

Abstract
Background
Reports of Ca2+ current (ICa) loss after injury to peripheral sensory neurons do not discriminate between axotomized and spared neurons. The spinal nerve ligation model separates axotomized from spared neurons innervating the same site. The authors hypothesized that ICa loss is a result of neuronal injury, so they compared axotomized L5 dorsal root ganglion neurons to spared L4 neurons, as well as neurons from rats undergoing skin incision alone.
Methods
After behavioral testing, dissociated neurons from L4 and L5 dorsal root ganglia were studied in both current and voltage patch clamp modes. The biophysical consequence of ICa loss on the action potential was confirmed using selective ICa antagonists. Data were grouped into small, medium, and large cells for comparison.
Results
Reduced ICa was predominantly a consequence of axotomy (L5 after spinal nerve ligation) and was most evident in small and medium neurons. ICa losses were associated with action potential prolongation in small and medium cells, whereas the amplitude and duration of after hyperpolarization was reduced in medium and large neurons. Blockade with Ca2+ channel antagonists showed that action potential prolongation and after hyperpolarization diminution were alike, attributable to the loss of ICa.
Conclusion
Axotomy is required for ICa loss. ICa loss correlated with changes in the biophysical properties of sensory neuron membranes during action potential generation, which were due to ICa loss leading to decreased outward Ca2+-sensitive K+ currents. Taken together, these results suggest that neuropathic pain may be mediated, in part, by loss of ICa and the cellular processes dependent on Ca2+.
FEW conditions resist effective treatment like pain resulting from nerve injury. For patients with limb trauma or chronic diseases such as diabetes or tumor formation, the cost of untreated pain is loss of function or total incapacity. Animal models in which the peripheral nerve is incompletely injured result in heightened reaction to mechanical stimulation indicative of neuropathic pain.1 Explaining the molecular and chemical bases of these changes will suggest means of better treatment.2
We have reported that chronic constriction injury3 of the sciatic nerve reduces calcium currents (ICa) in somata of primary afferent neurons in the dorsal root ganglia (DRG) proximal to the injury. These findings imply that decreased ICa may contribute to aberrant sensory processing, but it is not known whether ICa loss is solely a direct effect of axotomy or also an indirect effect of injury on adjacent, intact neurons. The spinal nerve ligation (SNL) model of neuropathic pain4 segregates the somata of axotomized L5 neurons from the adjacent L4 neurons that share the sciatic nerve, where intact L4 axons are exposed to inflammatory mediators induced by the degeneration of distal segments of L5 neurons.5–7 This model thus provides the opportunity to separately test the roles of neurons affected by these different pathogenic processes. Because the relative roles of axotomized and neighboring intact neurons are unresolved,8 we compared neurons from the ligated L5 DRG and the adjacent L4 DRG.9,10 We hypothesized that ICa loss is a generalized effect of painful sensory neuron injury and would be found after SNL as well as after chronic constriction injury. We further expected that the loss of current would be predominantly expressed in directly injured L5 neurons as compared with L4 neurons.
Voltage-activated Ca2+ channels contribute to shaping the action potential (AP) profile and neuronal excitability.11,12 To determine the biophysical role of decreased ICa, we measured the effects of injury on both the AP parameters and the underlying ICa in the same neurons. The normal AP of sensory somata is only a few milliseconds long, so the response to conventional patch clamp test pulses lasting hundreds of milliseconds may not accurately depict ICa during normal neuronal activity. By exposing each cell to a voltage command in the shape of an AP waveform, we measured total charge transfer as a representation of net Ca2+ entry into the cell under natural physiologic conditions. Finally, complex interactions of various currents make prediction of net effects of the change in any one current difficult to predict. Specifically, inward Ca2+ current depolarizes the membrane, but the arrival of Ca2+ in the cytoplasm triggers the opening of Ca2+-activated K+ channels that conduct an outward repolarizing current IK(Ca). The balance of these two actions dictates the outcome of decrease ICa on neuronal function. We therefore determined the direct consequences of ICa loss on AP characteristics and overall membrane currents using selective Ca2+ channel antagonists.
Materials and Methods
Animal Preparation
Two hundred forty-five cells were derived from 98 adult male Sprague-Dawley rats weighing 125–150 g provided by a single vendor (Charles River Laboratories Inc., Wilmington, MA), after approval of the Medical College of Wisconsin Institutional Animal Care and Use Committee (Milwaukee, Wisconsin). An additional 67 cells obtained from 21 adult male Sprague-Dawley rats (Taconic Farms, Inc., Hudson, NY) were studied in a separate group. Rats received either ligation and section of the right L5 and L6 spinal nerves (SNL) approximately 5 mm distal to the DRG4 or control surgery with skin incision only. After fully recumbent recovery from halothane anesthesia, rats were returned to individual cages under climate- and light-controlled conditions for 10 days before behavioral testing as previously described.9 Rats were weighed and placed on a 0.25-in wire grid in clear plastic enclosures and allowed to rest for 30 min. Five applications of a 22-gauge spinal needle were made to the plantar skin of each paw and repeated 3 min later, using a force adequate to indent the skin but not to puncture it. These mechanical stimuli produced either a normal brief flinch, or a hyperalgesic-type response characterized by sustained (> 2 s) paw lifting, shaking, and licking. Only SNL rats with an ipsilateral response rate of at least 20% averaged over 3 test days and normal contralateral responses were used for study. The validity of this approach to behavioral testing is supported by the observation that control rats never exhibited characteristic hyperalgesic responses.
Cell Isolation and Solutions
After a postsurgical interval of 15.8 ± 1.4 days, the L4 and L5 ganglia were removed after decapitation during halothane anesthesia, placed into separate 35-mm Petri dishes containing Ca2+ and Mg2+-free iced Hanks’ Balanced Salt Solution, and minced with iris scissors. Using separate sterile, silicone-coated (Sigmacote; Sigma, St. Louis, MO) Pasteur pipettes, each ganglion was placed into a 25-ml sterile tissue culture flask containing 0.0625% trypsin (Boehringer-Mannheim, Indianapolis, IN), 0.0125% DNase (Sigma), and 0.01% blendzyme 2 (Roche Molecular Biochemicals, Indianapolis, IN) in DMEM/F12 with glutaMAX (Invitrogen Corporation, Carlsbad, CA) for 1.5 h in a tissue shaking bath at 32°C and 70 rpm. Neurons were resuspended in adult neural basal media (1×; Invitrogen Corporation, Carlsbad, CA) containing 2% (vol:vol) B27 supplement (50 ×; Invitrogen Corporation), 0.5 mM glutamine, 0.02 mg/ml gentamicin, and 100 ng/ml nerve growth factor 7S (Alomone Labs, Ltd., Jerusalem, Israel). Dissociated L4 and L5 cells from each tube were plated onto separate poly-L-lysine (70–150 kd)-coated coverslips and placed in a 95:5 oxygen-carbon dioxide, water-jacketed incubator and allowed to plate for 2 h before study. All cells were studied within 8 h after plating.
Current clamp solutions duplicated natural cytosolic and extracellular conditions. A modified Tyrode solution was used externally in the bath during current clamp protocol, consisting of the following: 140 mM NaCl, 4 mM KCl, 2 mM CaCl, 2 mM MgCl, 10 mM D-glucose, and 10 mM HEPES at a pH of 7.4 with NaOH and an osmolarity of 300 mOsm. Internal pipette solution contained 120 mM KCl, 5 mM na-ATP, 0.4 mM na-GTP, 5 mM EGTA, 2.25 mM CaCl2, 5 mM MgCl2, and 20 mM HEPES at a pH of 7.4 with KOH and an osmolarity of 296 mOsm. Because internal pipette solutions cannot be changed after seal formation, ICa was isolated by external blockade of other voltage-sensitive currents. The following solution provided adequate separation from other currents: 2 mM BaCl2, 1 mM 4-aminophyridine, 10 mM HEPES, 160 mM tetraethylammonium chloride, and 0.1 mM tetrodotoxin at a pH of 7.4 with tetraethylammonium hydroxide and an osmolarity of 300 mOsm. Tetrodotoxin blocked tetrodotoxin-sensitive INa, and 4-aminophyridine, tetraethylammonium chloride, and Ba2+ blocked potassium currents. Because the change in external solutions results in a +4 mV shift in junction potential, no equivalent offset was added after switching from current clamp to voltage clamp. Cadmium (200 μM) was added at the end of some studies to verify the efficacy of ICa isolation. In a separate protocol examining AP parameters and whole cell currents before and after ICa blockade, Ca2+ channel antagonists were applied directly to the cell through a pressure regulated microperfusion system (ALA-BP8 system; ALA Scientific Instruments, Westbury, NY). Complete ablation of high voltage-activated Ca+ channels was achieved with a cocktail of antagonists that contained nisoldipine (200 nM), SNX-111 (200 nM), agatoxin IVA (200 nM), and SNX-482 (200 nM) to block L-, N-, P/Q-, and R-type calcium channel subtypes, respectively. Tetrodotoxin was obtained from Alomone Labs. SNX-111 was the kind gift of Dr. Scott Bowersox, Ph.D. (Department of Pharmacology, Neurex Corporation, Menlo Park, CA), agatoxin IVA was a gift of Dr. Nicholas Saccomano, Ph.D. (Assistant Director, Medical Chemistry Research at Pfizer, Groton, CT), and SNX-482 was purchased from Peptides International (Louisville, KY).
Recording Protocols
All cells were recorded in current clamp before changing to voltage clamp. Cells with resting membrane potentials more depolarized than −45 mV were excluded from study. Efficacy of potassium and sodium channel blockade was confirmed by observation of a reversal of negative membrane potential while still in current clamp and a lack of brief, inward currents during voltage clamp recording. Terminal application of Cd2+ to block ICa showed no residual voltage-activated currents. Membrane capacitance was measured and access resistance compensated from 80 to 95%. Cells with greater than 10 MΩ access resistance were rejected. Most recordings were made on a List EP-7 amplifier (ALA Scientific Instruments) modified by the addition of a 150-kΩ resistor to the current injection circuit to increase maximum stimulus to 5 nA. An A/D converter (Axon 1200B; Axon Instruments, Union City, CA) attached to a personal computer running pClamp 8 (Axon Instruments) was used to record data. A second series of experiments on large and medium cells with ICa of up to 72.5 nA were performed on an Axon 200B amplifier (Axon Instruments) with a different A/D converter (Axon 1320; Axon Instruments).
Current Clamp
Initial observation of resting membrane potential determined cell viability before evoked potentials. To determine AP characteristics, a 1-s step protocol was used with current sufficient (between 10 pA and 2 nA) to evoke an AP (fig. 1). AP voltage threshold (APthresh) was defined as the level of the inflection at the beginning of the rapid upstroke,13 whereas AP duration was measured at 50% of peak (AP50). Inflection on the repolarization phase of the AP was observed, and the number of APs during 1 s current injection was determined to characterize the neuron as either adapting or repetitive firing. Continuous current injection and positive feedback from patch clamp head stage amplification prolongs AP duration.14 Nevertheless, the relative effect of injury is still detectable. To avoid distortion from continuous current injection during measurement of the afterhyperpolarization (AHP), additional APs were evoked by a brief, 2-ms, 5-nA depolarizing pulse, and peak hyperpolarization (AHPampl) and duration at 50% amplitude (AHP50) were recorded, as well as the time constant for resolution of the AHP (AHPτ) by fitting the recovery phase to a single exponential (fig. 1).
Fig. 1.

(A) Measures of action potential (AP) produced by 1-s current injection, shown below the voltage trace. An inflection on the descending limb of the AP is demonstrated. Threshold for initiation of the AP. RMP = resting membrane potential; AP50 = AP duration at 50% of peak. (B) Determination of afterhyperpolarization (AHP) dimensions after an AP initiated by a 2-ms current injection, shown below the voltage trace. AHP amplitude is measured from RMP to peak. AHP50 = AHP duration at 50% recovery; AHPτ = time constant for AHP recovery. (C) Measures of Ca2+ current (ICa) response to voltage command in the form of an AP waveform. Inward ICa is measured at peak and as the integral of total inward current (Ca2+ charge transfer) during AP waveform.
Voltage Clamp
All cells were exposed to both a sustained square wave protocol and a standardized AP waveform voltage command protocol. The square wave protocol consisted of 200-ms commands from a holding potential of −90 to +50 mV in 10-mV increments with 5-s intervals between steps. Measured inward current was normalized by membrane capacitance, which results in a current density corrected for cell size. Peak inward current was determined, and linear leak was subtracted post hoc by fitting the first three steps before significant ICa activation to a linear function. Maximum conductance (Gmax) was determined by fitting peak inward current to a Boltzmann function, as previously described.15 Some large cells produced currents in excess of the bandwidth of the List EP-7 amplifier. Therefore, maximal ICa for large cells was calculated by linear fit of current-voltage peaks from the three traces before and at the reversal potential. Gmax determined by this procedure differed from that determined by Boltzmann fit by ±3% in 12 nonsaturating cells.
A standardized AP waveform command was used to reveal the effects of naturally rapid depolarization and repolarization on ICa across all cells. The basic features of the standardized wave were an inflection on the repolarization phase and a prolonged AHP to mimic standard features of a nociceptor (fig. 1). A modified P8 wave was used to subtract capacitance; namely, the AP waveform was inverted and divided by 8 to acquire membrane capacitance with minimal channel activation. Each P8 wave was subsequently multiplied by −8 and subtracted from the current in response to an AP waveform voltage command. Peak inward current and total charge transfer (current integrated over time) were determined by cursor measurements after current traces crossed 0 mV at the beginning and end of a wave (fig. 1). Charge transfer (coulombs) was normalized to cell area by converting cell capacitance to the specific capacitance of plasma membranes (0.01 pF/μm2) to compare Ca2+ flux between cells of different sizes. Because cells reliably produced a triangular current waveform (fig. 1), peak and area for large cells with saturating currents were calculated by linear extrapolation of the nonsaturating portion of the trace. The reliability of this method was tested on artificially truncated traces of 12 nonsaturating cells, with results varying ±3% from actual measured values.
Statistical Analyses
Cells were categorized according to cell size and surgical preparation. As noted elsewhere,16,17 cell size correlates roughly to neuronal types, so we classified cells as small (C type, < 30 μm), medium (Aδ type, 30–40 μm), or large (Aα/Aβ type, > 40 μm). Neurons were further grouped according to surgical preparation (control or SNL) and DRG level (L4 or L5). Because there were no differences in responses from L4 and L5 ganglia from control rats, these cells were combined, resulting in a control group, an SNL L4 group, and an SNL L5 group. Recordings were evaluated post hoc using Clampfit 8 (Axon Instruments), and data were summarized in Excel (Microsoft Co., Redmond, WA) and analyzed with Statistica 6 (StatSoft, Tulsa, OK) for analysis of variance and paired t tests or GraphPad Prism 4 for Mac (Graph-Pad Software, San Diego, CA) for correlations. Values are expressed as mean ± SEM. For each dependent variable, the main effect of a group was tested with standard univariate analysis of variance, followed by planned Bonferroni post hoc comparisons between groups. Normal distribution was not assumed for large cells in voltage clamp mode (confirmed by Kolmogorov-Smirnov test for normalcy) because of occasional outliers with very large currents. For this size group, a nonparametric Kruskal-Wallis analysis was used with the Dunn test for post hoc comparisons. Significance was estimated at P ≤ 0.05 versus control. Drug effects were evaluated with paired t tests for dependent samples before and after drug administration. Pearson tests were calculated to determine the coefficient of correlation between Gmax in voltage clamp and AP or AHP parameters in current clamp. Two-tailed P values and confidence levels were computed to determine whether covariance (r2) was different from zero.
Results
Current Clamp
Injury was associated with longer action potential duration (AP50) in small and medium neurons from L5 ganglia compared with control cells, whereas medium and large cells from the same ganglion were characterized by AHPs with lower amplitudes and shorter durations after injury (table 1 and fig. 2). The APthresh was significantly reduced only in small cells from the L5 DRG of SNL rats compared with control neurons.
Table 1.
Injury Effects on AP Parameters and ICa
| Command | Current Clamp
|
Voltage Clamp: Square Wave |
Voltage Clamp: AP Waveform
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cell Size | Injury Model |
n | APdur50, ms | APthresh, mV | AHPampl, mV | AHPdur50, ms | AHPτ, ms | Gmax, pS/pF | Ipeak, pA/pF | Charge Transfer, fC/μm2 |
| Small | Control | 20 | 4.4 ± 0.4 | −10.1 ± 2.0 | −15.8 ± 1.5 | 38.3 ± 4.8 | 55.7 ± 9.5 | 3.7 ± 0.3 | −249.9 ± 19.8 | 4.66 ± 0.20 |
| SNL L4 | 20 | 5.0 ± 0.5 | −11.5 ± 2.2 | −16.6 ±1.2 | 34.3 ± 7.3 | 41.0 ± 10.6 | 3.1 ± 0.4 | −220.3 ± 29.1 | 4.08 ± 0.52 | |
| SNL L5 | 19 | 14.8 ± 0.5* | −14.7 ± 1.8* | −14.9 ± 1.4 | 42.7 ± 7.2 | 53.2 ± 12.2 | 2.3 ± 0.2* | −143.1 ± 17.2* | 2.76 ± 0.40* | |
| Medium | Control | 20 | 3.4 ± 0.5 | −20.2 ± 2.0 | −15.3 ± 1.1 | 70.2 ± 10.4 | 121.2 ± 21.7 | 3.5 ± 0.4 | −180.0 ± 19.5 | 4.06 ± 0.47 |
| SNL L4 | 14 | 4.6 ± 1.1 | −22.4 ± 3.4 | −9.1 ± 1.8* | 45.4 ± 11.3 | 70.8 ± 15.9 | 2.6 ± 0.4 | −114.1 ± 21.6* | 2.52 ± 0.57* | |
| SNL L5 | 24 | 6.4 ± 1.1* | −19.4 ± 1.7 | −10.9 ± 0.8* | 20.8 ± 2.9* | 29.6 ± 4.9* | 2.2 ± 0.3* | −113.6 ± 16.5* | 2.54 ± 0.37* | |
| Large | Control | 16 | 3.6 ± 1.4 | −16.7 ± 3.2 | −14.9 ± 1.2 | 90.5 ± 14.1 | 181.6 ± 23.6 | 3.9 ± 0.5 | −192.2 ± 21.4 | 5.39 ± 1.64 |
| SNL L4 | 13 | 2.3 ± 0.3 | −20.8 ± 3.2 | −15.4 ± 1.8 | 53.0 ± 14.8* | 94.8 ± 29.5* | 2.4 ± 0.5* | −104.0 ± 30.4* | 2.05 ± 0.76* | |
| SNL L5 | 12 | 3.1 ± 0.5 | −17.7 ± 2.1 | −10.5 ± 1.3* | 57.7 ± 19.8* | 115.5 ± 46.3* | 2 3 ± 0 5* | −105.3 ± 26.1* | 1.87 ± 0.51* | |
Injury prolonged action potential (AP) in small and medium neurons, whereas afterhyperpolarization (AHP) was decreased in medium and large cells in response to current clamp command protocols. Every group lost Ca2+ current (ICa) in response to voltage clamp command protocols after injury. Neurons grouped by cell size and injury model were analyzed by comparison with “control” neurons from skin-sham operated rats. Means compared with analysis of variance and Bonferroni post hoc, except in large cell voltage clamp data, where nonparametric Kruskal-Wallis with Dunn post hoc comparisons were used to account for signal saturation. See Materials and Methods.
P < 0.05 vs. control; no other comparisons among injury groups were significant.
AHPampl = amplitude of hyperpolarization below resting membrane potential after AP; AHPdur50 = duration at 50% of AHP; AHPτ = time constant of recovery from AHP; APdur50 = duration at 50% of AP; APthresh = inflection on upstroke of AP; charge transfer = integral of area under current wave representing total Ca2+ charge transferred in response to standardized AP waveform expressed as coulombs per area of cell membrane; Gmax = maximum Boltzmann conductance in response to stepwise increments in 200-ms test pulse from holding potential of −90 mV; Ipeak = peak inward ICa in response to standardized AP waveform; SNL = spinal nerve ligation.
Fig. 2.
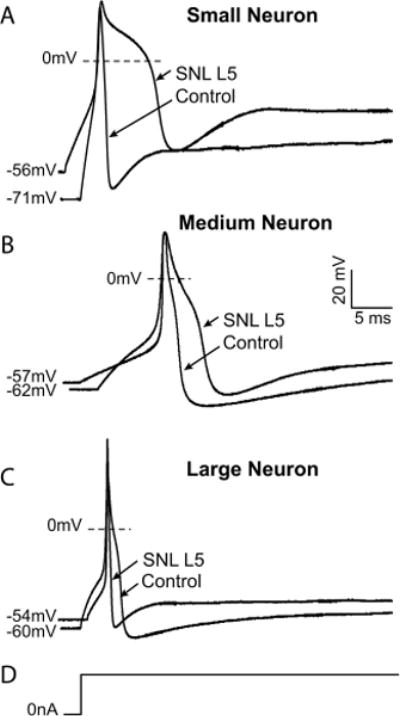
Exemplary traces showing effects of injury on action potential dimensions in control neurons and after ligation and axotomy of the fifth lumbar spinal nerve (SNL L5). (A) A typical small neuron displays prolonged action potential duration and a more depolarized resting membrane potential after injury. (B) A medium neuron exhibits prolonged action potential as well as decreased afterhyperpolarization amplitude and accelerated recovery of the afterhyperpolarization after spinal nerve ligation. (C) A large neuron develops decreased afterhyperpolarization amplitude and duration. (D) The stimulus in each case is a sustained current pulse of an amplitude just adequate for action potential generation. Scale bars apply to all traces.
Inflection of the descending limb of the AP identifies nociceptors.18 Overall, inflected cells had a longer AP50 (6.4 ± 0.6 vs. 33 ± 0.5 ms, P < 0.05) and depolarized threshold (−15.4 ± 0.8 vs. −23.2 ± 1.2 mV, P < 0.05). Comparing neurons with and without inflection did not show any difference in the response of AP parameters to injury in any surgical group.
Cells from adjacent, noninjured L4 ganglia after SNL were statistically indistinguishable from control neurons in most parameters, except for isolated changes after injury in AHP amplitude in medium neurons and AHP duration in large neurons. Therefore, cells from adjacent, nonligated ganglia did not show the consistent pattern of AP or AHP parameter changes demonstrated in cells from the L5 ganglia of injured rats.
Voltage Clamp
Voltage clamp studies were conducted in the same cells after fluid change. Efficacy of potassium and sodium channel blockade was confirmed by observation of elimination of the negative membrane potential while still in current clamp and lack of brief, inward currents during voltage clamp recording. In addition, terminal application of Cd2+ to block ICa showed no residual voltageactivated currents.
Spinal nerve ligation reduced Ca2+ conductance in L5 neurons across all size groups compared with neurons from control rats. Specifically, Gmax was reduced across all three size groups from 36 to 37% in cells from L5 SNL ganglia compared with neurons from control rats. Reduced ICa was also reflected in responses to AP waveform voltage commands. Specifically, peak current amplitude and the integral of Ca2+ flux over the course of the AP waveform (total charge transfer) fell between 37 and 60% in L5 ganglia from SNL rats compared with control. Less consistent decreases were found during voltage clamp examination of L4 neurons (table 1), which showed reduced Gmax only in large neurons and altered AP waveform responses only in medium and large neurons.
Only L5 neurons showed decreased ICa after injury in the small neuron group, whereas there were changes in both L4 and L5 neurons in the medium and large groups. However, this part of the study was not specifically designed to compare L4 and L5 neurons. To directly assess the possibly distinct effects of injury on L4 and L5 neurons, we devised additional studies to optimally compare currents in these two groups. In these experiments, we examined currents in paired L4 and L5 medium and large neurons isolated from the same animal under identical conditions. This focused examination also eliminated inconsistencies that accrue due to data collection over a span of time (2 yr for the data set above), including genetic drift in rat breeding colonies and different batches of dissociation enzymes. Further, we used a different amplifier with expanded bandwidth to clamp whole cell recordings with up to 72.5 nA peak inward current. These experiments identified contrasting injury responses in medium and large L4 and L5 neurons. Specifically, ICa in L4 neurons from medium cells was unchanged, whereas L5 neurons displayed reduced ICa compared with L4 and control neurons (fig. 3). ICa in large L4 and L5 neurons did not differ from control.
Fig. 3.

Voltage clamp data comparing paired sensory neurons from the L4 and L5 levels after spinal nerve ligation (SNL) and neurons from control animals, including large and medium groups. No differences were discerned in large cells (A). Medium cells demonstrated significant difference in maximum Ca2+ conductance (Gmax) in response to 200-ms, 10-mV, stepwise, square wave pulses (B), and in peak amplitude (C) and total charge transfer (D) in response to a standardized action potential waveform. n = 7–13 neurons for each group. Peak current is normalized for cell capacitance; charge transfer is normalized for cell surface area. § P < 0.05 versus both L4 and control, by Kruskal-Wallis nonparametric analysis of variance with Dunn multiple comparison test for post hoc differences.
Because reduced ICa may have diverging effects on the AP depending on whether the direct inward Ca2+ current or the induced outward IK(Ca) dominates, we correlated Gmax against AP parameters measured during current clamp in the same cells to determine the role of ICa. AP50 correlated negatively with Gmax in small cells from L5 of injured rats (fig. 4A) but positively in large control and SNL L5 neurons (fig. 4B), indicating opposite contributions of ICa to AP duration in these two neuronal groups. While Gmax did correlate positively with AP50 in large L5 neurons after SNL, this correlation was also seen in large cells from control ganglia without difference in means as determined by analysis of variance (table 1). Small control neurons showed minimal dependence of AP repolarization on Ca2+-dependent processes (fig. 4A). Gmax correlated negatively with AHPampl in large cells from L5 ganglia of injured rats (fig. 4C). Gmax did not correlate with any parameters in L4 and medium-sized neurons.
Fig. 4.

Correlations between neuronal Ca2+ conductance (Gmax) and action potential (AP) parameters. (A) Decreased Gmax after injury correlated with prolonged AP duration (AP50) in L5 dorsal root ganglia neurons of small cells (Pearson correlation r = −0.7, P = 0.0006), whereas Gmax and AP duration among neurons from control rats were not related (r = 0.3, P = 0.232). (B) AP duration positively correlated with Gmax in large cells from L4 and L5 dorsal root ganglia before injury (r = 0.78 ± 0.2, P = 0.0011), and this positive effect of increased Gmax on AP duration was not affected by injury in neurons from L5 ganglia (r = 0.8 ± 0.2, P = 0.0055). (C) Decreased Gmax after injury correlated to decreased afterhyperpolarization amplitude (AHPampl) in large neurons (r = −0.6, P = 0.0259), whereas changes in Gmax did not predict changes in AHPampl in neurons from control rats (r = −0.09, P = 0.7249). SNL = spinal nerve ligation.
ICa Blockade
To identify the biophysical consequences of reduced ICa, we duplicated the loss of ICa seen after injury by application of Ca2+ channel blockers and measured the effect of blockade on AP parameters and whole cell currents in the same cell (table 2). Medium or small neurons (n = 9) from nonoperated rats were studied in normal Tyrode solution before and after complete I2+ ablation with a cocktail of high voltage-activated Ca2+ channel antagonists containing nisoldipine (200 nM), SNX-111 (200 nm), agatoxin IVA (200 nM), and SNX-482 (200 nM) to block L-, N-, P/Q-, and R-type calcium channel subtypes, respectively. An AP was elicited with a brief (0.5-ms) pulse ranging from 1 to 3 nA. This same AP was used as a voltage command to elicit whole cell currents in the same cell. After capacity transients, an inward current representing calcium and sodium was followed by an outward current representing potassium (fig. 5). Calcium channel blockade significantly prolonged average AP50 and decreased AHPampl during current clamp recordings (fig. 5A), whereas underlying inward and outward currents were significantly reduced. It is noteworthy to observe the time course of ionic changes (fig. 5B). Reduced inward ICa correlated with the delayed pace of AP depolarization, whereas outward current, presumably IK(Ca), was reduced during the repolarization phase of the AP and the onset of AHP. This provides evidence of a critical role of ICa in production of outward currents during AP repolarization in sensory neurons.
Table 2.
Effect of Ca2+ Channel Antagonists
| Current Clamp
|
Voltage Clamp
|
||||
|---|---|---|---|---|---|
| n | APdur50, ms | AHPampl, mV | Ipeak Inward, pA/pF | Ipeak Outward, pA/pF | |
| Control | 9 | 3.4 ± 0.7 | −17.5 ± 1.4 | −239.5 ± 49.2 | 269.3 ± 38.0 |
| Blockade | 9 | 5.5 ± 1.3* | −12.2 ± 1.6* | −126.9 ± 26.6* | 147.5 ± 27.5* |
Ca2+ channel antagonists mimic injury effect. Blockade with specific high voltage-activated Ca2+ channels prolonged action potential (AP) duration and decreased afterhyperpolarization (AHP) amplitude in current clamp mode, whereas peak inward and outward currents were decreased in the same cell in voltage clamp mode.
P ≤ 0.01 vs. control, paired t test.
AHPampl = AHP amplitude; APdur50 = AP duration at 50%; Ipeak Inward, Outward = peak inward and outward voltage clamped currents in response to AP waveform from same cell.
Fig. 5.
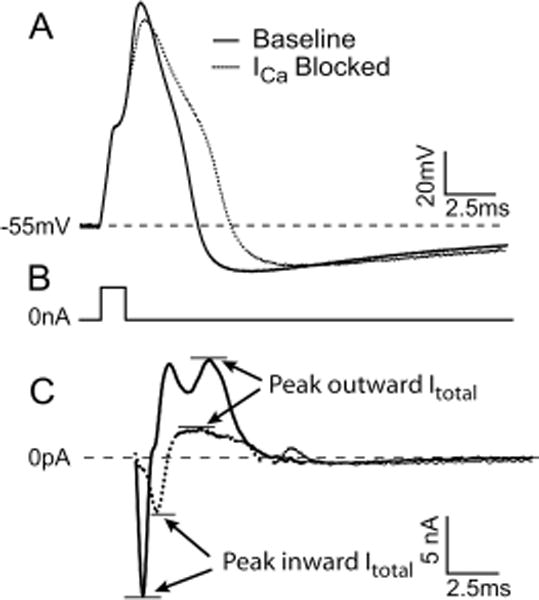
Action potential parameters and whole cell currents (Itotal) before and after Ca2+ channel (ICa) blockade. (A) Action potential elicited in response to a 3-nA, 1-ms pulse. (B) The stimulus artifact is removed for clarity. (C) Corresponding whole cell currents in response to the same action potential used as a voltage command. Reduction of depolarization rate after Ca2+ channel blockade is due to reduced inward Itotal, whereas action potential prolongation is mediated by delayed repolarization due to reduced outward Itotal. See table 2 for summary of results. Blockade cocktail included 200 nm SNX-111, 200 nm ω-Aga IVA, 200 nm SNX-482, and 200 nm nisoldipine.
Discussion
In this study, we have observed decreased ICa in the somata of sensory neurons after peripheral nerve injury by SNL. In combination with our previous findings after more peripheral sciatic injury by chronic constriction,19 this indicates that ICa is a general feature of nerve injury. The chronic constriction injury model of neuropathic pain is limited by the inability to distinguish the contribution of axotomy versus inflammatory mechanisms, because somata of axotomized neurons cannot be readily distinguished from those with intact axons. Incomplete injury is a prerequisite for provoked pain behavior, so we chose a model of neuropathic pain in which neurons injured by complete axonal transection might be compared with anatomically segregated neighboring neurons. We have found that reduced ICa is predominantly a consequence of axotomy (L5 after SNL) and is most evident in small and medium neurons.
Various voltage-activated currents, especially N and T types, inactivate during sustained depolarization,20,21 so dynamic aspects of voltage commands may substantially affect patterns of recorded ICa. Studies of ionic currents underlying neuronal membrane function typically rely on prolonged command pulses to detect voltage sensitivity and kinetic properties of channel gating. However, natural neuronal activity leads to opening of voltagegated channels in response to only brief membrane depolarization events, under which conditions the specific activation, inactivation, and channel closure after repolarization (deactivation) of various currents critically controls Ca2+ flux. We therefore used a voltage command in the form of an AP wave to determine whether an injury would produce a loss of ICa as has been observed with prolonged step protocols. We find that injury reduces ICa in response to an AP waveform to an even greater extent than ICa measured in response to 200-ms step pulses, which extends observations from traditional sustained square wave protocols and confirms that loss of ICa represents a true biophysical change associated with neuropathic pain.
We categorized neurons by size, whereas other studies have used AP characteristics, expression of membrane markers, or sensitivity to neuropeptides and algogenic agents. Clearly, sensory neurons show a high degree of heterogeneity, with some investigators describing seven subtypes in small cells alone.22 However, nerve injury causes shifts in all these markers,23,24 making their use problematic for categorizing neurons. A further limitation is that the exact electrophysiologic changes induced by injury in AP parameters are technique dependent. Specifically, intracellular recordings from nondissociated sensory neurons by us and others24,25 contrast with the current patch recordings from dissociated neurons in showing prolonged AP duration after injury in large but not small neurons. To address these limitations, the current study examined Ca2+ membrane currents together with AP characteristics in the same neuron, thus directly highlighting the role of ICa.
The functional implications of ICa loss are not fully understood. Previous reports have shown that Ca2+ contributes a large inward current during the repolarization phase of the AP, leading to a prolongation or inflection of the descending limb, whereas removing Ca2+ shortens AP duration.26–28 Accordingly, we have observed a direct relation between AP duration and ICa in large neurons (fig. 3B).
Among small and medium neurons, however, diminished ICa prolongs rather than shortens the AP (table 1 and fig. 4). Blockade with specific Ca2+ channel antagonists indicates that AP prolongation in these neurons is controlled by a shift in the balance of currents such that the loss of inward ICa is overwhelmed by a much greater loss of outward IK(Ca) (fig. 5). This relation by which decreased ICa leads to prolonged AP duration is mirrored in the findings of others who have shown that a net outward current can result from Ca2+ entry in neuronal cell types with a predominant expression of large conductance voltage- and Ca2+-gated K+ channels.29,30 Taken together, these results demonstrate the fine balance between ionic conductances and firing patterns which are altered in different ways by trauma. AP prolongation in small and medium neurons after injury may give rise to increased neurotransmitter release at synaptic junctions in the dorsal horn and thereby contribute to hyperalgesia.31
We have also shown that ICa loss is associated with AHP shortening in medium and large neurons after injury. Because currents through Ca2+-activated K+ channels contribute substantially to the AHP,32 our finding of shortened AHP provides further evidence of decreased outward K+ current secondary to ICa loss in these cell groups, although injury and inflammation could also have direct effects on these K+ channels. Blocking Ca2+ currents with specific toxins similarly caused a reduction of AHP amplitude and duration through diminished activation of Ca2+-dependent outward current. AHP loss primes the neuron for increased repetitive firing,33 as we have demonstrated in excised ganglia after injury.24 Therefore, the deficiency of ICa we have observed in sensory neurons after injury may substantially contribute to increased excitability and production of neuropathic pain. Further demonstration of this mechanism may be the antinociception provided by intrathecal Ca2+ injection in mice that is eliminated by blockers of calciumactivated potassium currents (IK(Ca))34 and the burning pain in humans after delivery of a solution containing the Ca2+ buffer EDTA to the DRG by epidural injection.35
Calcium signaling performs different functions in different neuronal types. Accordingly, our finding of decreased ICa in sensory neurons after nerve injury in rats with hyperalgesic behavior contrasts with relief of neuropathic pain in animal models by intrathecal administration of Ca2+ channel blockers.36,37 However, neuraxial blockade will affect both primary and secondary sensory neurons and interferes with neurotransmitter release in the spinal cord. Moreover, subarachnoid injection of blockers would not block sensory ganglia because of incomplete penetration into the DRG.38 Finally, loss of internal Ca2+ in the DRG has been shown to accelerate afferent conduction, whereas increased intracellular Ca2+ reduced AP propogation.39
We observed the greatest ICa loss in axotomized L5 neurons after SNL. Although these transected sensory neurons lack a peripheral receptive field, they nonetheless may be an important source of conditioning activity that produces dorsal horn hypersensitivity (fig. 6).8 Even in the absence of natural stimulation, axotomized neurons are particularly subject to chemical activation by catecholamines, proinflammatory cytokines, bradykinin and neurotrophins, and mechanical stimulation.40,41 Cross-excitation from adjacent intact neurons,42 such as those in the dorsal primary ramus of the segmental spinal nerve that remains intact after SNL, also excites the axotomized somata. Our findings indicating membrane hyperexcitability may amplify this induced L5 traffic, leading to hyperalgesic sensory events when naturally triggered L4 activity arrives in the hyperexcitable dorsal horn.
Fig. 6.

Schematic diagram of reduced Ca2+ current (ICa) contribution to neuropathic pain. Injured nerve tissue distal to the spinal nerve ligation (SNL) undergoes wallerian degeneration (dotted line), while neurons from the L5 dorsal root ganglion (DRG) are activated by movement, catecholamines, other algogenic agents and cross-excitation from adjacent intact neurons. Reduced ICa in Aα/Aß and Aδ fibers shortens afterhyperpolarizations (AHPs), which contributes to burst firing. Loss of ICa impairs natural signal filtering at the T-branch, where the stem axon splits into spinal nerve and dorsal root branches. Reduced ICa also prolongs action potential (AP) duration, which may increase excitatory synaptic transmission in the dorsal horn (DH). Spared nerves transmit activity evoked by stimulation in the receptive field, and these signals encounter DH neurons that are sensitized by L5 input. L4, L5 = neurons in the fourth and fifth lumbar ganglia.
Acknowledgments
The authors thank Ksenija Modric-Jednacak, M.D. (Resident), Mike Bode, B.A. (Technician), and Mark Poroli, B.S. (Research Technologist), for technical assistance (all from the Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin).
References
- 1.Hogan Q. Animal pain models. Reg Anesth Pain Med. 2002;27:385–401. doi: 10.1053/rapm.2002.33630. [DOI] [PubMed] [Google Scholar]
- 2.Elmslie KS. Calcium channel blockers in the treatment of disease. J Neurosci Res. 2004;75:733–41. doi: 10.1002/jnr.10872. [DOI] [PubMed] [Google Scholar]
- 3.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 4.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 5.Sommer C, Schafers M. Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res. 1998;784:154–62. doi: 10.1016/s0006-8993(97)01327-9. [DOI] [PubMed] [Google Scholar]
- 6.Ramer MS, French GD, Bisby MA. Wallerian degeneration is required for both neuropathic pain and sympathetic sprouting into the DRG. Pain. 1997;72:71–8. doi: 10.1016/s0304-3959(97)00019-5. [DOI] [PubMed] [Google Scholar]
- 7.Stoll G, Jander S, Myers RR. Degeneration and regeneration of the peripheral nervous system: From Augustus Waller’s observations to neuroinflammation. J Peripher Nerv Syst. 2002;7:13–27. doi: 10.1046/j.1529-8027.2002.02002.x. [DOI] [PubMed] [Google Scholar]
- 8.Gold MS. Spinal nerve ligation: What to blame for the pain and why. Pain. 2000;84:117–20. doi: 10.1016/s0304-3959(99)00309-7. [DOI] [PubMed] [Google Scholar]
- 9.Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB. Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology. 2004;101:476–87. doi: 10.1097/00000542-200408000-00030. [DOI] [PubMed] [Google Scholar]
- 10.Pitcher GM, Ritchie J, Henry JL. Nerve constriction in the rat: Model of neuropathic, surgical and central pain. Pain. 1999;83:37–46. doi: 10.1016/s0304-3959(99)00085-8. [DOI] [PubMed] [Google Scholar]
- 11.Gerard J, Borst G, Helmchen F. In: Calcium influx during an action potential, Methods in Enzymology. Conn PM, editor. San Diego: Academic Press; 1998. pp. 352–71. [DOI] [PubMed] [Google Scholar]
- 12.Blair NT, Bean BP. Role of tetrodotoxin-resistant Na+ current slow inactivation in adaptation of action potential firing in small-diameter dorsal root ganglion neurons. J Neurosci. 2003;23:10338–50. doi: 10.1523/JNEUROSCI.23-32-10338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Study RE, Kral MG. Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain. 1996;65:235–42. doi: 10.1016/0304-3959(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 14.Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: Are they genuine? Trends Neurosci. 1996;19:530–4. doi: 10.1016/s0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- 15.McCallum JB, Kwok WM, Mynlieff M, Bosnjak ZJ, Hogan QH. Loss of T-type calcium current in sensory neurons of rats with neuropathic pain. Anesthesiology. 2003;98:209–16. doi: 10.1097/00000542-200301000-00032. [DOI] [PubMed] [Google Scholar]
- 16.Harper AA, Lawson SN. Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol. 1985;359:31–46. doi: 10.1113/jphysiol.1985.sp015573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J Physiol. 1992;445:639–58. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harper AA, Lawson SN. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J Physiol. 1985;359:47–63. doi: 10.1113/jphysiol.1985.sp015574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, Bosnjak ZJ. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- 20.Swandulla D, Carbone E, Lux HD. Do calcium channel classifications account for neuronal calcium channel diversity? Trends Neurosci. 1991;14:46–51. doi: 10.1016/0166-2236(91)90018-p. [DOI] [PubMed] [Google Scholar]
- 21.McNaughton NC, Bleakman D, Randall AD. Electrophysiological characterisation of the human N-type Ca2+ channel II: activation and inactivation by physiological patterns of activity. Neuropharmacology. 1998;37:67–81. doi: 10.1016/s0028-3908(97)00153-6. [DOI] [PubMed] [Google Scholar]
- 22.Petruska JC, Napaporn J, Johnson RD, Gu JG, Cooper BY. Subclassified acutely dissociated cells of rat DRG: Histochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol. 2000;84:2365–79. doi: 10.1152/jn.2000.84.5.2365. [DOI] [PubMed] [Google Scholar]
- 23.Averill S, Davis DR, Shortland PJ, Priestley JV, Hunt SP. Dynamic pattern of reg-2 expression in rat sensory neurons after peripheral nerve injury. J Neurosci. 2002;22:7493–501. doi: 10.1523/JNEUROSCI.22-17-07493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH. Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology. 2005;103:360–76. doi: 10.1097/00000542-200508000-00020. [DOI] [PubMed] [Google Scholar]
- 25.Ma C, Shu Y, Zheng Z, Chen Y, Yao H, Greenquist KW, White FA, LaMotte RH. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J Neurophysiol. 2003;89:1588–602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- 26.Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–63. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heyer EJ, Macdonald RL. Calcium- and sodium-dependent action potentials of mouse spinal cord and dorsal root ganglion neurons in cell culture. J Neurophysiol. 1982;47:641–55. doi: 10.1152/jn.1982.47.4.641. [DOI] [PubMed] [Google Scholar]
- 28.Matsuda Y, Yoshida S, Yonezawa T. A Ca-dependent regenerative response in rodent dorsal root ganglion cells cultured in vitro. Brain Res. 1976;115:334–8. doi: 10.1016/0006-8993(76)90519-9. [DOI] [PubMed] [Google Scholar]
- 29.Scholz A, Gruss M, Vogel W. Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol. 1998;513:55–69. doi: 10.1111/j.1469-7793.1998.055by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–90. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–35. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sah P. Ca(2+)-activated K+ currents in neurones: Types, physiological roles and modulation. Trends Neurosci. 1996;19:150–4. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 33.Belmonte C, Gallego R. Membrane properties of cat sensory neurones with chemoreceptor and baroreceptor endings. J Physiol. 1983;342:603–14. doi: 10.1113/jphysiol.1983.sp014871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welch SP, Stevens DL, Dewey WL. A proposed mechanism of action for the antinociceptive effect of intrathecally administered calcium in the mouse. J Pharmacol Exp Ther. 1992;260:117–27. [PubMed] [Google Scholar]
- 35.Stevens RA, Urmey WF, Urquhart BL, Kao TC. Back pain after epidural anesthesia with chloroprocaine. Anesthesiology. 1993;78:492–7. doi: 10.1097/00000542-199303000-00013. [DOI] [PubMed] [Google Scholar]
- 36.Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994;269:1117–23. [PubMed] [Google Scholar]
- 37.Malmberg AB, Yaksh TL. Voltage-sensitive calcium channels in spinal nociceptive processing: Blockade of N- and P-type channels inhibits formalininduced nociception. J Neurosci. 1994;14:4882–90. doi: 10.1523/JNEUROSCI.14-08-04882.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abram SE, Yi J, Fuchs A, Dean-Bernhoft C, Hogan QH. Drug Access to the Dorsal Root Ganglion. Washington, D.C.: Society for Neuroscience; 2005. [Google Scholar]
- 39.Lüscher C, Lipp P, Lüscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J Physiol. 1996;490:319–24. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen M, Segond von Banchet G, Heppelmann B, Koltzenburg M. Nerve growth factor regulates the expression of bradykinin binding sites on adult sensory neurons via the neurotrophin receptor p75. Neuroscience. 1998;83:161–8. doi: 10.1016/s0306-4522(97)00374-6. [DOI] [PubMed] [Google Scholar]
- 41.Petersen M, Zhang J, Zhang JM, Lamotte RH. Abnormal spontaneous activity and responses to norepinephrine in dissociated dorsal root ganglion cells after chronic nerve constriction. Pain. 1996;67:391–7. doi: 10.1016/0304-3959(96)03146-6. [DOI] [PubMed] [Google Scholar]
- 42.Devor M, Wall PD. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J Neurophysiol. 1990;64:1733–46. doi: 10.1152/jn.1990.64.6.1733. [DOI] [PubMed] [Google Scholar]