

Abstract
The physiological functions of hydrogen sulfide (H2S) include vasorelaxation, stimulation of cellular bioenergetics, and promotion of angiogenesis. Analysis of human colon cancer biopsies and patient-matched normal margin mucosa revealed the selective up-regulation of the H2S-producing enzyme cystathionine-β-synthase (CBS) in colon cancer, resulting in an increased rate of H2S production. Similarly, colon cancer-derived epithelial cell lines (HCT116, HT-29, LoVo) exhibited selective CBS up-regulation and increased H2S production, compared with the nonmalignant colonic mucosa cells, NCM356. CBS localized to the cytosol, as well as the mitochondrial outer membrane. ShRNA-mediated silencing of CBS or its pharmacological inhibition with aminooxyacetic acid reduced HCT116 cell proliferation, migration, and invasion; reduced endothelial cell migration in tumor/endothelial cell cocultures; and suppressed mitochondrial function (oxygen consumption, ATP turnover, and respiratory reserve capacity), as well as glycolysis. Treatment of nude mice with aminooxyacetic acid attenuated the growth of patient-derived colon cancer xenografts and reduced tumor blood flow. Similarly, CBS silencing of the tumor cells decreased xenograft growth and suppressed neovessel density, suggesting a role for endogenous H2S in tumor angiogenesis. In contrast to CBS, silencing of cystathionine-γ-lyase (the expression of which was unchanged in colon cancer) did not affect tumor growth or bioenergetics. In conclusion, H2S produced from CBS serves to (i) maintain colon cancer cellular bioenergetics, thereby supporting tumor growth and proliferation, and (ii) promote angiogenesis and vasorelaxation, consequently providing the tumor with blood and nutritients. The current findings identify CBS-derived H2S as a tumor growth factor and anticancer drug target.
The endogenous gasotransmitter hydrogen sulfide (H2S) is a stimulator of vasorelaxation (1–3), angiogenesis (3–5), and cellular bioenergetics (6, 7). H2S is generated from l-cysteine by two pyridoxal-5′-phospate–dependent enzymes, cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE), and by the combined action of cysteine aminotransferase and 3-mercaptopyruvate sulfurtransferase (3-MST) (8–10). H2S exerts its cellular actions via multiple mechanisms (1–15), including activation of potassium channels (1–3), stimulation of kinase pathways (4, 11, 12), and inhibition of phosphodiesterases (3, 15).
Both ATP generation and angiogenesis are vital factors for the growth and proliferation of tumors (16–19). Using human colon cancer tissues and cancer-derived cell lines, we have now conducted a series of in vitro and in vivo studies to explore whether endogenous, tumor cell-derived H2S plays a role as a tumor-derived survival factor. The results show that CBS is selectively overexpressed in colon cancer, and that H2S produced by it serves to maintain the tumor's cellular bioenergetics and to promote tumor angiogenesis.
Results
CBS Is Overexpressed in Human Colon Adenocarcinomas.
Comparison of human colon cancer specimens with patient-matched normal mucosa tissue revealed the selective up-regulation of the H2S-producing enzyme CBS in the cancers (Fig. 1 A and B). In contrast, the expression of the other two H2S-producing enzymes, CSE, and 3-MST remained unchanged (Fig. 1A). Similar to colon tumors, colon adenocarcinoma-derived cell lines (HCT116, HT-29, LoVo) exhibited the selective up-regulation of CBS, compared with the nonmalignant colonic epithelial cell line (NCM356) (Fig. 1 C and D). Homogenates of the patient-derived colon tumor specimens, as well as homogenates of the colon cancer-derived cell lines exhibited increased rates of H2S production, which was blocked by the CBS inhibitor aminooxyacetic acid (AOAA), but not by the CSE inhibitor propargylglycine (PAG) (Fig. 1 E and F and Fig. S1).
Fig. 1.
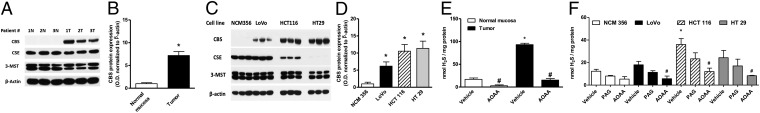
CBS is overexpressed in human colorectal cancer. (A) Representative Western blot of CBS, CSE, and 3-MST protein expression in human colorectal cancer specimens, paired with the corresponding normal mucosa tissues. PVDF membranes were probed with rabbit polyclonal antibodies against CBS, CSE, and 3-MST. (B) Densitometric analyses of CBS expression, in seven pairs of human colorectal cancers and the patient-matched normal mucosa, showed an approximately sevenfold increase in CBS protein expression in colon cancer (arbitrary relative densitometric units were normalized with β-actin using image analysis software) (*P < 0.05 vs. normal mucosa). (C and D) CBS was highly expressed in three different colon cancer cell lines (LoVo, HCT116, and HT29); low expression was detected in the nontumorigenic normal colon mucosa cells (NCM356) (arbitrary relative densitometric units were normalized with β-actin using image analysis software) (*P < 0.05 vs. NCM356 cells). H2S production was measured in human colorectal cancer specimens (E) and in colon cancer cell lines (F) by the methylene blue method. H2S production was stimulated in tissue or cell lysates by incubation at 37 °C (30 min) in presence of the CBS substrates l-cysteine (3 mM) and l-homocysteine (0.5 mM). CBS activity was significantly higher in colon cancer tissues, compared with their corresponding controls. AOAA (1 mM) blocked the H2S-producing activity of CBS in the tissue extracts (*P < 0.05 vs. corresponding from normal mucosa and #P < 0.05 vs. vehicle), whereas PAG (3 mM) had no significant effect. HCT116 cells exhibited the highest rate of H2S production, as measured by the methylene blue method in cell lysates (*P < 0.05 vs. corresponding values in NCM356 and #P < 0.05 vs. vehicle). Western blots show representatives of at least n = 3 experiments; H2S measurements represent mean ± SEM of at least n = 3 determinations.
CBS Is Associated with Colon Cancer Cell Mitochondria.
Cell fractionation studies showed that a significant portion of the total amount of cellular CBS was associated with the mitochondria in HCT116 cells (Fig. 2A). A trypsin digestion assay on isolated mitochondria (Fig. 2B) showed that CBS was primarily associated with the outer mitochondrial membrane.
Fig. 2.

Presence of CBS in cytosolic and mitochondrial fractions of HCT116 cells. (A) CBS was detected in whole cell lysates as well as in mitochondrial and cytoplasmic cell fractions harvested from HCT116 cells. (B) Limited trypsin digestion of isolated mitochondria (30–60 min) reduced mitochondrial CBS, as well as the mitochondrial outer membrane protein Tom20, while enriching complex IV (an inner membrane protein). Each Western blot is representative of at least three independent experiments.
CBS-Derived H2S Stimulates Colon Cancer Cell Proliferation, Migration, and Invasion in Vitro.
Gene specific short-hairpin RNA (shRNA) sequences in lentiviral vectors were used to suppress the expression of either CBS or CSE in HCT116 cells (Fig. 3A). Densitometric analyses of Western blots revealed an ∼50% decrease in CBS expression with comparable reductions in both cell proliferation (Fig. 3 A and B) and H2S production (Fig. 3C). In contrast to CBS, silencing of CSE or the pharmacological CSE inhibitor PAG (3 mM) did not significantly affect either HCT116 cell proliferation or H2S production (Fig. 3 A–E). To further define the function of CBS in colon cancer cells, we treated both NCM356 (which express CSE, but only low levels of CBS, Fig. 1C) and HCT116 cells (which overexpress CBS) with the pharmacological CBS inhibitor, AOAA. Consistent with the CBS knockdown experiments, AOAA treatment inhibited the growth of HCT116 colon cancer cells, but did not affect the proliferation of the slower-growing nonmalignant NCM356 cell line (Fig. 3 D and E). Conversely, forced overexpression of CBS in NCM356 cells significantly increased their basal rate of proliferation (Fig. 3 F and G). AOAA treatment also suppressed the migration (Fig. 3H) and invasion (Fig. 3I) of HCT116 cells, whereas the presence of the H2S donor NaHS (30 µM) in the lower chamber slightly enhanced HCT116 cell migration and invasion in serum-free media (Fig. 3 H and I). Inhibition of CBS with AOAA reduced endothelial cell migration in colon cancer/endothelial cell cocultures (Fig. 3J).
Fig. 3.

ShRNA mediated down-regulation of CBS or pharmacological inhibition by AOAA inhibits proliferative, migratory, and invading activity of HCT116 cells in vitro. (A and B) The lentiviral shRNA vectors targeting CBS (shCBS) and CSE (shCSE) were transfected into HCT116 cells. A nontargeting sequence was used as control (shNT). The shRNA approach inhibited the expression of both CBS and CSE genes at the protein level, as shown by Western blotting (Inset). Following CBS and CSE silencing, cells were seeded at the density of 3,000 cells per well in xCELLigence plates and proliferation was monitored for 36 h. Down-regulation of CBS, but not CSE, significantly reduced HCT116 proliferation rate (*P < 0.05 vs. shNT). (C) shCBS but not shCSE yielded lower H2S production in cellular homogenates (P < 0.05 vs. shNT). (D and E) HCT116 cells exhibited a significantly higher proliferation rate, compared with NCM356 cells. AOAA (1 mM) did not affect NCM356 growth, but markedly reduced HCT116 cell proliferation (*P < 0.05 vs. NCM356 and #P < 0.05 vs. vehicle), whereas PAG (3 mM) did not affect cell proliferation. (F and G) Adenoviral-mediated CBS overexpression enhances the proliferation rate of NCM356 cells. The NCM356 cells were infected overnight with a CBS expressing adenovirus (Ad-CBS, 10 multicitiplies of infection) or its control, a green fluorescent protein (Ad-GFP). The culture media was then replaced and cells were seeded in XCELLigence plates at 3,000 cells per well. Cell proliferation was then measured in real-time over 36 h. Effective overexpression of CBS was detected within 12–24 h following infection (Inset). Adenoviral-mediated CBS overexpression significantly enhanced NCM356 cell proliferation (*P < 0.05 vs. Ad-GFP). (H and I) The effect of AOAA and NaHS was also tested on HCT116 cell migration (H) and invasion (I). Cells were pretreated with either vehicle or AOAA (1 mM) and seeded in serum-free DMEM (0.1% albumin) in the upper chamber of a Transwell insert uncoated (migration assay) or coated with growth factor reduced matrigel (invasion assay). Migration and invasion were stimulated by fibroblast-conditioned media in the lower chamber for 6 and 24 h, respectively. NaHS (30 µM) in the lower chamber slightly enhanced HCT116 cell migration and invasion in serum-free media. AOAA markedly reduced fibroblast derived growth factor-induced HCT116 cell migration and invasion (*P < 0.05 and #P < 0.05). (J) In a coculture of human endothelial cells (EAhy926) and colon cancer cells (HCT116), HCT116 cells were seeded in the lower chamber of a Transwell insert and cultured to confluence. Endothelial cells were then seeded into the upper chamber in serum-free DMEM and allowed to migrate for 4 h at 37 °C. The presence of HCT116 in the lower chamber markedly increased the number of migrated endothelial cells, and this effect was reduced by AOAA (*P < 0.05). Western blots are representative of at least three independent experiments; proliferation/migration/invasion assays and H2S measurements represent mean ± SEM of n = 3 determinations.
CBS-Derived H2S, Produced by Human Colon Cancer Cells, Stimulates Cancer Cell Bioenergetics in Vitro.
In agreement with the physiological role of endogenous H2S in promoting cellular bioenergetics in various cell types including colonocytes (6, 7, 20, 21), low concentrations of H2S caused a stimulation of mitochondrial function in mitochondria isolated from HCT116 cells (Fig. S2). Moreover, shRNA-mediated silencing of CBS (but not of CSE) expression, or CBS inhibition with AOAA reduced basal cellular respiration, suppressed the calculated ATP synthesis, and attenuated the spare respiratory capacity (Fig. 4A and Fig. S3). CBS silencing or CBS inhibition also reduced glycolytic functions (Figs. S3 and S4). This latter effect may be attributed, at least in part, to inhibition of GAPDH activity: GAPDH activity was quantified in HCT116 cells, and it was reduced by CBS silencing. (GAPDH activity in wild-type and CBS silenced cells amounted to 6.2 ± 0.2 units/mL and 4.0 ± 0.1 units/mL, respectively, n = 3, P < 0.01). AOAA only exerted minor residual effects in shCBS HCT116 cells (Fig. S5). In addition, l-cysteine stimulated bioenergetics more substantially in HCT116 cells than in NCM356 cells (Fig. S6). Likewise, in mitochondria prepared from HCT116 cells, l-cysteine stimulated mitochondrial electron transport; this effect was attenuated in mitochondria prepared from shCBS cells (Fig. 4C).
Fig. 4.

ShRNA-mediated down-regulation of CBS suppresses cellular bioenergetics in HCT116 cells. (A) Oxygen consumption rate (OCR) in HCT116 cells subjected to either nontargeting (shNT, control) or stable lentiviral silencing of CBS or CSE (shCBS, shCSE). shCBS enzyme significantly decreased basal OCR, calculated ATP production, maximal respiration, and spare respiratory capacity (*P < 0.05 or **P < 0.01 vs. shNT), whereas CSE silencing had no effect on the bioenergetic profile. (B) Coupling experiments show that l-cysteine (30 nM) elevates OCR in state 3 and state 3u respiration in mitochondria isolated from control shNT cells (*P < 0.05), but not in mitochondria isolated from shCBS cells (#P < 0.05 or ##P < 0.01). Data represent mean ± SEM of n = 4–5 determinations.
l-Cystathionine Is Not Responsible for the Proliferative/Bioenergetic Role of CBS.
In addition to producing H2S, CBS also produces cystathionine. l-Cystathionine content of NCM356 cells was 1.0 ± 0.1 mM/mg protein basally, and 33.9 ± 4.1 mM/mg after stimulation with l-homocysteine/l-cysteine (n = 3). Consistently with their higher CBS expression, HCT116 cells had higher basal and stimulated l-cystathionine levels (6.4 ± 0.1 and 52.4 ± 4.2, respectively, n = 3; P < 0.05). Addition of 1 mM l-cystathionine to either NCM356 or HCT116 cells failed to affect their proliferation rate; proliferation rate of NCM356 and HCT116 cells was 102 ± 3% and 101 ± 2% of their respective vehicle control at 36 h (n = 3). Similarly, 1 mM cystathionine failed to stimulate cellular bioenergetics: carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP)-induced oxygen consumption rate (OCR) in the presence of cystathionine amounted to 91 ± 7% or 84 ± 10% of their respective vehicle control in NCM356 and HCT117 cells (n = 6).
CBS Stimulates Tumor Xenograft Growth, Angiogenesis, and Peritumoral Vascular Tone.
Consistent with the in vitro findings, shRNA-mediated knockdown of CBS expression significantly reduced the growth rate and size (i.e., volume) of HCT116 tumor xenografts (Fig. 5 A and B). In contrast, silencing of CSE did not affect tumor growth (Fig. 5 A and B). As expected, silencing of CBS or CSE in the tumor cells did not alter circulating H2S levels in tumor-bearing mice (Fig. 5C). CBS suppression caused a significant reduction in the density of CD31-positive blood vessels within the tumor tissue (Fig. 5D) as well as the prevalence of larger blood vessels and the extent of vessel branching (Fig. 5E). These data are consistent with the hypothesis that CBS-derived H2S acts locally in a paracrine manner to stimulate tumor angiogenesis.
Fig. 5.

ShRNA-mediated down-regulation of CBS or pharmacological inhibition by AOAA (9 mg/kg·d−1) inhibits colon cancer growth and tumor angiogenesis in vivo. Effects of shRNA-mediated gene silencing of CBS (shCBS) and CSE (shCSE) on HCT116 tumor xenograft: (A) growth rate (shNT = nontargeting shRNA control), (B) tumor volume at harvest (*P = 0.04), and (C) plasma levels of H2S. (D) Quantification of the effects of CBS silencing on CD31-positive blood vessel density in HCT116 tumor xenografts (*P < 0.0001). (E) Photomicrographs of representative sections (10 μm) from control (shNT) and CBS knockdown (shCBS) xenografts showing CD31-positive blood vessels (brown). Note the increased density of blood vessels in shNT vs. shCBS. Arrows indicate larger vessels and bracket indicates areas of necrosis with shCBS xenograft. Effects of AOAA or vehicle (PBS) on HCT116 tumor xenografts: (F) growth rate, (G) tumor volume (*P = 0.02), (H) wet weight (*P = 0.001), and (I) plasma concentrations of H2S (*P = 0.0005). (J) Photomicrographs of H&E stained formalin-fixed paraffin-embedded sections (5 μm) of the primary colon adenocarcinoma from a patient with stage III disease and Kras mutation (PT), and the corresponding patient-derived tumor xenograft (PDTX). Note the similar morphology of both specimens. (K) Western blot comparing the relative levels of expression of CBS in tissue extracts from the patient’s tumor (T) shown in J and adjacent normal (N) mucosa. (L and M) Effects of AOAA and PBS treatment of growth rates of PDTXs from patient 1 and patient 2, respectively. (N) Summary data from two independent experiments showing the effects of AOAA and PBS on the change in PDTX volume over a 7-d course of treatment (*P = 0.07). Photomicrographs of histological sections are representative of at least n = 6 sections; tumor volume/weight data and H2S measurements represent mean ± SEM of tumors/plasma values obtained from n = 6 mice.
Because small-molecule inhibitors are potentially more amenable to therapeutic translation than shRNA-based strategies, we next tested the effects of AOAA and PAG on tumor growth in vivo. Similar to CBS knockdown, AOAA treatment significantly slowed the rate of HCT116 xenograft growth (Fig. 5F), resulting in tumors with reduced size and weight at the time of harvest (Fig. 5 G and H). A reduction in the plasma concentration of H2S confirmed the inhibitory effect of AOAA on systemic H2S production (Fig. 5I). Also, in accordance with the role of H2S as a local vasodilator, direct injection of AOAA into the tumor parenchyma significantly reduced, whereas intratumor administration of the CBS substrate l-cysteine increased peritumor flow (Fig. 6). Similar to the lack of inhibition of tumor growth by CSE silencing (Fig. 5 A and B), the CSE inhibitor PAG failed to reduce tumor growth in vivo (Fig. S7).
Fig. 6.
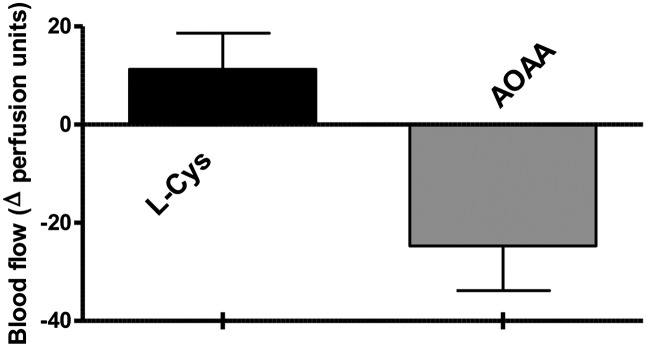
l-cysteine increases, whereas AOAA reduces microvessel blood flow in tumor-bearing mice. Tumor-bearing mice were anesthetized by i.p. injections of ketamine-xylazine and a laser Doppler (PeriFlux system 5000) was placed on top of the tumor for microvessel blood flow measurements. After a stabilization period, l-cysteine or AOAA were injected s.c. in proximity of the tumor. AOAA caused a marked decrease of the skin microvessel blood flow. Data represent mean ± SEM of n = 6 determinations.
The patient-derived tumor xenograft (PDTX) is an emerging preclinical model for anticancer drug development, because it more closely recapitulates both the genetic and cellular heterogeneity of the patient’s original tumor tissue (22) (Fig. 5J). To begin testing the concept that CBS inhibition could be an effective strategy for future therapeutic development, we evaluated the effects of AOAA treatment on mice bearing PDTXs derived from the primary adenocarcinoma of a patient with pathological stage III disease with an activating Kras mutation and with a marked up-regulation of endogenous CBS (Fig. 5K). Following randomization, xenograft-bearing mice were treated daily either with AOAA or vehicle (PBS). AOAA-treated animals showed a reduced rate of PDTX growth (Fig. 5L). Importantly, the growth inhibitory effects of AOAA on PDTX growth were also observed using tumor tissue from a second patient with stage II disease that also possessed a mutant Kras allele (Fig. 5M). Summary data from two independent experiments using the PDTXs from the different patients showed that AOAA effectively reduced the rate of PDTX growth over the time course of the treatment (Fig. 5N).
Discussion
Several prior studies have demonstrated that H2S donors can stimulate mitochondrial electron transport and ATP generation in various cell lines in vitro (6, 7). Furthermore, H2S, via sulfhydration, has been shown to increase the catalytic activity of the glycolytic enzyme GAPDH (14). In line of these findings, the current study shows that both oxidative and glycolytic tumor cell metabolism is suppressed by shRNA-mediated silencing of CBS or by the pharmacological CBS inhibitor AOAA. Tumor bioenergetics relies both on oxidative phosphorylation, as well as glycolysis (even in the presence of normal oxygen supply, a phenomenon known as the Warburg effect) (16, 17). Because proliferation, migration, and invasion are energetically demanding processes, we hypothesize that CBS inhibition/silencing reduces the intracellular levels of H2S, which, in turn, contributes to the energy starvation of the tumor, impairing its growth (Fig. 7). In support of a role for CBS in regulating the proliferation of colon cancer cells, forced overexpression of CBS was sufficient to increase the rate of proliferation of nontumorigenic NCM365 cells in vitro. In addition to bioenergetic effects, endogenous H2S may also contribute to tumor growth by additional mechanisms including activation of phosphoinositide-3-kinase and Akt kinase (11, 12), inhibition of phosphatases (12, 23), and the regulation of the expression of genes involved in the control of the cell cycle (24).
Fig. 7.

Proposed mechanisms for H2S induced colon cancer growth. As a result of the CBS overexpression, H2S is overproduced in colon cancer cells. H2S serves as an inorganic electron donor, stimulating mitochondrial electron transport, increasing ATP turnover. In addition, it increases the glycolytic activity of the tumor cell. Via these autocrine bioenergetic effects, H2S stimulates cancer cell proliferation, migration, and invasion. In addition, H2S diffuses into the surrounding cells and tissues, stimulating angiogenesis, as well as acting as a vascular relaxant. Via these paracrine effects, H2S promotes the supply of blood and nutrients to the tumor.
Tumor growth and metastasis require the development of new blood vessels (angiogenesis) in and around the tumor to facilitate the delivery of nutrients and oxygen as well as removal of waste products, thereby enhancing tumor proliferation and dissemination (18, 19). Although H2S has previously been shown to regulate angiogenesis in the context of organ ischemia (25, 26), its potential role in tumor angiogenesis has not yet been explored. The current study, showing that knockdown of CBS expression significantly reduces both the number and complexity of tumor blood vessels, indicates that tumor-derived H2S, in addition to its action as a bioenergetic factor, also supports tumor growth via promotion of angiogenesis (Fig. 7). Our postulate is further supported by in vitro data showing that the migration of endothelial cells toward HCT116 colon cancer in coculture is attenuated by the CBS inhibitor AOAA (Fig. 3J).
Tumors are known to secrete multiple angiogenic factors, including VEGF, which, in turn, stimulate the host to develop neovessels (18, 19). In vitro studies indicate that intracellular H2S production, by CSE, contributes to VEGF-stimulated endothelial cell migration (3, 5, 27). Although it has not been explored in the current study, it is conceivable that host-derived H2S, produced by endothelial CSE, may also contribute to tumor angiogenesis in vivo. On the other hand, the lack of suppression of tumor growth by intratumor CSE silencing (Figs. 3 and 5) and the lack of effect of PAG on HCT116 proliferation in vitro and in vivo indicates that in the tumor tissue itself, CBS (and not CSE) plays the primary role in the support of growth, proliferation, and angiogenesis.
The focus of the current study was to delineate the functional role of endogenous H2S production by tumor cells in vitro and in vivo. There are multiple prior in vitro reports with tumor cells subjected to exogenously applied H2S donors, demonstrating either proliferation-promoting or, at higher concentrations, antiproliferative effects (28–30). These latter findings are not inconsistent with the current results and are likely related to the well-known bell-shaped or biphasic biological character of H2S: whereas lower levels of H2S exert multiple physiological, cytoprotective, antioxidant, and anti-inflammatory functions, at higher local concentrations, H2S can become prooxidant, cytostatic, and cytotoxic (6–10, 30).
There are several lines of prior studies demonstrating that volatile sulfur compounds, including H2S, are increased in patients with certain forms of cancer (31, 32), although its sources or biological functions remained unexplored. In addition, several prior studies demonstrated an increase in CBS expression in various forms of human cancer (33–35), although the functional consequence of these observations remained unclear. Finally, a prior study showed that treatment of tumor-bearing mice with AOAA suppresses the growth of breast cancer (36), although these findings have not been linked to pathways related to CBS or H2S. Indeed, one of the limitations of AOAA, a commonly used CBS inhibitor, is that it also inhibits a variety of additional pyridoxal-5′-phosphate–dependent enzymes thereby inhibiting, among others, GABA synthesis, kynurenine synthesis, and glutamic-oxaloacetic transaminase, a component of the tricarboxylic acid cycle and of the malate-aspartate shuttle (36–39). In the current study the combined use of lentiviral CBS silencing and AOAA was used to overcome this limitation.
In summary, the current study—concluding that colon cancer cells selectively overexpress CBS, which produces H2S to (i) maintain cellular bioenergetics, thereby supporting tumor growth, and (ii) promote angiogenesis and vasorelaxation—puts all of these prior data into a unified concept and identifies CBS-derived H2S as an endogenous tumor-promoting factor and anticancer drug target.
Materials and Methods
Collection and Analysis of Samples from Human Colon Cancers.
Freshly resected human colorectal tumor tissue, along with a specimen of patient-matched normal mucosa (wide margin), was collected under an institutional review board, approved protocol for discarded tissue not requiring informed patient consent, and snap frozen in liquid N2. Clinicopathologic information of the samples collected is provided in Table S1.
Colon Cancer Cell Lines.
The colon cancer cells line HT-29 (ATCC) and the nontumorigenic colon epithelial cell line derived from the normal margin of a rectal cancer specimen, NCM356 (40) were cultured in DMEM supplemented with 10% (vol/vol) FBS. HCT116 and LoVo cells (ATCC) were cultured in McCoy’s 5A and F-12, respectively.
Subcellular Fractionation and Western Blotting.
Western blotting for CSE, CBS, and 3-MST was conducted in cell and tissue homogenates as described (3, 5, 7). HCT116 cells were subjected to subcellular fractionation by centrifugation (41). Mitochondrial and cytosolic fractions were analyzed by Western blotting; membranes were probed overnight with anti-CBS (60 kDa), anti–β-actin (43 kDa), antilactate dehydrogenase (35 kDa), anti-Tom20 (20 kDa), and anti-complex IV (17 kDa) antibodies. In a separate set of experiments, the mitochondrial outer membrane was subjected to limited trypsin digestion (42), followed by Western blotting.
H2S/l-Cystathionine Measurements.
For the measurement of H2S production, the methylene blue assay (43) was used, and for the measurement of L-cystathionine production, the ninhydrin assay (44) was used.
ShRNA-Mediated Silencing of CBS and CSE and Adenoviral-Mediated Overexpression of CBS.
Colon cancer cell lines were transduced with either a lentiviral vector containing shRNA sequences targeting CBS (SHCLNV, clone TRCN0000045359) or shCSE (clone TRCN0000078263). A nontargeting control shRNA sequence (shNT) was used to control off-target effects (SHC002V, MISSION shRNA; Sigma-Aldrich). HCT116 cells were infected at a multiplicity of infection (MOI) of 3 with hexadimethrine bromide (8 µg/mL). Transduced cells were selected and maintained in McCoy’s 5A media supplemented with 10% (vol/vol) FBS and puromycin (2 µg/mL) (45). To overexpress CBS in NCM356 cells, gene transfer was accomplished using an adenovirus at a MOI of 10, with a green fluorescent protein–expressing adenovirus used as a control (46).
Proliferation, Migration, Invasion, and Endothelial Cell Proliferation Assays in Vitro.
Cellular migration and invasion assays were performed as described (47). A total of 105 cells were suspended in serum-free media containing 0.1% BSA and then added to 6.5 mm Transwell chambers (8 mm pore) uncoated (migration assay) or coated with a thin layer (45 µL) of growth factor reduced matrigel (cell invasion assay). Cells were allowed to migrate toward the bottom chamber containing fibroblast (National Institutes of Health 3T3 cells) conditioned media at 37 °C for 6 or 24 h for cell migration and invasion, respectively. Migrated and invaded cells were then fixed in Carson’s fixative, stained in 0.33% toluidine blue, and quantified by visual counting. For the HCT116–EAhy926 coculture experiment, the cancer cell line was grown to confluence on the bottom wells of the Transwell chambers containing 600 µL of DMEM media. Endothelial cells were grown until 60–70% confluent and then trypsinized, suspended at a density of 105 cells/mL in DMEM, and added to the upper chamber (100 µL of the cell suspension). Cell migration was then assessed as described (3). For assessment of cell proliferation, the xCELLigence system (Roche) was used (25).
Bioenergetic Analysis in Intact Cells.
The XF24 Extracellular Flux Analyzer (Seahorse Bioscience) was used to measure bioenergetic function as described (7, 43, 44). OCR after oligomycin (1.5 µM) was used to assess ATP production rate and OCR after FCCP (0.5 µM) to assess maximal mitochondrial respiratory capacity. Glycolytic dependency was determined by the use of 2-deoxyglucose (100 μM) and antimycin A (2 µg/m) and rotenone (2 µM) were used to inhibit the flux of electrons through complex III and I. For the measurement of glycolytic parameters, the changes in proton production rate (PPR) were monitored in response to the sequential administration of d-glucose (10 mM), oligomycin (1.5 µM), and 2-deoxyglucose (100 mM), to assess glycolysis, maximal glycolytic capacity, glycolytic reserve capacity, and nonglycolytic acidification rate, respectively. GAPDH activity was determined by a kinetic assay (ScienCell).
Bioenergetic Analysis in Isolated Mitochondria.
Bioenergetic measurements in mitochondria isolated from shNT and shCBS HCT116 cells were conducted in the absence or presence of l-cysteine (30 nM) as described (7, 43). Basal respiration (state 2) in the presence of succinate and rotenone was measured, followed by state 3 (phosphorylating respiration), in the presence of ADP. State 4o was determined after the addition of oligomycin, and maximal uncoupler-stimulated respiration (state 3u) was assessed using FCCP.
Human Tumor Xenograft Studies in Immune Compromised Mice.
The animal studies were approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch. Nu/nu Balb/C female mice (8–10 wk) were injected s.c. in the right and left dorsum (106 cells per side with HCT116 cells). One week later, the mice were randomized and s.c. injection of PBS, AOAA (9 mg/kg⋅d−1) or PAG (50 mg/kg⋅d−1) was performed 6 d/wk. Tumor diameters were measured transcutaneously using calipers and tumor volumes were calculated using the formula V = (π/6)hd2. T-test comparisons of the tumor volumes and final weights at harvest were performed. Xenotrials with PDTXs were performed on nu/nu mice implanted s.c. with a piece of passage 2 tumor measuring 2–5 mm3 each. When a small palpable tumor was evident at 7–10 d, the mice with tumor take were randomized and subjected to s.c. injection of vehicle or AOAA (9 mg/kg⋅d−1) 6 d/wk. Specimens were fixed with 10% formalin and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin. Each in vivo experiment was repeated at least twice.
Quantification of Blood Vessel Density.
Tissue preparation, immunostaining of CD31, and evaluation of microvessel density with the Chalkley reticle was performed as described (47).
Measurement of Microvascular Blood Flow.
Skin microvascular blood flow was measured using a PeriFlux 5000 laser-Doppler flowmeter. Mice bearing PDTX were anesthetized by ketamine-xylazine (i.p.). The laser-Doppler probe was placed on top of the tumors and a stable basal signal was recorded. Changes in the s.c. microcirculatory perfusion were detected following intratumoral injections of either l-cysteine (1 mg/kg) or AOAA (9 mg/kg).
Statistical Analysis.
All data are presented as means ± SEM and were analyzed using GraphPad Prism software. Statistical analyses included Student t test or one-way ANOVA followed by Bonferroni’s multiple comparisons.
Supplementary Material
Acknowledgments
This work was supported, in part, by the Sealy Memorial Endowment Fund of the University of Texas Medical Branch (Drs. Hellmich and Szabo). K.M. is supported by a McLaughlin fellowship of the University of Texas Medical Branch. C. Coletta is supported by a postdoctoral fellowship from the American Heart Association. C. Chao is supported by the National Institutes of Health (K08CA125209-01A2). This work has been cofinanced by the European Science Foundation and Greek national funds through the Operational Program Aristeia 2011 (1436) (to A.P.).
Footnotes
Conflict of interest statement: C.S. and M.R.H. are founders of and C. Chao is consultant at CBS Therapeutics, a start-up company involved in research and development of cystathionine-β-synthase inhibitors.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306241110/-/DCSupplemental.
References
- 1.Yang G, et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mustafa AK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109(11):1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coletta C, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;109(23):9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai WJ, et al. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76(1):29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 5.Papapetropoulos A, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA. 2009;106(51):21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goubern M, Andriamihaja M, Nübel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21(8):1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 7.Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27(2):601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 8.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6(11):917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 9.Kimura H. Hydrogen sulfide: Its production, release and functions. Amino Acids. 2011;41(1):113–121. doi: 10.1007/s00726-010-0510-x. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 11.Cai WJ, Wang MJ, Ju LH, Wang C, Zhu YC. Hydrogen sulfide induces human colon cancer cell proliferation: Role of Akt, ERK and p21. Cell Biol Int. 2010;34(6):565–572. doi: 10.1042/CBI20090368. [DOI] [PubMed] [Google Scholar]
- 12.Manna P, Jain SK. Hydrogen sulfide and L-cysteine increase phosphatidylinositol 3,4,5-trisphosphate (PIP3) and glucose utilization by inhibiting phosphatase and tensin homolog (PTEN) protein and activating phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase (AKT)/protein kinase Cζ/λ (PKCζ/λ) in 3T3l1 adipocytes. J Biol Chem. 2011;286(46):39848–39859. doi: 10.1074/jbc.M111.270884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucci M, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30(10):1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- 14.Mustafa AK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2(96):ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul BD, Snyder SH. H₂S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 16.Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010;1797(6-7):1225–1230. doi: 10.1016/j.bbabio.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook KM, Figg WD. Angiogenesis inhibitors: Current strategies and future prospects. CA Cancer J Clin. 2010;60(4):222–243. doi: 10.3322/caac.20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lagoutte E, et al. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797(8):1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Mimoun S, et al. Detoxification of H(2)S by differentiated colonic epithelial cells: Implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid Redox Signal. 2012;17(1):1–10. doi: 10.1089/ars.2011.4186. [DOI] [PubMed] [Google Scholar]
- 22.Tentler JJ, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4(203):ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deplancke B, Gaskins HR. Hydrogen sulfide induces serum-independent cell cycle entry in nontransformed rat intestinal epithelial cells. FASEB J. 2003;17(10):1310–1312. doi: 10.1096/fj.02-0883fje. [DOI] [PubMed] [Google Scholar]
- 25.Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br J Pharmacol. 2011;164(3):853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qipshidze N, Metreveli N, Mishra PK, Lominadze D, Tyagi SC. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int J Biol Sci. 2012;8(4):430–441. doi: 10.7150/ijbs.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pupo E, et al. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic Biol Med. 2011;51(9):1765–1773. doi: 10.1016/j.freeradbiomed.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Lee ZW, et al. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE. 2011;6(6):e21077. doi: 10.1371/journal.pone.0021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu YC, et al. Hydrogen sulfide lowers proliferation and induces protective autophagy in colon epithelial cells. PLoS ONE. 2012;7(5):e37572. doi: 10.1371/journal.pone.0037572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baskar R, Bian J. Hydrogen sulfide gas has cell growth regulatory role. Eur J Pharmacol. 2011;656(1-3):5–9. doi: 10.1016/j.ejphar.2011.01.052. [DOI] [PubMed] [Google Scholar]
- 31.Yamagishi K, et al. Generation of gaseous sulfur-containing compounds in tumour tissue and suppression of gas diffusion as an antitumour treatment. Gut. 2012;61(4):554–561. doi: 10.1136/gutjnl-2011-300721. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, et al. Selected ion flow tube-MS analysis of headspace vapor from gastric content for the diagnosis of gastro-esophageal cancer. Anal Chem. 2012;84(21):9550–9557. doi: 10.1021/ac302409a. [DOI] [PubMed] [Google Scholar]
- 33.De Vos J, et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene. 2002;21(44):6848–6857. doi: 10.1038/sj.onc.1205868. [DOI] [PubMed] [Google Scholar]
- 34.Hansel DE, et al. Identification of novel cellular targets in biliary tract cancers using global gene expression technology. Am J Pathol. 2003;163(1):217–229. doi: 10.1016/S0002-9440(10)63645-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo H, et al. (2012) Characterization of hydrogen sulfide and its synthases, cystathionine β-synthase and cystathionine γ-lyase, in human prostatic tissue and cells. Urology. 79:483.e1–5. [DOI] [PubMed]
- 36.Thornburg JM, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10(5):R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry TL, et al. Failure of aminooxyacetic acid therapy in Huntington disease. Neurology. 1980;30(7 Pt 1):772–775. doi: 10.1212/wnl.30.7.772. [DOI] [PubMed] [Google Scholar]
- 38.Turski WA, Dziki M, Urbanska E, Calderazzo-Filho LS, Cavalheiro EA. Seizures induced by aminooxyacetic acid in mice: Pharmacological characteristics. Synapse. 1991;7(3):173–180. doi: 10.1002/syn.890070302. [DOI] [PubMed] [Google Scholar]
- 39.Støttrup NB, et al. Inhibition of the malate-aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc Res. 2010;88(2):257–266. doi: 10.1093/cvr/cvq205. [DOI] [PubMed] [Google Scholar]
- 40.Chao C, et al. CCK2 receptor expression transforms non-tumorigenic human NCM356 colonic epithelial cells into tumor forming cells. Int J Cancer. 2010;126(4):864–875. doi: 10.1002/ijc.24845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2(2):287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 42.Szczesny B, Hazra TK, Papaconstantinou J, Mitra S, Boldogh I. Age-dependent deficiency in import of mitochondrial DNA glycosylases required for repair of oxidatively damaged bases. Proc Natl Acad Sci USA. 2003;100(19):10670–10675. doi: 10.1073/pnas.1932854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Módis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013;433(4):401–407. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- 44.Kriebitzsch C, et al. 1,25-dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3-E1 cells by direct regulation of cystathionine β-synthase. J Bone Miner Res. 2011;26(12):2991–3000. doi: 10.1002/jbmr.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim SO, et al. Raf-1 kinase inhibitory protein (RKIP) mediates ethanol-induced sensitization of secretagogue signaling in pancreatic acinar cells. J Biol Chem. 2012;287(40):33377–33388. doi: 10.1074/jbc.M112.367656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chao C, Lotz MM, Clarke AC, Mercurio AM. A function for the integrin alpha6beta4 in the invasive properties of colorectal carcinoma cells. Cancer Res. 1996;56(20):4811–4819. [PubMed] [Google Scholar]
- 47.Wu M, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292(1):C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.