

Abstract
A high proportion of human B cells carry B-cell receptors (BCRs) that are autoreactive. Inhibitory receptors such as CD22 can downmodulate autoreactive BCR responses. With its extracellular domain, CD22 binds to sialic acids in α2,6 linkages in cis, on the surface of the same B cell or in trans, on other cells. Sialic acids are self ligands, as they are abundant in vertebrates, but are usually not expressed by pathogens. We show that cis-ligand binding of CD22 is crucial for the regulation of B-cell Ca2+ signaling by controlling the CD22 association to the BCR. Mice with a mutated CD22 ligand-binding domain of CD22 showed strongly reduced Ca2+ signaling. In contrast, mice with mutated CD22 immunoreceptor tyrosine-based inhibition motifs have increased B-cell Ca2+ responses, increased B-cell turnover, and impaired survival of the B cells. Thus, the CD22 ligand-binding domain has a crucial function in regulating BCR signaling, which is relevant for controlling autoimmunity.
Keywords: B-lymphocyte differentiation, B-lymphocyte signaling, Siglecs
B cells constitutively express inhibitory coreceptors, which can down-modulate B-cell receptor (BCR) signaling. Among the B-cell inhibitory receptors are receptors of the sialic acid-binding Ig-like lectin (Siglec) family (1). B cells express the Siglec CD22 (Siglec-2), which binds with its extracellular domain to sialic acids in α2,6 linkages (2,6Sia). Sialic acids are abundant in humans and mice, but are usually absent in microorganisms (2). Thus, CD22 recognizes self structures and triggers inhibitory signals, which may be relevant for suppression of autoimmune B-cell responses. How ligand recognition of the Siglecs is mechanistically linked to the inhibitory function of these molecules is not clear. Loss of both CD22 and Siglec-G in B cells leads to spontaneous autoimmunity in mice (3). Additionally, mutations in an acetyl sialic acid esterase gene, coding for an enzyme, which modifies Siglec ligands, lead to autoimmunity (4).
CD22 has seven extracellular Ig-like domains, of which the first Ig domain is responsible for ligand binding (2). CD22 inhibits BCR signaling via immunoreceptor tyrosine-based inhibition motifs (ITIMs) located in the intracellular tail. Upon BCR cross-linking, CD22 is rapidly phosphorylated by the tyrosine kinase Lyn (5). Of the six tyrosines in the murine CD22 cytoplasmic tail, three tyrosines are found in ITIMs (5–7). The tyrosine phosphatase SHP-1, which is involved in negative signaling of many inhibitory receptors (2), is recruited to phosphorylated ITIMs of CD22. The inositol phosphatase SHIP can also bind via the adaptors Grb2 and Shc to the CD22 tail (8); however, the biological relevance of this interaction is unclear. It is controversial whether CD22 carries activating signaling motifs as well. By generation of CD22−/− mice it has been clearly demonstrated that CD22 inhibits BCR-induced Ca2+ signaling (9–12).
Whereas it is well established that a crucial function of CD22 is the inhibition of B-cell signaling, the important question remains of how the inhibition of CD22 is regulated. Several studies suggest that ligand binding may play an important role. The expression of 2,6Sia is abundant on soluble proteins (13, 14), but also on the surface of many cells, including lymphocytes. The ligand expression on the B-cell surface appears to “mask” most of the CD22 molecules and therefore limits their availability to trans-ligands (15, 16). The proximity of CD22 to the BCR is crucial for its inhibitory function. However, this interaction seems not to depend on 2,6Sia binding but rather consists of protein–protein interactions. Instead, CD22 itself is a prominent cis-ligand forming CD22 homooligomers, distinct from the BCR (17). Mice with a deficiency in ST6GalI, the enzyme that creates the 2,6Sia CD22 ligands, show increased CD22–BCR association and an increased inhibition of Ca2+ signaling, which correlates with a loss of CD22 homooligomer formation (18). Although most of the CD22 molecules are masked by cis-ligands on resting B cells, CD22 can nevertheless also be engaged in trans cell–cell interactions, as reviewed in ref. 2. These trans-interactions can induce coligation of CD22 and the BCR on the B cell, leading to enhanced CD22-mediated inhibition (19). Also, trans-ligand binding has been implicated in recirculation of mature B cells to the bone marrow, as 2,6Sia ligands are expressed specifically on bone marrow sinusoidal endothelium (20). Recently it was shown that trans-ligand binding of CD22 can induce tolerance and prevent antibody responses (21).
CD22-deficient mice show a generally normal B-cell development (9–12), but mature recirculating B cells in the bone marrow and marginal zone (MZ) B cells in the spleen are severely reduced (22). Despite the loss of an inhibitory receptor, CD22−/− mice on a C57BL/6 background do not develop spontaneous autoimmunity (9). This is different on a mixed 129 × C57BL/6 background (23) or when CD22−/− mice are crossed to Siglec-G−/− mice, which suggests some redundancy, as Siglec-G is a similar inhibitory receptor on B cells (3). One study showed that mice with a mutated CD22 ligand-binding domain had similar phenotypes to the CD22-deficient mice, without affecting Ca2+ signaling. However, this study was hampered by the fact that the introduced mutations resulted in reduced surface expression of CD22 (24).
Open questions in the field are about how the inhibitory function of CD22 is regulated by ligand binding and whether the ITIMs are the only crucial inhibiting signaling motifs. We addressed these questions by generating three CD22 knockin mouse lines with two or three mutated ITIM motifs, respectively, or with a mutation in the ligand-binding domain of CD22, which leads to loss of ligand binding but does not affect cell surface expression.
Results
Generation of CD22 Knockin Mice with Mutated Ligand-Binding or Signaling Domains.
To analyze functional domains of CD22 in vivo, knockin mice with either mutated ITIM signaling domains or a mutated ligand-binding domain were generated. For the ITIM mutations, a targeting vector was designed to mutate the three ITIM motifs. One ES clone lost one mutation, so a CD22-Y5,6F mouse (for homozygous mice carrying mutations in the Y843 and Y863, the fifth and sixth tyrosines of CD22) with mutations in two ITIMs resulted. From another ES clone, the CD22-Y2,5,6F mouse (for homozygous mice carrying tyrosine to phenylalanine mutations in Y783, Y843, and Y863, the second, fifth, and sixth tyrosines of CD22) with all mutations in three ITIMs was generated (Fig. S1). Mice with a mutated CD22 ligand-binding domain were created by introducing a mutation in arginine 130 (R130) of CD22. The R130E mutation, replacing the positively charged arginine by a negatively charged glutamate destroys all ligand binding in vitro (25). The resulting homozygous mice were named CD22-R130E (Fig. S2).
We first confirmed that all three CD22 knockin strains showed an unchanged CD22 surface expression (Fig. 1A). The functional loss of 2,6Sia ligand binding of CD22-R130E mice was analyzed by a probe with 2,6Sia coupled to streptavidin-alkaline phosphatase (2,6 Sia-SAAP) (16). This probe can only stain CD22 on B cells, if cis-ligands are removed by sialidase treatment or if 2,6Sia ligands are destroyed genetically, such as in ST6GalI−/− mice. Whereas sialidase-pretreated WT B cells or ST6GalI−/− B cells were readily stained with this probe, CD22-R130E B cells only showed background staining, similar to CD22-deficient B cells (Fig. 1B). Because the loss of 2,6Sia binding in CD22-R130E cells is comparable to CD22-deficient B cells, we conclude a loss of the ligand-binding function of CD22 in CD22-R130E mice. The consequence of the ITIM mutations was tested by CD22 immunoprecipitation (IP) of CD22. Whereas WT B cells showed readily detectable CD22 phosphorylation after anti-IgM stimulation, this phosphorylation was strongly reduced in B cells of CD22-Y5,6F mice and not present in B cells of CD22-Y2,5,6F mice (Fig. 1C). Similarly, SHP-1 was coprecipitated with CD22 in stimulated WT B cells, but was not detectable in CD22-Y5,6F or CD22-Y2,5,6F B cells (Fig. 1C). Interestingly, both CD22 tyrosine phosphorylation, as well as SHP-1 recruitment was increased in CD22-R130E B cells (Fig. 1D). We conclude that the ITIM mutations led to the expected reduction of CD22 tyrosine phosphorylation and that CD22-Y2,5,6F mice were more severely affected than CD22-Y5,6F mice.
Fig. 1.
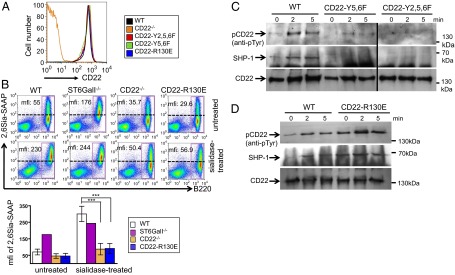
CD22-R130E B cells show strongly impaired ability to bind sialic acids and tyrosine phosphorylation of CD22 is strongly reduced in CD22-Y5,6F and CD22-Y2,5,6F B cells. (A) CD22 expression is shown for splenic B220+ lymphocytes of CD22 knockin mice, CD22−/− mice, and wild-type control. One typical example of 10 independent experiments is shown. (B) Splenic cells of WT, ST6GalI−/−, CD22−/−, or CD22-R130E mice were stained for B220 and the sialic acid probe Neu5Gc-α2,6Gal-SAAP. Lower samples were treated with sialidase. A quantification of four mice each (for WT, CD22−/−, and CD22-R130E) and one ST6GalI−/− mouse is shown (Lower). mfi, mean fluorescence intensity for cells in box. (C) Splenic B cells of CD22-Y5,6F, CD22-Y2,5,6F, and control mice or of CD22-R130E mice (D) were stimulated for indicated time points with anti-IgM [F(ab′)2]. After CD22 IP, CD22 phosphorylation was analyzed with antiphosphotyrosine antibody, SHP-1 binding and CD22 total amount (loading control) were analyzed with specific antibodies. One typical example of four experiments is shown for both C and D.
B-Cell Development in CD22 Knockin Mice.
To study whether the changes of B-cell subpopulations seen in CD22-deficient mice are caused by the lack of inhibitory signaling or by the missing ligand-binding function of CD22, B-cell development was analyzed in the CD22 knockin mice. In the bone marrow, there was no defect in early B-cell differentiation, similar to CD22-deficient mice (Table S1). The lack of mature, recirculating B cells in the bone marrow was found in CD22-Y2,5,6F and in CD22-Y5,6F mice, but not in CD22-R130E mice (Fig. 2A), suggesting that the signaling domain, rather than the ligand-binding domain of CD22 plays a functional role for this B-cell population. In the spleen, CD22-Y2,5,6F mice show a reduction of transitional type 1 (T1) and T2 B cells, similar to CD22-deficient mice. This phenotype is not observed in the other two knockin mouse lines and seems to depend on a fully mutated inhibitory signaling domain (Fig. S3). Finally, clearly reduced MZ B-cell populations were detected both in CD22 knockin mice with mutated ligand-binding or with mutated ITIM domains (Fig. 2B). This suggests that both of these CD22 domains are necessary for generating a normal number of MZ B cells in the spleen.
Fig. 2.

Mice with mutated ITIM domains have reduced numbers of recirculating B cells in the bone marrow. Marginal zone B cells are decreased in all CD22 knockin mice. (A, Left) Representative analysis of B220, IgM, and IgD expression in the bone marrow. Numbers give percentages of immature B cells (B220loIgMlo-hi) and recirculating B cells (B220hiIgMlo and B220hiIgDhi) in bone marrow. (A, Right) shows summarized absolute cell numbers displayed as means ±SD. (B) Splenic lymphocytes are analyzed by the indicated markers and total cell numbers are given for MZ B cells (CD1d+B220+ and CD21+CD23−) and follicular B cells (CD1d−B220+ and CD21+CD23+). *P < 0.05; **P < 0.005; ***P < 0.001. Each experiment was done at least 10–12 times.
To address whether the defect of bone marrow recirculating B cells results from defective migration or from defective survival of the cells, we first performed a cell transfer migration experiment of CFSE-labeled splenic B cells. Compared with wild-type B cells, all CD22 knockin B cells migrated to the spleen equally well. However, there was a defective migration of CD22-Y2,5,6F B cells to the bone marrow, whereas CD22-Y5,6F and CD22-R130E B cells migrated to the bone marrow normally (Fig. 3A). To further examine the mechanism, we analyzed B-cell turnover in vivo by BrdU incorporation. Mature B cells (IgD+) in the bone marrow of both CD22-deficient and CD22-Y2,5,6F mice show a clearly increased BrdU incorporation (Fig. 3B). In contrast, the BrdU incorporation in mature B cells of CD22-Y5,6F or CD22-R130E mice was normal or only mildly increased (Fig. 3B). Similar results were observed in B cells of the spleen. Particularly in mature B cells, there was a higher BrdU incorporation in CD22-deficient or CD22-Y2,5,6F mice at later time points than in wild-type mice (Fig. S4 A and B), indicating a higher B-cell turnover. To investigate whether this higher turnover is due to higher apoptosis, B cells of CD22-deficient or CD22-Y2,5,6F mice were cultivated in vitro and spontaneous apoptosis was measured. The rate of spontaneous cell death in vitro was higher in B cells of CD22-deficient or CD22-Y2,5,6F mice than in B cells of control mice (Fig. S5). A higher turnover of B cells may affect B-cell numbers in the blood. Indeed, both CD22-deficient and CD22-Y2,5,6F mice show decreased B-cell numbers in the blood, whereas the other CD22 knockin mice do not show this phenotype (Table S1). Together, these data strongly indicate that the reduction of the mature B-cell population in CD22-deficient mice can be attributed to shorter survival and higher turnover caused by lacking ITIM motifs, but not to missing CD22 ligand interactions in bone marrow tissues.
Fig. 3.

B cells from CD22 Y2,5,6F mice show impaired migration to the bone marrow and a higher turnover rate. (A) Splenic B cells of indicated mice were labeled with CFSE, i.v. injected into wild-type mice and stained with B220 after 24 h. (Left) Examples of bone marrow stainings after transfer. The relative amount of CFSE-positive cells from B220-positive cells of spleen and bone marrow is shown (Right). Three independent experiments are shown in combined analysis. n = 28, WT; n = 14, CD22-R130E; n = 12, CD22-Y5,6F; and n = 15, CD22-Y2,5,6F. (B) BrdU incorporation of bone marrow B cells in mice, which were fed for indicated time periods with BrdU. BrdU+ cells are shown in line diagrams for mature B cells (B220hiIgDhi) in the bone marrow. One typical example of two independent experiments is shown. Statistics are summarized results of three mice each. *P < 0.05; **P < 0.01; ***P < 0.001.
Reciprocal Regulation of Ca2+ Signaling by the Ligand-Binding Domain and ITIM Sequences of CD22.
CD22-deficient mice showed enhanced BCR-induced Ca2+ signaling. Similarly, CD22-Y2,5,6F and CD22-Y5,6F mice show elevated Ca2+ responses, as expected, as the ITIM motifs are mutated (Fig. 4A). Interestingly, the responses are not increased to the same extent as in CD22-deficient mice. Also, at lower anti-IgM antibody concentrations, the response of CD22-Y2,5,6F B cells is higher than that of CD22-Y5,6F B cells (Fig. 4A). These results suggest that all three ITIMs are needed for efficient inhibition. Surprisingly, the Ca2+ signaling response in B cells of CD22-R130E mice was clearly decreased, compared with wild-type B cells, at all anti-IgM concentrations tested (Fig. 4B).
Fig. 4.

Ca2+ signaling is enhanced in B cells of CD22-Y5,6F and CD22-Y2,5,6F mice and impaired in B cells of CD22-R130E mice. Intracellular calcium mobilization of Indo-1 loaded splenic B cells (CD11b−CD5−) of wild-type and (A) CD22-Y5,6F and CD22Y2,5,6F mice and (B) CD22-R130E mice, stimulated with anti-IgM [F(ab′)2]. Results are plotted as medians of bound/unbound Indo-1 over time. One of eight experiments with similar results is shown.
The CD22 Ligand-Binding Mutant Affects the CD22/BCR Association.
Because the R130E mutated form of CD22 inhibited BCR signaling more strongly than wild-type CD22 and this may be due to a changed association of CD22 to the BCR, we analyzed this association in R130E and control mice. We used a proximity ligation assay (PLA), which measures protein associations in situ by Fab fragment of antibodies directed against CD22 and IgM with attached oligonucleotides that can only hybridize when the two proteins are in close proximity and are detected by a rolling circle PCR (26, 27). The R130E mutant of CD22 showed a significantly higher association to IgM in the unstimulated B cells compared with wild-type B cells (Fig. 5A). Whereas there was more CD22 recruited to IgM in pervanadate-stimulated wild-type cells, the association of CD22 with IgM was not increased in B cells of CD22-R130E mice (Fig. 5A). Treatment with latrunculin A, which disrupts actin polymerization, was used as a positive control, showing the maximal association of IgM and CD22 in both wild-type and CD22-R130E cells. In contrast to CD22-R130E mice, B cells of CD22-Y2,5,6F mice showed no changes in CD22–BCR association compared with wild-type B cells (Fig. S6). Anti-IgD stimulations gave similar results to pervanadate stimulation (Fig. S6).
Fig. 5.

Association of CD22 with IgM in B cells of CD22 knockin mice analyzed by PLA and by anti-Ig kappa co-IP. (A) Association of IgM and CD22 in situ was analyzed by proximity ligation assay (PLA) with B cells of CD22-R130E or WT control mice. PBS is the unstimulated control; cells were stimulated with pervanadate or with latrunculin for 5 min. An example is shown on the Left, quantitative analysis is shown on the Right. One typical experiment out of three is shown. (B) Splenic B cells of indicated mice were stimulated for indicated time points with anti-IgM [F(ab′)2]. After IP with anti-kappa antibody, coprecipitated CD22 was analyzed with antiphosphotyrosine antibody, followed by total CD22 and kappa light chain antibodies (loading control). (Below) quantification of band intensities (pCD22 or CD22 bands divided by kappa loading control) in arbitrary units, means from four experiments each ±SD *P < 0.05; **P < 0.01; ***P < 0.001.
CD22–IgM associations were also examined by anti-Ig kappa IP with coprecipitated CD22. Whereas B cells of CD22-Y2,5,6F and CD22-Y5,6F mice showed a similar amount of total coprecipitated CD22 to B cells of wild-type mice, the amount of total CD22 coprecipitated with the kappa chain of CD22-R130E mice was clearly reduced (Fig. 5B). Interestingly, despite this, the fraction of associated tyrosine-phosphorylated CD22 was higher in CD22-R130E mice after anti-IgM stimulation. In contrast, CD22-Y5,6F and CD22-Y2,5,6F B cells did show much reduced or no CD22 phosphorylation, respectively (Fig. 5B). Thus, in this assay, less total, but more phosphorylated CD22 was detected in association with the BCR of CD22-R130E mice. Both assays clearly show that the association of the ligand-binding–deficient CD22 with the BCR is affected by the R130E mutation.
CD22 ITIM Motifs Suppress Early Thymus-Dependent Antibody Responses.
Mice with mutated CD22 ligand-binding or signaling domains did not show large changes in serum Ig levels (Fig. S7A). Thymus-independent (TI) type 2 antigen Trinitrophenyl (TNP)-Ficoll responses in all CD22 mutant mice were quite normal (Fig. S7B). When immunized with the thymus-dependent (TD) antigen, Nitrophenyl (NP)-KLH, there were no consistent changes in the IgM response. However, CD22-Y2,5,6F mice showed clearly enhanced IgG1 responses 7 or 14 d after each immunization or rechallenge with the antigen (Fig. S7C). In contrast, responses of the two other CD22 knockin mouse lines were largely normal. This suggests that the TD antibody response is normally repressed by the ITIMs of the cytoplasmic tail of CD22.
Discussion
This study uniquely dissects the two functional domains of CD22, the extracellular and intracellular domains, by site-specific mutations in mice in vivo. Our results show that the first Ig domain of CD22, which binds α2,6-linked sialic acids, has a distinct and often opposing function to the inhibitory signaling domain of CD22.
Whereas CD22 is not crucial for early B-cell development in the bone marrow, several groups observed a strong reduction of IgD+ recirculating B cells in the bone marrow of CD22-deficient mice (11, 12, 20). Because a high expression of 2,6Sia on endothelial cells of the bone marrow was found, it was suggested that CD22 could be a homing receptor for recirculating B cells in the bone marrow (20). An alternative explanation was that the reduced life span of B cells in CD22-deficient mice led to the reduction of this population in the bone marrow (9, 11, 24). The results of this study here clearly provide an answer to this question. Whereas CD22-R130E mice surprisingly do not show the mature B-cell defect in the bone marrow, mice with mutations in the CD22 ITIM motifs show a similar defect to that in CD22-deficient mice. Thus, mature B-cell numbers in the bone marrow are controlled by inhibitory signal functions of CD22 and not by ligand interactions to bone marrow 2,6Sia ligands. Mechanistically, the reduction of mature B-cell numbers in the bone marrow of mice with three mutated ITIM domains can be explained by an increased turnover of their B cells, coinciding with a higher apoptosis rate as demonstrated in vitro. As a consequence, mature B-cell populations in the blood and bone marrow are reduced.
The mutated CD22 ITIMs also led to a reduction of the immature transitional populations in the spleen similar to CD22-deficient mice (28). Again, CD22-R130E mice were not affected. Apparently increased B-cell signaling in CD22 ITIM-mutated mice can lead to a more efficient maturation from transitional to mature B cells. The CD22-dependent defect of MZ B cells in the spleen was observed in both ITIM-mutated as well as ligand-domain–mutated mice. Hence, both CD22–ligand interactions, which could be trans-interactions to other cells in the spleen, as well as a correct BCR signaling strength, is needed to generate the right number of B cells in the marginal zone. For CD22-deficient MZ B cells, an increased chemokine responsiveness has been shown, compared with wild-type cells (22). This may affect the localization or homeostasis of MZ B cells and could be an explanation of why B cells with mutated CD22 ITIMs show the same phenotype.
The inhibition of BCR-induced Ca2+ signaling by CD22 is mediated via ITIM motifs by recruited SHP-1, as studies in vitro have shown (6, 7, 29). Our in vivo study confirms this, as CD22-Y2,5,6F and CD22-Y5,6F mice showed increased Ca2+ responses. Interestingly, the increase in Ca2+ signaling was less pronounced when only the two C-terminal ITIMs were mutated. Structural studies had shown that SHP phosphatases need binding with both of their SH2 domains to phosphorylated ITIM motifs to get fully activated (30). The results presented here show that efficient inhibition by CD22 requires all three ITIMs. Notably, CD22-deficient B cells showed an even stronger BCR Ca2+ response than CD22-Y2,5,6F B cells, suggesting that mutating all three ITIM sequences is not enough to remove the complete inhibitory function of CD22. Either the other three tyrosines or another part of the CD22 tail must be responsible.
In contrast, B cells of CD22-R130E mice showed strongly reduced Ca2+ signaling, compared with wild-type B cells. Surprisingly, this was not observed in another study where either R130 and R137 of CD22 were mutated to alanines or both first Ig domains of CD22 were deleted (24). However, in that study of CD22 ligand-mutant mice a 50% reduced CD22 surface expression was detected, in contrast to our results (24). In that case, the reduced CD22 surface expression may have been caused unintentionally by the gene targeting approach. Our results are fully compatible with ST6GalI−/− mice, where ligands of CD22 are missing on the B-cell surface, which also shows reduced Ca2+ responses (18, 31). The higher inhibitory function of R130E CD22 can be explained by an enhanced association to the B-cell receptor. This was detected by our PLA assay in the resting state. After pervanadate or anti-IgD stimulation, the association of R130E-mutated CD22 with the BCR was not increased, in contrast to WT CD22. Also, R130E-mutated CD22 was higher tyrosine phosphorylated and recruited more SHP-1 after activation.
Surprisingly, the anti-kappa IP did not show increased coprecipitation of total R130E-mutated CD22, but enriched tyrosine-phosphorylated CD22. This may be due to technical reasons and may be explained by a new model. CD22 is normally associated to clathrin-coated pits where the protein forms oligomers via 2,6Sia binding on other CD22 molecules (18). The BCR is usually minimally associated with these microdomains. In ST6GalI-deficient mice, which lack CD22 ligands, more colocalization of the BCR to CD22 in fused raft/clathrin-coated pit domains has been detected (18). This increased colocalization of the BCR and CD22 apparently also happens in CD22-R130E mice as detected by the in situ PLA assay. The higher amount of CD22 that is colocalized with the BCR are likely single CD22 molecules that can be detected by the PLA, but not by the kappa-IP assay (see model in Fig. S8). Kappa IP leads to a lower amount of CD22 being coprecipitated with the kappa light chain, as this technique requires lysis of the cells and would therefore lead to loss of CD22 molecules, which are not bound in CD22 clusters anymore. One would predict that these single molecules are higher tyrosine phosphorylated in the case of CD22-R130E, exactly as detected and explained by our new model (Fig. S8). Therefore, our results show that there is a crucial cis-ligand binding function of CD22, which normally keeps CD22 away from the BCR.
What biological sense would it make to regulate the inhibition of BCR signaling by CD22 via cis-ligand binding? The 2,6Sia expression on the B-cell surface is not static but seems to be regulated by sialic acid transferases, sialidases, and enzymes that modify sialic acids (4). Activated B cells show stronger CD22 unmasking, i.e., a down-regulation of cis-ligands (32). Germinal center (GC) B cells can be stained by peanut agglutinin, a lectin, which stains the core carbohydrate structures lacking sialic acids. Thus, GC B cells potentially express fewer CD22 ligands in cis, leading to a potentially stronger CD22–BCR association and likely a stronger BCR signal inhibition. Recently it was shown that GC B cells are not able to signal via their BCR, due to high association of SHP-1 to the BCR (33). Whether CD22 recruitment to the BCR is involved in this, has to be determined, but it is an interesting possibility. Additionally, trans-ligand interactions with other cells also determine the association of CD22 with the BCR, as has been shown (19, 21). This could also influence the level of CD22 inhibition by stronger recruitment to the BCR in cases when cells are densely packed like in B-cell follicles (29). Additionally, trans-CD22 ligands can tolerize B cells when attached next to antigens on a carrier (21). Thus, recruitment of CD22 molecules to the BCR by trans-CD22 ligands could be important to suppress autoimmune responses.
Despite reduced MZ B-cell populations, all CD22 knockin mice did not produce lower TI type 2 responses. Because MZ B cells participate in these responses, the decreased TI type 2 response of CD22-deficient mice was explained by this defect (22). In mice with a mutated ligand-binding domain or mutated ITIM motifs, apparently other B cells can replace the missing MZ B cells in these responses, or the defect of the MZ population is not as profound as in CD22-deficient mice. Several groups have found normal TD antibody responses to various antigens in CD22-deficient mice (9, 11, 12). However, here we clearly identify higher IgG1 responses of the CD22-Y2,5,6F mice with mutated ITIM motifs to NP-KLH at almost all time points both in primary responses, as well as on rechallenge. This means that the CD22 ITIM motifs do inhibit this response. When CD22−/− mice were crossed to anti-NP transgenic quasimonoclonal mice (34), an earlier and stronger germinal center response was detected compared with transgenic controls. These results fit well with our observations in CD22-Y2,5,6F mice.
In conclusion we have found that the ligand-binding domain of CD22 is important for regulation of B-cell Ca2+ signaling. This control is mediated by ligand-dependent regulation of the association with the BCR. In contrast, the CD22 ITIM motifs are crucial for a direct inhibition of BCR Ca2+ signaling. The regulation of BCR signaling by the ligand-binding domain of CD22 has implications on regulation of autoimmunity, as sialic acids are self ligands, being mainly expressed in vertebrates.
Materials and Methods
Mice.
CD22 knockin mice were generated as described in SI Materials and Methods. All mice were on a mixed C57BL/6 × 129 background. Wild-type controls were littermates or age matched to the same background. Animal experiments were approved by a local ethics committee (Regierung Mittelfranken).
Flow Cytometry.
Staining was done with the antibodies (conjugated with APC, biotin, FITC, PerCP-cy5, or PE): anti-B220 (RA3-6B2; BD Biosciences), anti-IgM (11/41; eBioscience), anti-IgD (11-26C; our hybridoma), anti-CD5 (53-7-3; BD Biosciences), anti-CD23 (B3B4; eBioscience), anti-CD21 (7G6; our hybridoma), anti-AA4.1 (eBioscience), anti-MHC-II (I-Ab, AF6-120.1; BD Biosciences), anti-CD1d (233; our hybridoma), anti-CD22 (Cy34.1; BD Biosciences), and Fc-Block (clone 2.4G2; our hybridoma). Biotinylated antibodies were stained with steptavidin PE-cy5 (BD Biosciences) or PerCP-cy5.5 (eBioscience). Sialidase treatment: 5 × 106 cells were incubated in 150 µL PBS with 1.5 µL sialidase (0.015 units) at 37 °C for 1 h. After addition of 1.5 mM sialidase inhibitor (50 mM a2,3-dehydroneuraminic acid) and incubation for 2 min at 37 °C, cells were washed. Staining was carried out with B220-PE and Neu5Gc-a2,6-Gal-SAAP-FITC.
Calcium Mobilization Assays in Primary Mouse Cells.
Splenic cells were loaded with Indo-1 as described (35). Cells were extracellularly stained with anti-CD5 and anti-CD11b and unstimulated cells were analyzed for 40 s using an LSR II (Becton Dickinson). Cells were stimulated with anti-IgM [F(ab′)2], (Jackson ImmunoResearch) and cells were further analyzed as described (35).
BCR Stimulation and Immunoblot Analysis.
T-cell removal from splenic cell suspensions was achieved as described (35). After 1 h of starvation, cells were stimulated with 10 µg/mL anti-IgM [F(ab′)2] at 37 °C. Then cells were lysed with Brij lysis buffer for 30 min on ice. CD22 or Ig-kappa were precipitated as described (36). Rabbit anti-CD22 (gift of P. Crocker, Dundee University, Dundee, UK), anti-κ (Southern Biotech), anti-p-Tyr (4G10; our hybridoma), anti–SHP-1 (Millipore), and antiactin (Cedarlane) were used in 1:1,000 or 1:4,000 concentrations. Proteins were detected with anti-mouse IgG HRP (Jackson Dianova), anti-rabbit IgG HRP (Cell Signaling), anti-goat IgG HRP (Cell Signaling), and the ECL detection kit (Amersham).
Cell Apoptosis Measurement and BrdU Incorporation Measurements.
The cell apoptosis assay and the BrdU assay were performed as described (35).
Adoptive Transfer of CFSE-Labeled Cells.
After T-cell lysis, splenic cells of donor mice were labeled with 0.5 µM carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes) for 5 min at room temperature (RT). Cells were washed in supplemented 10% (vol/vol) FCS RPMI 1640. After incubation for an additional 30 min at RT in 10% FCS RPMI 1640, cells were then washed twice with PBS and 1 × 107 cells were resuspended in 100 µL PBS and i.v. injected into recipient mice (1 × 107 cells per mouse). Twenty-four hours later, mice were killed. Bone marrow cells and splenic cells were stained for B220 and IgD and analyzed for percentage of CFSE-labeled cells by flow cytometry.
Proximity Ligation Assay.
Antibodies and chemicals for PLA.
For PLA probes, the following antibodies were used: IgD (11-26c.2a; BioLegend), IgM (R33.24.12; our hybridoma), and CD22 (Cy34.1). Latrunculin A (Cayman Chemicals). Pervanadate was prepared from sodium orthovanadate by reacting with H2O2 at equimolar ratio.
PLA probe preparation and experiment.
Specific oligos (Eurofins MWG) were conjugated to purified Fab fragments as described (37). Fab fragments were prepared by Pierce Fab preparation kit. The purified Fabs (20–50 µg) were labeled with C6-N-succinimidyl-6-hydrazino-nicotinamide (C6-s-HyNic) and the oligos were labeled with C6-N-succinimidyl-4-formylbenzamide (C6-s-4FB), (Solulink). HyNic modified Fab and the 5′ 4FB modified oligo were purified and reacted together. All of the Fab-PLA probes have average protein: oligo coupling ratio within the range of 1–1.5. PLA reaction was performed following the protocol described (26). PLA results were analyzed by fluorescence microscope and quantified (27).
Microscopy and image analysis.
Fluorescence microscopy was performed with a Zeiss 510 Meta confocal microscope (Carl Zeiss). At least 100 cells from several images were analyzed by BlobFinder software and the average number of PLA dots per cell was calculated.
Supplementary Material
Acknowledgments
We thank Anne Urbat and Martina Döhler for excellent technical support; all colleagues who contributed to early phases of this project, i.e., Drs. Claus-Peter Danzer, Jörg Klein, Anton van der Merwe, and Christina Piperi; and the Consortium for Functional Glycomics for the biotinylated Neu5Gc-α2,6Gal construct. This work was supported by the Deutsche Forschungsgemeinschaft (NI 549/6-2, SFB643, and TRR130).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304888110/-/DCSupplemental.
References
- 1.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7(4):255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 2.Jellusova J, Nitschke L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front Immunol. 2011;2:96. doi: 10.3389/fimmu.2011.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol. 2010;184(7):3618–3627. doi: 10.4049/jimmunol.0902711. [DOI] [PubMed] [Google Scholar]
- 4.Surolia I, et al. Functionally defective germline variants of sialic acid acetylesterase in autoimmunity. Nature. 2010;466(7303):243–247. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith KGC, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: A requirement for Lyn. J Exp Med. 1998;187(5):807–811. doi: 10.1084/jem.187.5.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blasioli J, Paust S, Thomas ML. Definition of the sites of interaction between the protein tyrosine phosphatase SHP-1 and CD22. J Biol Chem. 1999;274(4):2303–2307. doi: 10.1074/jbc.274.4.2303. [DOI] [PubMed] [Google Scholar]
- 7.Otipoby KL, Draves KE, Clark EA. CD22 regulates B cell receptor-mediated signals via two domains that independently recruit Grb2 and SHP-1. J Biol Chem. 2001;276(47):44315–44322. doi: 10.1074/jbc.M105446200. [DOI] [PubMed] [Google Scholar]
- 8.Poe JC, Fujimoto M, Jansen PJ, Miller AS, Tedder TF. CD22 forms a quaternary complex with SHIP, Grb2, and Shc. A pathway for regulation of B lymphocyte antigen receptor-induced calcium flux. J Biol Chem. 2000;275(23):17420–17427. doi: 10.1074/jbc.M001892200. [DOI] [PubMed] [Google Scholar]
- 9.Nitschke L, Carsetti R, Ocker B, Köhler G, Lamers MC. CD22 is a negative regulator of B-cell receptor signalling. Curr Biol. 1997;7(2):133–143. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- 10.O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274(5288):798–801. doi: 10.1126/science.274.5288.798. [DOI] [PubMed] [Google Scholar]
- 11.Otipoby KL, et al. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature. 1996;384(6610):634–637. doi: 10.1038/384634a0. [DOI] [PubMed] [Google Scholar]
- 12.Sato S, et al. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: Altered signaling in CD22-deficient mice. Immunity. 1996;5(6):551–562. doi: 10.1016/s1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- 13.Adachi T, et al. CD22 serves as a receptor for soluble IgM. Eur J Immunol. 2012;42(1):241–247. doi: 10.1002/eji.201141899. [DOI] [PubMed] [Google Scholar]
- 14.Hanasaki K, Powell LD, Varki A. Binding of human plasma sialoglycoproteins by the B cell-specific lectin CD22. Selective recognition of immunoglobulin M and haptoglobin. J Biol Chem. 1995;270(13):7543–7550. doi: 10.1074/jbc.270.13.7543. [DOI] [PubMed] [Google Scholar]
- 15.Collins BE, et al. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc Natl Acad Sci USA. 2004;101(16):6104–6109. doi: 10.1073/pnas.0400851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danzer CP, Collins BE, Blixt O, Paulson JC, Nitschke L. Transitional and marginal zone B cells have a high proportion of unmasked CD22: implications for BCR signaling. Int Immunol. 2003;15(10):1137–1147. doi: 10.1093/intimm/dxg114. [DOI] [PubMed] [Google Scholar]
- 17.Han S, Collins BE, Bengtson P, Paulson JC. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat Chem Biol. 2005;1(2):93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 18.Collins BE, Smith BA, Bengtson P, Paulson JC. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat Immunol. 2006;7(2):199–206. doi: 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- 19.Lanoue A, Batista FD, Stewart M, Neuberger MS. Interaction of CD22 with alpha2,6-linked sialoglycoconjugates: Innate recognition of self to dampen B cell autoreactivity? Eur J Immunol. 2002;32(2):348–355. doi: 10.1002/1521-4141(200202)32:2<348::AID-IMMU348>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 20.Nitschke L, Floyd H, Ferguson DJ, Crocker PR. Identification of CD22 ligands on bone marrow sinusoidal endothelium implicated in CD22-dependent homing of recirculating B cells. J Exp Med. 1999;189(9):1513–1518. doi: 10.1084/jem.189.9.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duong BH, et al. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J Exp Med. 2010;207(1):173–187. doi: 10.1084/jem.20091873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samardzic T, et al. Reduction of marginal zone B cells in CD22-deficient mice. Eur J Immunol. 2002;32(2):561–567. doi: 10.1002/1521-4141(200202)32:2<561::AID-IMMU561>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.O’Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med. 1999;189(8):1307–1313. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poe JC, et al. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nat Immunol. 2004;5(10):1078–1087. doi: 10.1038/ni1121. [DOI] [PubMed] [Google Scholar]
- 25.van der Merwe PA, et al. Localization of the putative sialic acid-binding site on the immunoglobulin superfamily cell-surface molecule CD22. J Biol Chem. 1996;271(16):9273–9280. [PubMed] [Google Scholar]
- 26.Söderberg O, et al. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45(3):227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 27.Infantino S, et al. Arginine methylation of the B cell antigen receptor promotes differentiation. J Exp Med. 2010;207(4):711–719. doi: 10.1084/jem.20091303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerlach J, et al. B cell defects in SLP65/BLNK-deficient mice can be partially corrected by the absence of CD22, an inhibitory coreceptor for BCR signaling. Eur J Immunol. 2003;33(12):3418–3426. doi: 10.1002/eji.200324290. [DOI] [PubMed] [Google Scholar]
- 29.Doody GM, et al. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269(5221):242–244. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 30.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92(4):441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh S, Bandulet C, Nitschke L. Regulation of B cell development and B cell signalling by CD22 and its ligands alpha2,6-linked sialic acids. Int Immunol. 2006;18(4):603–611. doi: 10.1093/intimm/dxh402. [DOI] [PubMed] [Google Scholar]
- 32.Razi N, Varki A. Cryptic sialic acid binding lectins on human blood leukocytes can be unmasked by sialidase treatment or cellular activation. Glycobiology. 1999;9(11):1225–1234. doi: 10.1093/glycob/9.11.1225. [DOI] [PubMed] [Google Scholar]
- 33.Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. 2012;336(6085):1178–1181. doi: 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onodera T, Poe JC, Tedder TF, Tsubata T. CD22 regulates time course of both B cell division and antibody response. J Immunol. 2008;180(2):907–913. doi: 10.4049/jimmunol.180.2.907. [DOI] [PubMed] [Google Scholar]
- 35.Ackermann JA, Radtke D, Maurberger A, Winkler TH, Nitschke L. Grb2 regulates B-cell maturation, B-cell memory responses and inhibits B-cell Ca2+ signalling. EMBO J. 2011;30(8):1621–1633. doi: 10.1038/emboj.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waisman A, et al. IgG1 B cell receptor signaling is inhibited by CD22 and promotes the development of B cells whose survival is less dependent on Ig alpha/beta. J Exp Med. 2007;204(4):747–758. doi: 10.1084/jem.20062024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dirksen A, Dirksen S, Hackeng TM, Dawson PE. Nucleophilic catalysis of hydrazone formation and transimination: implications for dynamic covalent chemistry. J Am Chem Soc. 2006;128(49):15602–15603. doi: 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.