffer (7.5 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate, 1 mM PMSF, 1 µM aprotinin, 1 µM pepstatin A, and 10 mM Tris-HCl [pH 8.8]). After removal of cell debris by centrifugation at 20,000× g at 4°C for 20 min, proteins in the cell lysates were labeled with CyDye DIGE Fluors minimal dyes (GE Healthcare Bio-Sciences, Little Chalfont, UK). The labeled proteins were subjected to two-dimensional difference gel electrophoresis (2D-DIGE) using the Ettan DALTsix Large Electrophoresis System (GE Healthcare Bio-Sciences), as described in the manufacturer's protocol. Protein samples obtained from three independent experiments were subjected to proteomic analysis. Spots with significant differences in intensity between the samples (>20% upregulation or downregulation, P≤0.011) were excised from the gel using the Ettan Spot Picker (GE Healthcare Bio-Sciences). These criteria for spot selection were determined based on previous studies, in which the repression of cellular protein synthesis by miRNA overexpression was typically mild [21], [22]. Proteins in the spots were subjected to digestion with 10 ng/µl trypsin (Sigma, St. Louis, MO, USA) at 37°C, followed by elution in an acetonitrile buffer. The eluted proteins were then identified using a liquid chromatography-tandem mass spectrometry mass spectrometer (LCQ DECA XP Plus; Thermo-Finnigan, San Jose, CA, USA). Mass spectrometry data were analyzed using the MASCOT software (Matrix Science, London, England). Specificity of trypsin digestion was used as the cutting enzyme with allowing three missed cleavages. Peptide mass tolerance and fragment mass tolerance were set to ±1.2 Da and ±0.6 Da, respectively. Carbamidomethyl of cysteine was chosen as the fixed modification and oxidation of methionine was searched as the variable modification. Identity or extensive homology is indicated for peptides with individual ion scores >42 (p<0.05).
Western blotting
HuCCT1 cells in culture were rinsed in PBS and lysed in M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Rockford, lL, USA) containing Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific). The lysates were centrifuged at 20,000× g at 4°C for 3 min to remove debris, and supernatants were subjected to western blotting as follows: the lysates were separated on SDS-PAGE gels and transferred to Sequi-Blot polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). Blots were then incubated at 4°C overnight with primary antibodies diluted as recommended in the manufacturer's instructions. This was followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Signals were detected using Immobilon reagent (Millipore, Billerica, MA, USA) and visualized using an LAS-4000 Lumino image analyzer (Fujifilm, Tokyo, Japan). The intensity of visualized signals was quantitated using the Multigauge software (Fujifilm). A monoclonal GRB2 antibody (catalog number 61011) was purchased from Becton Dickinson (Franklin Lakes, NJ, USA). A monoclonal antibody for beta actin (ACTB) was purchased from Sigma (catalog number A5316).
Real-time PCR
Real-time PCR for miRNAs was carried out using TaqMan Gene Expression Assays (Applied Biosystems) in a 7300 Real-Time PCR System (Applied Biosystems) or a 7900 FAST Real-Time PCR System (Applied Biosystems), according to the manufacturer's protocols. U6 snRNA (RNU6B) was used as an endogenous internal control. To quantify mRNA levels, SYBR Premix Ex Taq (Perfect Real Time) (Takara Bio) was applied. To normalize mRNA expression levels, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an endogenous internal control. Primers used for RT-PCR of GRB2, IL1B, MMP9 were as follows: GRB2-Foward: CCATCGCCAAATATGACTTCAAA; GRB2-Reverse: CTTCGTTCAAAACCTTGAGGATGT; IL1B -Forward: CAACAAGTGGTGTTCTCCATGT; IL1B-Reverse: GACAAATCGCTTTTCCATCTTC; MMP9-Foward: ACTTTGACAGCGACAAGAGGTGG; MMP9-Reverse: CCGGCACTGAGGAATGATCTAA.
3′-UTR reporter assay
To construct a reporter plasmid, we first cloned the 3′-UTR of the human GRB2 into pMIR-REPORT vector (Applied Biosystems). Total RNA, isolated from HuCCT1 cells, was reverse-transcribed to cDNA using PrimeScript reverse transcriptase (Takara Bio). The 3′-UTR of GRB2 mRNA was then amplified from the cDNA using the following primers: GAACTAGTGAGTCAAGAAGCAATTATTTAAAGAAAGTGAA (SpeI site underlined) and GAAAGCTTTGAAGAATTCATTGTGTATTTATTATTCACAG (HindIII site underlined). The PCR product was cloned into a pCR4-Blunt-TOPO vector for sequence verification. The GRB2 3′-UTR was then cloned into pMIR-REPORT via the SpeI and HindIII restriction sites. This final construct was designated pMIR-GRB2. To construct a reporter plasmid with a mutated miR-376c recognition site of GRB2 3′-UTR, an inverse PCR method was used. The primers used for the inverse PCR were: GRB2 3′UTRmutinv-Foward: ggtattcgtatctctcaaaaTGTCTGTTTTAGTTGGAATG; GRB2 3′UTRmutinv-Reverse: ttttgagagatacgaataccAGCCCACTTGGTTTCTTGTT (complementary sequence shown in lowercase), and were designed to introduce the mutation. PCR amplification was carried out using the previously cloned vector pCR4-Blunt-TOPO containing the GRB2 3′-UTR. Plasmid DNA was digested by DpnI. Amplified DNA was transformed into Escherichia coli. After sequence verification, the mutated 3′-UTR sequence was cloned into pMIR-REPORT via SpeI and HindIII restriction sites. The final construct was designated pMIR-GRB2mt. HuCCT1 cells were transfected with the pMIR-GRB2, pMIR-GRB2mt, or the control vector pRL-TK (Renilla luciferase expression plasmid), together with Pre-miR-376c or the Pre-miR-negative control at 10 nM, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in 24-well plates. Twenty-four hours after transfection, luciferase assays were performed using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA). Firefly luciferase activity was normalized to Renilla luciferase activity.
Cell migration assay
HuCCT1 cell migration analysis was carried out using Transwell inserts (8.0 µm pore size, Costar, Cambridge, MA, USA) [23]. Cells were transfected with the Pre-miR molecule or GRB2-siRNA at final concentrations of 30 nM using Lipofectamine 2000 (Invitrogen). Two siRNAs for GRB2, siGRB2-1 (Hs_GRB2_5; sense: GUUUGGAAACGAUGUGCAGTT; antisense: CUGCACAUCGUUUCCAAACTT) and siGRB2-2 (Hs_GRB2_8; sense: AGAACUACAUAGAAAUGAATT; antisense UUCAUUUCUAUGUAGUUCUTG) were used (Qiagen, Valencia, CA, USA). A nonspecific non-silencing siRNA (AllStars Negative Control siRNA, Qiagen) was used as a negative control. After 24 h of transfection, cells (5×104) in 250 µl of serum-free medium RPMI 1640 were seeded onto filters in 24-well plates. The medium (750 µl) containing RPMI 1640 supplemented with 0.1% fetal bovine serum was placed in the lower chamber in the presence of epidermal growth factor (EGF) (5 ng/ml, R&D Systems, Minneapolis, USA). After incubation for 24 h, non-invading cells on the top of each Transwell were scraped off with a cotton swab. Cells that had migrated to the other side were fixed with 2.5% glutaraldehyde (Wako, Tokyo, Japan) and stained with crystal violet (Wako). The number of migrated cells was manually counted with a light microscope (KX4, Olympus, Tokyo, Japan) under 200× magnification. The sum of the numbers of cells in five areas was used as the migrated cell number, and expressed as a percentage of the control value. These experiments were repeated at least three times, and significant differences among treatments were assessed by ANOVA followed by Tukey's test. In our preliminary experiments of cells exposed to different concentrations of EGF (0.1, 0.5, 1, 5, and 10 ng/ml), EGF regulation of cell migration was in a dose-dependent manner. HuCCT1 cell migration peaked at the 5 ng/ml concentration but then decreased at 10 ng/ml (data not shown). The preliminary experiments indicate that the optimized concentration of EGF for cell migration assay was 5 ng/ml.
Gene expression analysis
HuCCT1 cells were treated with EGF as described above. Total RNA was extracted from Pre-miR-376c- and siGRB2-2-transfected HuCCT1 cells and from individual transfection controls with RNAiso Plus (Takara Bio), and its concentration was measured using a NanoDrop ND-2000 spectrometer (Thermo Scientific, Wilmington, DE, USA). Of the total RNA obtained, 200 ng was used in a labeling reaction, using a Low-Input QuickAmp Labeling Kit, One-Color (Agilent Technologies, Santa Clara, CA, USA), and the quality and yield of labeled cRNA was evaluated on an Agilent 2100 Bioanalyzer (Agilent). Gene expression profiling was conducted using an Agilent microarray (Human GE 4×44 K, v2). The resulting signals were normalized to the 75th percentile signal intensity and the processed data were filtered for an at least twofold change using the GeneSpring GX software (ver. 11.5; Agilent). The array data were imported into the Ingenuity Pathway Analysis software (IPA, Ingenuity Systems, Redwood City, CA, USA). Functional analyses of down-regulated genes were conducted using the IPA software (Ingenuity Systems, http://www.ingenuity.com). A right-tailed Fisher's exact test was used to calculate a p-value for the probability of an association between the gene expression data and the knowledge-based biological function. The mRNA array data are publicly available (Gene Expression Omnibus accession number GSE47186, http://www.ncbi.nlm.nih.gov/geo/).
Bisulfite conversion of DNA and bisulfite sequencing
Genomic DNA was extracted using a DNA pure kit (Takara Bio), according to the manufacturer's protocol. Bisulfite conversion of DNA (1 µg) was performed using a Methyl Easy Xceed Rapid DNA modification kit (Takara Bio) according to the manufacturer's protocol. PCR amplification was performed using Ex Taq HS (Takara Bio). Primer sequences were TTGAATATTTTTGAGAGGAAGGTTAGT and TCCTAAAAAACATAAACCTAAACACAAT. PCR products were ligated into pCR4 TOPO (Invitrogen). At least 20 clones were sequenced per cell.
Treatment with 5-AZA-dCR and TSA
HuCCT1 cells were seeded at a density of 5×104/well in six-well plates, cultured for 24 h and then treated with 10 µM DNA-demethylating agent 5-aza-2′-deoxycytidine (5-AZA-dCR, Sigma-Aldrich, St. Louis, MO, USA) for 3 days, during which the drug-containing medium was replaced daily. After 3 days, the histone deacetylase (HDAC) inhibitor trichostatin A (TSA, Sigma-Aldrich; 0.1, 0.5, or 1.0 µM) was added and cells were cultured for a further 24 h.
Results
MiR-376c is downregulated in CC cell lines including HuCCT1
We previously performed small RNA sequencing using HuCCT1 and HIBEpiC to reveal differential miRNA expression [18]. Small RNA sequencing showed that miR-376c was found exclusively in HIBEpiC and not in HuCCT1 (i.e., cloning frequency >0.1% in HIBEpiC and 0.0% in HuCCT1) [18]. For further characterization of miR-376c expression in CC cell lines, the ICC cell lines TKKK, Huh28, and IHGGK, and the extrahepatic CC cell line TFK-1 were examined. Real-time PCR analysis revealed that miR-376c, which was highly expressed in HIBEpiC, was significantly downregulated in all CC cell lines examined in this study (Figure 1A). It is likely that miR-376c is downregulated specifically in bile duct carcinoma cell lines.
Figure 1. Downregulation of miR-376c expression levels in bile duct carcinoma cell lines, and proteomic analysis of miR-376c-overexpressing HuCCT1.

(A) Real-time PCR assay of miR-376c in HIBEpiC, HuCCT1, Huh28, IHGGK, TKKK, and TFK1. Expression levels were normalized to RNU6B, and the expression level in HIBEpiC cells was defined as 1. The significance of differences among cells was assessed by ANOVA followed by Tukey's test (*P<0.05). (B) Representative 2D-DIGE images of miR-376c-overexpressing HuCCT1 cells. Cells were harvested 72 h after the initiation of transfection of Pre-miR-376c or the Pre-miR-negative control, and subjected to proteomic analysis. A spot downregulated by treatment with Pre-miR-376c is indicated by the arrow, which was later shown by mass spectrometry to be GRB2. (C) Quantitative analysis of the fluorescence intensity of the GRB2 protein spot shown in B (peak outlined in red).
MiR-376c target identification by proteomic analysis
We performed a proteomic analysis to identify proteins whose expression was altered in miR-376c-overexpressing HuCCT1 cells, some of which may be targets of miR-376c. Proteomic analysis was performed by 2D-DIGE (Figure 1B). We found 41 protein spots with significant differences in intensity between controls and Pre-miR-376c-transfected HuCCT1 cells (upregulation or downregulation by more than 1.2-fold, P≤0.011). Of these 41 spots, 18 and 31 spots exhibited upregulation and downregulation of intensity, respectively. These spots were isolated and subjected to liquid chromatography-tandem mass spectrometry analysis. We focused on the downregulated proteins as potential direct targets of miR-376c (Table 1). Using the online software TargetScan (http://www.targetscan.org) and miRbase (http://mirbase.com), we identified only GRB2 protein as an in silico candidate; its expression level was decreased by 44% (Table 1 and Figure 1C).
Table 1. Proteins downregulated by miR-376c overexpression in HuCCT1 cells in proteomic analysis.* .
| FC | P-value | Protein Name | Gene Name |
| −2.22 | 0.011 | cytoskeleton-associated protein 4 | CKAP4 |
| Alpha-2-HS-glycoprotein precursor | AHSG | ||
| −2.21 | 0.011 | cofilin 1 (non-muscle) | CFL1 |
| −1.85 | 0.0027 | lamin A/C isoform 1 precursor | LMNA |
| ERO1-like | ERO1L | ||
| leukotriene A4 hydrolase | LTA4H | ||
| Alpha-2-HS-glycoprotein precursor | AHSG | ||
| very-long-chain acyl-CoA dehydrogenase | ACADVL | ||
| 80K-H protein, protein kinase C substrate 80K-H isoform 1 | G19P1 | ||
| −1.58 | 0.0097 | calpain, small subunit 1 | CAPNS1 |
| −1.52 | 0.0086 | Keratin, type I cytoskeletal 19 | KRT19 |
| −1.51 | 0.0092 | lamin A/C isoform 2 | LMNA |
| ERO1-like | ERO1L | ||
| very-long-chain acyl-CoA dehydrogenase | ACADVL | ||
| heterogeneous nuclear ribonucleoprotein L | HNRPL | ||
| tubulin 5-beta | TUBB4 | ||
| −1.51 | 0.01 | transferase, HG phosphoribosyl | HPRT |
| −1.48 | 0.0033 | cathepsin D preproprotein | CTSD |
| splicing factor, arginine/serine-rich 9 | SFRS9 | ||
| −1.44 | 0.0094 | growth factor receptor-bound protein 2 isoform 1 | GRB2 |
| Ras family small GTP binding protein TC21 | RRAS2 | ||
| −1.42 | 0.0024 | Annexin A2 | ANXA2 |
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | ||
| serine dehydratase-like | SDSL | ||
| −1.42 | 0.006 | annexin A2 isoform 2 | ANXA2 |
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | ||
| ribosomal protein P0 | RPLP0 | ||
| F-actin capping protein beta subunit | CAPZB | ||
| capping protein alpha | CAPZA2 | ||
| −1.4 | 0.0062 | LASP1 protein | LASP1 |
| heterogeneous nuclear ribonucleoprotein D-like | HNRPDL | ||
| annexin A2 isoform 2 | ANXA2 | ||
| −1.39 | 0.0097 | Annexin A2 | ANXA2 |
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | ||
| ribosomal protein P0 | RPLP0 | ||
| −1.35 | 0.0041 | cytokeratin 8 (279 AA) | KRT8 |
| tyrosine 3/tryptophan 5 -monooxygenase activation protein, zeta polypeptide | YWHAZ | ||
| GRP78 precursor, BiP protein | |||
| tumor protein D52-like 2 isoform a | TPD52L2 | ||
| proteasome alpha 3 subunit isoform 1 | PSMA3 | ||
| rho GDP dissociation inhibitor (GDI) | ARHGDIA | ||
| −1.35 | 0.0044 | cathepsin D preproprotein | CTSD |
| mitochondrial ribosomal protein L46 | MRPL46 | ||
| platelet-activating factor acetylhydrolase, isoform Ib, beta subunit 30 kDa | PAFAH1B2 | ||
| −1.35 | 0.011 | ERO1-like | ERO1L |
| Ras-GTPase-activating protein SH3-domain-binding protein | G3BP1 | ||
| lamin A/C | LMNA | ||
| −1.34 | 0.0065 | cathepsin D preproprotein | CTSD |
| eukryotic translation elongation factor 1 gamma, isoform CRA a | EEF1G | ||
| −1.32 | 0.0061 | myosin light chain 3 | MYL3 |
| ATP synthase, H+ transporting, mitochondrial F1 complex, delta subunit precursor | ATP5D | ||
| −1.31 | 0.0017 | Annexin A2 | ANXA2, ANXA2P1 |
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | ||
| heterogeneous nuclear ribonucleoprotein D-like | HNRPDL | ||
| fibrillarin | FBL | ||
| torsin family 1, member A (torsin A) | TOR1A | ||
| palmitoyl-protein thioesterase 1 | PPT1 | ||
| −1.28 | 0.002 | apolipoprotein L2 | APOL2 |
| mitochondria import inner membrane translocase subunit TIM50 precursor | TIMM50 | ||
| Annexin A2 | ANXA2 | ||
| heterogeneous nuclear ribonucleoprotein D-like | HNRPDL | ||
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | ||
| −1.26 | 0.0034 | annexin A2 isoform 2 | ANXA2 |
| carboxyl terminal LIM domain protein | CLIM1 | ||
| mitochondrial ribosomal protein L39 isoform b | MRPL39 | ||
| aflatoxin aldehyde reductase AFAR | AKR7A2 | ||
| glyceraldehyde-3-phosphate dehydrogenase | GAPDH | ||
| −1.23 | 0.0025 | keratin | |
| −1.21 | 0.0023 | ERO1-like | ERO1L |
| lamin A/C isoform 2 | LMNA | ||
| chaperonin containing TCP1, subunit 5 (epsilon) | CCT5 | ||
| Beta-galactosidase precursor | GLB1 | ||
| heterogeneous nuclear ribonucleoprotein K isoform a | HNRPK | ||
| copine I | CPNE1 | ||
| Ras-GTPase-activating protein SH3-domain-binding protein | G3BP1 | ||
| DHX9 protein | DHX9 |
The protein spots that had significant differences in intensity between the control- and Pre-miR-376c-transfected HuCCT1 cells (down-regulation by more than 1.2-fold change, p≤0.011) are listed in proteomic analysis. FC: fold change. The GRB2 protein predicted by TargetScan as a target of miR-376c is shown in bold.
Validation of GRB2 as a miR-376c Target
To confirm the 2D-DIGE results, we performed Western blotting analysis of HuCCT1 transfected with Pre-miR-376c. As shown in Figure 2A, the GRB2 protein level was markedly decreased in Pre-miR-376c-transfected cells compared to control cells. Furthermore, as shown in Figure 2B, the GRB2 mRNA level was also significantly decreased by Pre-miR-376c overexpression.
Figure 2. Validation of GRB2 as a miR-376c Target.
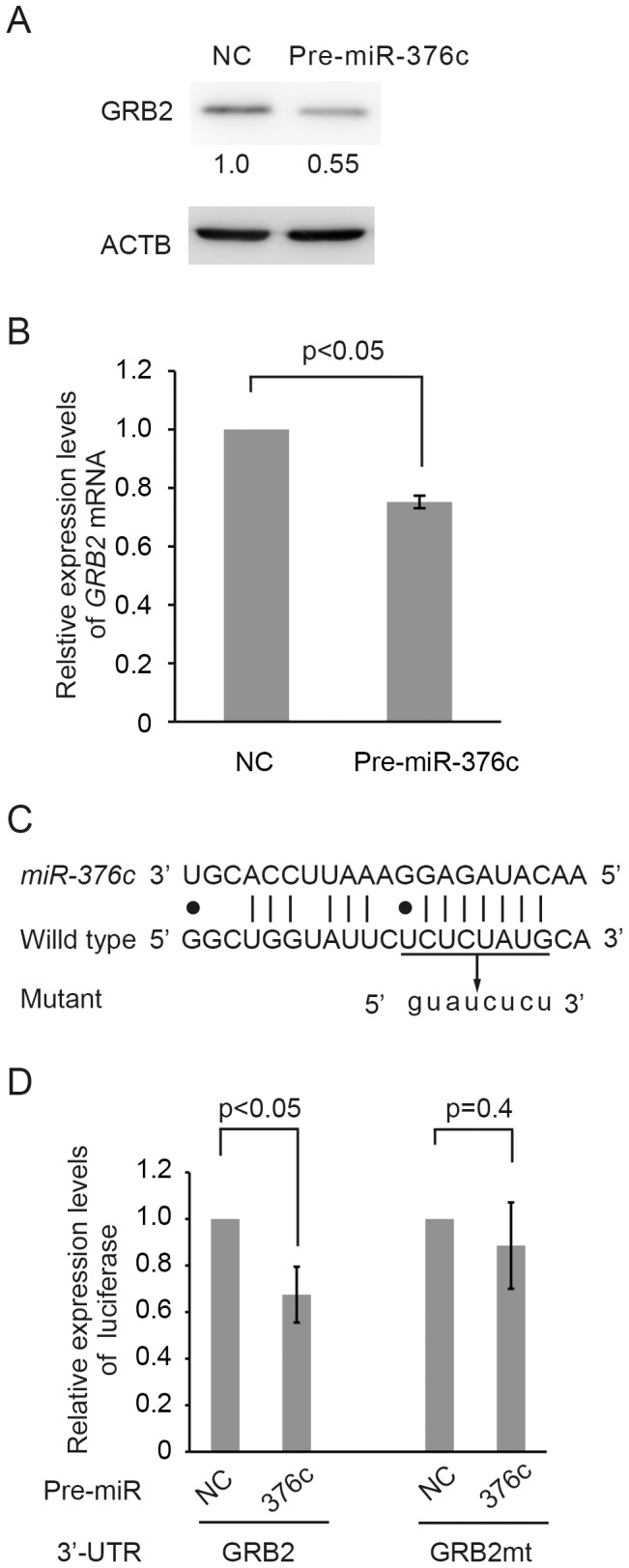
(A) Western blotting analysis of GRB2 protein levels in HuCCT1 cells transfected with Pre-miR-376c or the Pre-miR negative control (NC). ACTB was monitored as an internal control. Relative GRB2 expression levels were calculated and are indicated below the bands. (B) GRB2 mRNA expression levels in HuCCT1 cells transfected with Pre-miR-376c or the Pre-miR negative control (NC). The GRB2 expression level was normalized to GAPDH. The expression level of the NC sample was defined as 1. The significance of differences between means was determined by Student's t-test. (C) The sequences of the mature miR-376c and its putative target site in the 3'-UTR of GRB2. The target site corresponding to the seed sequence of miR-376c was converted via mutation; the mutation introduced into the miR-376c recognition site of GRB2 3'-UTR in the reporter plasmid is also shown. (D) GRB2 3'-UTR luciferase reporter assay. Reporter vector (pMIR-GRB2 [GRB2] or pMIR-GRB2mt [GRB2mt]) and Pre-miR molecule (Pre-miR-376c [376c] or Pre-miR negative control [NC]) were co-transfected into HuCCT1 cells. Renilla luciferase vector pRL-TK was used as an internal control. Luciferase expression levels of Pre-miR negative control (NC) were set to 1.0. The significance of differences between means was determined by Student's t-test.
Next, to determine whether the 3′-UTR of GRB2 is a direct target of miR-376c, we performed a luciferase reporter assay. As shown in Figure 2D, Pre-miR-376c overexpression decreased significantly the luciferase activity in HuCCT1 cells cotransfected with pMIR-GRB2 (by ∼30% compared to the negative control), but not significantly in cells cotransfected with pMIR-GRB2mt, a reporter plasmid mutated in the putative miR-376c recognition site of the GRB2 3′-UTR (Figure 2C). Taken together, these results suggest that GRB2 is a target of miR-376c in HuCCT1 cells.
MiR-376c regulates epidermal growth factor (EGF)-dependent migration of HuCCT1 via GRB2
GRB2 is known to be a key molecule in intracellular signal transduction through the EGF receptor (EGFR), and can be overexpressed in tumors of breast and bladder cancers [24]–[26]. Molecules that bind to GRB2 protein and inhibit signal transduction have been demonstrated to reduce motility in vitro and to decrease cancer metastasis in animal models [24], [27]–[29]. GRB2 plays a critical role in EGFR signal transmission and internalization [30]–[32]. EGF stimulation increases motility in multiple cell lines [32]. Therefore, we assessed the effect of miR-376c on migration of HuCCT1 cells stimulated with EGF in transwell assays. In the presence of EGF, the migration of HuCCT1 cells transfected with the Pre-miR-negative control was induced (Figure 3A), indicating that such migration is EGF dependent. Treatment with Pre-miR-376c significantly decreased EGF-induced migration (to 57% of the control value, Figure 3A). Furthermore, to confirm whether EGF-induced migration was dependent on GRB2, we analyzed migration by cells in which GRB2 was downregulated by siRNA-mediated knockdown. We used two siRNA duplexes specific for GRB2 (designated siGRB2-1 and siGRB2-2). First, we evaluated the efficiency of siRNA-mediated knockdown by Western blotting and real-time PCR. In HuCCT1 cells, siGRB2-1 and siGRB2-2 inhibited expression of both GRB2 protein (Figure 3B) and mRNA (Figure 3C). Next, we assayed migration of HuCCT1 cells treated with siRNAs against GRB2. After GRB2 knockdown, EGF-dependent cell migration was reduced significantly (Figure 3D). These results suggest that miR-376c regulates EGF-dependent cell migration through repression of GRB2 translation.
Figure 3. MiR-376c represses cell migration via GRB2 reduction.

(A) Transwell migration assay of HuCCT1 cells transfected with Pre-miR-376c. Medium containing 5 ng/ml EGF in the lower chamber served as a chemoattractant. After 24 h of transfection, migrating cells were stained and counted. Data are presented as the ratio of the number of migrating Pre-miR-376c (376c)-transfected cells to that of cells transfected with the Pre-miR-negative control (NC), in the presence or absence of EGF. Cell migration of NC in the presence of EGF was set to 1.0. The significance of differences among treatments was assessed by ANOVA followed by Tukey's test (* p<0.05). (B) Western blotting analysis of the GRB2 protein level in HuCCT1 cells transfected with the siRNAs. Two siRNA molecules targeting GRB2 (siGRB2-1 and siGRB2-2) and negative control siRNA (NC) were used. ACTB was used as an internal control. (C) Real-time PCR analysis of the GRB2 mRNA level in HuCCT1 cells transfected with the siRNAs. The expression level of cells transfected with negative control siRNA (NC) was set to 1.0. The GRB2 expression levels were normalized to GAPDH. (D) Transwell migration assay of HuCCT1 cells transfected with the siRNAs. Data are presented as numbers of migrating siRNA-transfected cells relative to cells transfected with the negative control siRNA (NC), in the presence or absence of EGF. Migration of the negative controls in the presence of EGF was set to 1.0. The significance of differences among treatments was assessed by ANOVA followed by Tukey's test (* p<0.05).
In addition, we assessed the effect of Pre-miR-376c and siGRB2 transfection on cell growth (Figure S1). No significant differences in cell growth were identified between transfected and non-transfected cells. This indicates that GRB2 modulates migration but not growth of HuCCT1 cells.
Network analysis relevant to GRB2-mediated HuCCT1 migration
To elucidate key molecules in EGF-dependent HuCCT1 migration, in which GRB2 upregulation is caused by abnormal suppression of miR-376c, we compared expression profiles of mRNAs affected by Pre-miR-376c and siGRB2 (siGRB2-2) in EGF-treated HuCCT1 cells. Microarray analysis showed that changes in expression of 1148 and 2733 genes were induced by Pre-miR-376c and siGRB2-2, respectively. In the microarray data, 85 genes were common to both sets (Figure 4A). Of these, 41 genes were downregulated by both treatments, and 44 were upregulated by both treatments (Table S1 and S2). We next conducted a network pathway analysis, using Ingenuity Pathway Analysis (IPA) to identify molecules relevant to GRB2-mediated cell migration. A network of 85 genes derived from the microarray data with c-Jun N-terminal kinase 1 (JNK1, also known as MAPK8)-mediated EGFR signaling was generated (Figure 4B). Ten of the 85 genes were associated with the EGFR pathway. From the viewpoint of the cellular function of miR-376c, two down-regulated genes; i.e., interleukin-1 beta (IL1B) and matrix metallopeptidase 9 (MMP9), were associated primarily with several cellular-movement networks in the Ingenuity Knowledge Base (Table S3). These data suggest that IL1B and MMP9 function downstream of GRB2 signaling in EGF-dependent cell migration by HuCCT1. Thus, we evaluated the gene suppression of IL1B and MMP9 in HuCCT1 cells treated with Pre-miR-376c and siGRB2-2. Pre-miR-376c significantly decreased (to 66%) expression of IL1B (Figure 4C) and MMP9 (to 52%, Figure 4D), compared to Pre-miR-negative controls. Use of siGRB2-2 also resulted in significant reductions in the expression of these genes, to 83% and 43% relative to controls, respectively (Figure 4C and 4D).
Figure 4. Network analysis relevant to GRB2-mediated HuCCT1 migration.

(A) Venn diagrams showing the significantly different mRNA expression levels of Pre-miR-376c and siGRB2-2 transfectants of HuCCT1 relative to appropriate controls. Expression profiles of mRNAs affected by Pre-miR-376c and siGRB2-2 in EGF-treated HuCCT1 cells were conducted by microarray analysis. The numbers of genes regulated by Pre-miR-376c and siGRB2-2 are indicated. (B) The network of the identified molecules regulated by both Pre-miR-376c and siGRB2-2 in this study were connected with EGF, EGFR and GRB2 by IPA analysis. Numbers below the upregulation (red) and downregulation (green) symbols represent the fold changes by Pre-miR-376c treatment; numbers in parentheses represent the fold changes by siGRB2-2 treatment. Solid and dotted lines indicate direct and indirect gene relationships, respectively. (C and D) Real-time PCR analysis of IL1B (C) and MMP9 (D) expression levels in EGF-treated HuCCT1 cells after transfection with Pre-miR-376c and siGRB2-2. Pre-miR-negative control and nonspecific non-silencing siRNA were used as negative controls (NC). For quantitative comparisons, expression levels were normalized to GAPDH. The expression levels in negative controls were set to 1.0. The significance of differences between means was determined by Student's t-test. * p<0.05.
MiR-376c expression is regulated by epigenetic control in HuCCT1
To determine whether miR-376c expression was dysregulated by epigenetic alterations in HuCCT1 cells, we first assessed the methylation status of the 6 CpG sites (sites I–VI) located 85 to 202 bp upstream of miR-376c; i.e., in the putative promoter region, in HIBEpiC and HuCCT1 cells by bisulfite genome sequencing (Figure 5A). A mutation in CpG site III was found in the genome sequence of HuCCT1. We quantified the relative level of methylation of the remaining CpG sites in HIBEpiC and HuCCT1 (Figure 5B). Three sites (I, II, and VI) in HuCCT1 cells were fully methylated, and the unmethylated levels at sites IV and V were 5% and 10%, respectively (Figure 5B). In contrast, in HIBEpiC cells the nonmethylation levels of sites I–VI were 14, 21, 4, 3.5, 35, and 45%, respectively (Figure 5B). These bisulfite sequencing data show that, with the exception of site IV, the levels of methylation at CpG sites in HuCCT1 cells are higher than those in HIBEpiC cells.
Figure 5. Methylation of miR-376c.

(A) Location of the six CpG sites (sites I–VI) upstream of miR-376c, in the putative promoter region of the gene. (B) Bisulfite sequencing analysis of these CpG sites in HIBEpiC and HuCCT1. The unmethylated levels of the six sites were expressed as percentages of methylation reference values. A mutation in the genome sequence of HuCCT1 was found at CpG site III. (C) Real-time PCR analysis of miR-376c expression levels in HuCCT1 cells treated with the DNA-demethylating agent 5-AZA-dCR and/or the HDAC inhibitor TSA. After treatment with 10 µM of 5-AZA-dCR for 3 days, HuCCT1 cells were incubated with TSA (0.1, 0.5, or 1.0 µM) for a further 24 h. Expression levels were normalized to RNU6B. The expression level in untreated HuCCT1 cells was defined as 1 (lane 1). Differences among treatments were tested by ANOVA followed by Tukey's test (* p<0.05).
DNA methylation and histone acetylation play critical roles in the expression of miRNA genes [33]–[35]. We next treated HuCCT1 cells with the DNA-demethylating agent 5-AZA-dCR and/or the HDAC inhibitor TSA. MiR-376c expression was increased slightly by 5-AZA-dCR or TSA treatment alone (Figure 5C, number 4 and 5). Combination treatment with 5-AZA-dCR and TSA showed that miR-376c was significantly upregulated (Figure 5C, number 7 and 8). These results indicate that the synergistic effects of DNA demethylation and histone acetylation are responsible for regulation of miR-376c expression.
Discussion
In the present study, we revealed that epigenetic repression of miR-376c accelerated EGF-dependent cell migration by targeting GRB2 in HuCCT1 cells. The investigation of differential expression profiles between cancer and normal cells is valuable for understanding both carcinogenesis and cancer progression. We previously performed miRNA sequencing analysis of HuCCT1 and HIBEpiC [18]. MiR-376c was cloned exclusively from the normal cell line HIBEpiC, and not from HuCCT1. Here, we focused on functional studies of miR-376c, which is significantly downregulated in CC cell lines. MiR-376c belongs to the miR-376 cluster gene family, containing miR-376a, miR-376a*, and miR-376b. Kawahara et al. showed that miR-376 transcripts were subject to RNA editing, converting adenosine to inosine in a tissue-dependent manner, leading to a change in the silenced gene set [36]. Recently Ye et al. reported that activin receptor-like 7 (ALK7), which induces apoptosis via its ligand Nodal, was a target, and that it was downregulated in ovarian cancer cells [37]. Two reports of miR-376c upregulation in a subset of acute myeloid leukemia samples have been published [38], [39]. However, the target genes and functions of miR-376c in biliary epithelial cells have been not reported. In silico prediction of target mRNA candidates is a useful tool [40]; however, the specification of functional targets in cells by computational prediction only is generally thought to be difficult. MiRNAs are able to interact with the 3′-UTRs of many mRNAs and primarily repress their translation. Thus, we performed proteomic analysis, which is often employed to elucidate direct targets of miRNAs [41], [42]. Our results demonstrated that GRB2, an in silico miR-376c target candidate, was significantly downregulated in miR-376c-overexpressing HuCCT1 cells. Moreover, a 3′-UTR reporter assay confirmed that miR-376c repressed the translation of GRB2 in a seed-sequence-dependent manner in HuCCT1 cells (Figure 2).
In migration experiments using Pre-miR-376c and siGRB2s, downregulation of GRB2 protein was shown to reduce EGF-dependent migration of HuCCT1 cells. Down-regulation of miR-376c allowed an increase in the amount of GRB2 protein, resulting in acceleration of HuCCT1 migration. Conversely, in normal intrahepatic biliary epithelial cells, the expression of miR-376c might regulate the amount of GRB2 protein and cell migration. GRB2 is a key adapter protein, involved in signal transduction via binding to various other proteins, including the EGF receptor [24], [43], [44]. GRB2 plays essential roles in mediating EGFR signaling. A recent report showed that GRB2 interacted directly with Ras homolog family member U (RhoU) and with activated EGFR, leading to augmented JNK1 pathway activity and cell migration [45]. Although the expression and function of RhoU in bile duct cells are unknown, miR-376c may modulate EGF-mediated signaling through physical interaction between RhoU and GRB2. GRB2 is also well known to activate the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway [46]–[48]. In cancer cells, this pathway is also activated and stimulates cell growth [47], [48]. We did not, however, observe an effect of miR-376c on cell growth in this cancer cell type (Figure S1).
From microarray profiling and knowledge-based network analysis, we identified IL1B and MMP9 as putative factors correlated with cell migration in HuCCT1 (Figure 4). Furthermore, in this study, we demonstrated that IL1B and MMP9 were significantly downregulated in HuCCT1 cells treated with Pre-miR-376c or siGRB2. MMP9 is a matrix metallopeptidase that has been shown to be involved in degradation of basement membrane proteins in the extracellular matrix [49]. This MMP forms a homodimer, and this structure is required in the mechanism of MMP9-mediated cell migration [50]. Intriguingly, it has been reported that IL1B regulates MMP9 expression in human umbilical vein endothelial cells [51] and in extravillous trophoblasts [52]. Taken together with previous observations, our findings lend support to the idea that IL1B may be downstream of EGF signaling, such that EGF can accelerate the expression of IL1B and subsequent activation of MMP9 in HuCCT1 cells. We did not fully explore the signaling pathways of EGF-dependent cell migration in HuCCT1. Further studies will elucidate the detailed mechanisms of metastasis in ICC.
Mechanisms controlling miR-376c gene expression have not been reported. In this study, combination treatment with 5-AZA-dCR and TSA significantly induced the expression of miR-376c in HuCCT1 cells (Figure 5C), suggesting epigenetic, including histone, modification. The synergistic effect of DNA demethylation and HDAC inhibition is essential for strong expression of some genes [53], [54]. Saito et al. reported the miRNA expression profile of T24 bladder cancer cells treated with 5-AZA-dCR and the HDAC inhibitor 4-phenylbutyric acid, and showed that miR-376c was one of the miRNAs upregulated after treatment [35]. This is consistent with our results. We also found that CpG sites upstream of the miR-376c gene were highly methylated in HuCCT1 cells. Our data suggest that DNA methylation and HDAC modification suppress the transcription of miR-376c in these cells.
In summary, we have shown that GRB2-targeted miR-376c is epigenetically repressed in HuCCT1 cells, resulting in acceleration of cell migration by EGF signaling enhancement via GRB2. Since metastasis is the major cause of death in ICC, elucidating the mechanisms underlying EGF-dependent cell migration could facilitate the development of novel anti-cancer strategies for ICC using miRNAs. Although our experiments were conducted with an ICC cell line model in vitro, our findings on miR-376c function provide a new insight into pathophysiology of ICC. Further functional and pathological studies are needed to understand the mechanism[s] of miRNA-mediated posttranscriptional regulation in ICC.
Supporting Information
The effects of Pre-miR-376c and siGRB2s transfection on cell growth. HuCCT1 cells (5×103) were seeded in 96-well plates in a serum-containing medium 1 day prior to transfection. Cells were transfected with the Pre-miRNA or siGRB2s at final concentrations of 30 nM using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 24 h, the medium was changed to a medium containing RPMI 1640 supplemented with 0.1% fetal bovine serum in the presence or absence of EGF. After a further 24 h, cell growth was assayed using CellTiter-Glo (Promega, USA) according to the manufacturer's protocol. Graph shows relative cell growth. Values for cells transfected with Pre-miR-negative control (Pre-miR Cont) or nonspecific non-silencing siRNA (siRNA Cont) (AllStars Negative Control siRNA, Qiagen) were set to 1.0. No statistically significant differences between groups were detected (ANOVA followed by Tukey's test).
(TIF)
Genes downregulated by Pre-miR-376c and siGRB2-2.
(XLS)
Genes upregulated by Pre-miR-376c and siGRB2-2.
(XLS)
Molecular and cellular functions of downregulated genes associated with cellular movement in Ingenuity's Knowledge Base.
(XLS)
Acknowledgments
We thank Mr. Takuji Kosuge and Ms. Yoshimi Hinohara for their technical assistance. We also thank Dr. Tomoko Ishikawa for her help in proteomic analysis and Dr. Atsushi Watanabe for GeneSpring analysis.
Funding Statement
This work was supported by Grants-in-Aids for Scientific Research and Private University Strategic Research Foundation Support Program from the Ministry of Education, Culture, Sports, Science and Technology/Japan Society for the Promotion of Science, Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, et al. (2004) Global identification of human transcribed sequences with genome tiling arrays. Science 306: 2242–2246. [DOI] [PubMed] [Google Scholar]
- 2. Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, et al. (2005) The transcriptional landscape of the mammalian genome. Science 309: 1559–1563. [DOI] [PubMed] [Google Scholar]
- 3. Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, et al. (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308: 1149–1154. [DOI] [PubMed] [Google Scholar]
- 4. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu L, Belasco JG (2008) Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell 29: 1–7. [DOI] [PubMed] [Google Scholar]
- 6. Shenouda SK, Alahari SK (2009) MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev 28: 369–378. [DOI] [PubMed] [Google Scholar]
- 7. Wang XW, Heegaard NH, Orum H (2012) MicroRNAs in liver disease. Gastroenterology 142: 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, et al. (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102: 13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, et al. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10: 593–601. [DOI] [PubMed] [Google Scholar]
- 10. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, et al. (2005) RAS is regulated by the let-7 microRNA family. Cell 120: 635–647. [DOI] [PubMed] [Google Scholar]
- 11. Malhi H, Gores GJ (2006) Cholangiocarcinoma: modern advances in understanding a deadly old disease. J Hepatol 45: 856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakanuma Y, Curado MP, Franceschi S, Gores G, Paradis V, et al.. (2010) Intrahepatic cholangiocarcinoma. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, editors. WHO Classification of Tumours of the Digestive System. Lyon: IARC Press. pp. 217–224.
- 13.Anthony PP (2002) Tumours and tumour-like lesions of the liver and the biliary tract: etiology, epidemiology and pathology. In: MacSween RNM, Burt AD, Portmann BC, Ishak KG, Scheuer PJ, Anthony PP, editors. Pathology of the Liver. London: Churchill Livingstone. pp. 711–775.
- 14. Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD (2005) Cholangiocarcinoma. Lancet 366: 1303–1314. [DOI] [PubMed] [Google Scholar]
- 15. Patel T (2006) Cholangiocarcinoma. Nat Clin Pract Gastroenterol Hepatol 3: 33–42. [DOI] [PubMed] [Google Scholar]
- 16. Olnes MJ, Erlich R (2004) A review and update on cholangiocarcinoma. Oncology 66: 167–179. [DOI] [PubMed] [Google Scholar]
- 17. Shirabe K, Shimada M, Harimoto N, Sugimachi K, Yamashita Y, et al. (2002) Intrahepatic cholangiocarcinoma: its mode of spreading and therapeutic modalities. Surgery 131: S159–S164. [DOI] [PubMed] [Google Scholar]
- 18. Kawahigashi Y, Mishima T, Mizuguchi Y, Arima Y, Yokomuro S, et al. (2009) MicroRNA profiling of human intrahepatic cholangiocarcinoma cell lines reveals biliary epithelial cell-specific microRNAs. J Nippon Med Sch 76: 188–197. [DOI] [PubMed] [Google Scholar]
- 19. Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, et al. (2006) Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology 130: 2113–2129. [DOI] [PubMed] [Google Scholar]
- 20. Luo SS, Ishibashi O, Ishikawa G, Ishikawa T, Katayama A, et al. (2009) Human villous trophoblasts express and secrete placenta-specific microRNAs into maternal circulation via exosomes. Biol Reprod 81: 717–729. [DOI] [PubMed] [Google Scholar]
- 21. Baek D, Villén J, Shin C, Camargo FD, Gygi SP, et al. (2008) The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, et al. (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63. [DOI] [PubMed] [Google Scholar]
- 23. Giehl K, Skripczynski B, Mansard A, Menke A, Gierschik P (2000) Growth factor-dependent activation of the Ras-Raf-MEK-MAPK pathway in the human pancreatic carcinoma cell line PANC-1 carrying activated K-ras: implications for cell proliferation and cell migration. Oncogene 19: 2930–2942. [DOI] [PubMed] [Google Scholar]
- 24. Giubellino A, Burke TR Jr, Bottaro DP (2008) Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets 12: 1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daly RJ, Binder MD, Sutherland RL (1994) Overexpression of the Grb2 gene in human breast cancer cell lines. Oncogene 9: 2723–2727. [PubMed] [Google Scholar]
- 26. Verbeek BS, Adriaansen-Slot SS, Rijksen G, Vroom TM (1997) Grb2 overexpression in nuclei and cytoplasm of human breast cells: a histochemical and biochemical study of normal and neoplastic mammary tissue specimens. J Pathol 183: 195–203. [DOI] [PubMed] [Google Scholar]
- 27. Atabey N, Gao Y, Yao ZJ, Breckenridge D, Soon L, et al. (2001) Potent blockade of hepatocyte growth factor-stimulated cell motility, matrix invasion and branching morphogenesis by antagonists of Grb2 Src homology 2 domain interactions. J Biol Chem 276: 14308–14314. [DOI] [PubMed] [Google Scholar]
- 28. Gay B, Suarez S, Weber C, Rahuel J, Fabbro D, et al. (1999) Effect of potent and selective inhibitors of the Grb2 SH2 domain on cell motility. J Biol Chem 274: 23311–23315. [DOI] [PubMed] [Google Scholar]
- 29. Giubellino A, Gao Y, Lee S, Lee MJ, Vasselli JR, et al. (2007) Inhibition of tumor metastasis by a growth factor receptor bound protein 2 Src homology 2 domain-binding antagonist. Cancer Res 67: 6012–6016. [DOI] [PubMed] [Google Scholar]
- 30. Sorkin A (2001) Internalization of the epidermal growth factor receptor: role in signalling. Biochem Soc Trans 29: 480–484. [DOI] [PubMed] [Google Scholar]
- 31. Yamazaki T, Zaal K, Hailey D, Presley J, Lippincott-Schwartz J, et al. (2002) Role of Grb2 in EGF-stimulated EGFR internalization. J Cell Sci 115: 1791–1802. [DOI] [PubMed] [Google Scholar]
- 32. Jiang X, Huang F, Marusyk A, Sorkin A (2003) Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell 14: 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chuang JC, Jones PA (2007) Epigenetics and microRNAs. Pediatr Res 2007 61: 24R–29R. [DOI] [PubMed] [Google Scholar]
- 34. Sato F, Tsuchiya S, Meltzer SJ, Shimizu K (2011) MicroRNAs and epigenetics. FEBS J 278: 1598–1609. [DOI] [PubMed] [Google Scholar]
- 35. Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, et al. (2006) Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9: 435–443. [DOI] [PubMed] [Google Scholar]
- 36. Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, et al. (2007) Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ye G, Fu G, Cui S, Zhao S, Bernaudo S, et al. (2011) MicroRNA 376c enhances ovarian cancer cell survival by targeting activin receptor-like kinase 7: implications for chemoresistance. J Cell Sci 124: 359–368. [DOI] [PubMed] [Google Scholar]
- 38. Dixon-McIver A, East P, Mein CA, Cazier JB, Molloy G, et al. (2008) Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS One 3: e2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Z, Lu J, Sun M, Mi S, Zhang H, et al. (2008) Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA 105: 15535–15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saito T, Saetrom P (2010) MicroRNAs–targeting and target prediction. N Biotechnol 27: 243–249. [DOI] [PubMed] [Google Scholar]
- 41. Muniyappa MK, Dowling P, Henry M, Meleady P, Doolan P, et al. (2009) MiRNA-29a regulates the expression of numerous proteins and reduces the invasiveness and proliferation of human carcinoma cell lines. Eur J Cancer 45: 3104–3118. [DOI] [PubMed] [Google Scholar]
- 42. Yang F, Zhang L, Wang F, Wang Y, Huo XS, et al. (2011) Modulation of the unfolded protein response is the core of microRNA-122-involved sensitivity to chemotherapy in hepatocellular carcinoma. Neoplasia 13: 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70: 431–442. [DOI] [PubMed] [Google Scholar]
- 44. Rozakis-Adcock M, Fernley R, Wade J, Pawson T, Bowtell D (1993) The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 363: 83–85. [DOI] [PubMed] [Google Scholar]
- 45. Zhang JS, Koenig A, Young C, Billadeau DD (2011) GRB2 couples RhoU to epidermal growth factor receptor signaling and cell migration. Mol Biol Cell 22: 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dance M, Montagner A, Salles JP, Yart A, Raynal P (2008) The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal 20: 453–459. [DOI] [PubMed] [Google Scholar]
- 47. Scaltriti M, Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12: 5268–5272. [DOI] [PubMed] [Google Scholar]
- 48. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, et al. (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773: 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, et al. (1993) Matrix metalloproteinases: a review. Crit Rev Oral Biol Med 4: 197–250. [DOI] [PubMed] [Google Scholar]
- 50. Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J (2010) Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J Biol Chem 285: 35944–35956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Qin W, Lu W, Li H, Yuan X, Li B, et al. (2012) Melatonin inhibits IL1beta-induced MMP9 expression and activity in human umbilical vein endothelial cells by suppressing NF-kappaB activation. J Endocrinol 214: 145–153. [DOI] [PubMed] [Google Scholar]
- 52. Lockwood CJ, Oner C, Uz YH, Kayisli UA, Huang SJ, et al. (2008) Matrix metalloproteinase 9 (MMP9) expression in preeclamptic decidua and MMP9 induction by tumor necrosis factor alpha and interleukin 1 beta in human first trimester decidual cells. Biol Reprod 78: 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB (1999) Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 21: 103–107. [DOI] [PubMed] [Google Scholar]
- 54. Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, et al. (2003) A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 31: 141–149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effects of Pre-miR-376c and siGRB2s transfection on cell growth. HuCCT1 cells (5×103) were seeded in 96-well plates in a serum-containing medium 1 day prior to transfection. Cells were transfected with the Pre-miRNA or siGRB2s at final concentrations of 30 nM using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 24 h, the medium was changed to a medium containing RPMI 1640 supplemented with 0.1% fetal bovine serum in the presence or absence of EGF. After a further 24 h, cell growth was assayed using CellTiter-Glo (Promega, USA) according to the manufacturer's protocol. Graph shows relative cell growth. Values for cells transfected with Pre-miR-negative control (Pre-miR Cont) or nonspecific non-silencing siRNA (siRNA Cont) (AllStars Negative Control siRNA, Qiagen) were set to 1.0. No statistically significant differences between groups were detected (ANOVA followed by Tukey's test).
(TIF)
Genes downregulated by Pre-miR-376c and siGRB2-2.
(XLS)
Genes upregulated by Pre-miR-376c and siGRB2-2.
(XLS)
Molecular and cellular functions of downregulated genes associated with cellular movement in Ingenuity's Knowledge Base.
(XLS)