

Abstract
The antibiotic blasticidin S (BlaS) is a potent inhibitor of protein synthesis in bacteria and eukaryotes. We have determined a 3.4-Å crystal structure of BlaS bound to a 70S⋅tRNA ribosome complex and performed biochemical and single-molecule FRET experiments to determine the mechanism of action of the antibiotic. We find that BlaS enhances tRNA binding to the P site of the large ribosomal subunit and slows down spontaneous intersubunit rotation in pretranslocation ribosomes. However, the antibiotic has negligible effect on elongation factor G catalyzed translocation of tRNA and mRNA. The crystal structure of the antibiotic–ribosome complex reveals that BlaS impedes protein synthesis through a unique mechanism by bending the 3′ terminus of the P-site tRNA toward the A site of the large ribosomal subunit. Biochemical experiments demonstrate that stabilization of the deformed conformation of the P-site tRNA by BlaS strongly inhibits peptidyl-tRNA hydrolysis by release factors and, to a lesser extent, peptide bond formation.
Keywords: peptidyl transfer, ribosome crystal structure, termination inhibitor, translation inhibitor, translation termination
The growing problem of pathogen resistance to existing antibacterials prompts a search for alternative modes of inhibiting bacterial growth. The development of new drugs can be facilitated by understanding the mechanisms of action of known antibiotics (1). Because of its central role in cell metabolism, the ribosome is the target of numerous inhibitors that bind to various sites on the ribosome and interfere with different steps of protein synthesis. One of the predominant modes of action of ribosomal antibiotics is the inhibition of peptide bond formation. A majority of peptidyl-transferase inhibitors, including the widely used antibacterials chloramphenicol, linezolid, and the lincosamides lincomycin and clindamycin, bind to the A site of the large ribosomal subunit at the peptidyl-transferase center of the ribosome (2–4). By contrast, blasticidin S (BlaS) binds to the P site of the large subunit (5) and inhibits the peptidyl-transferase reaction through a distinct mechanism, which is still poorly understood.
BlaS, produced by some Streptomyces species, is a nucleoside analog consisting of a cytosine bonded to a pyranose ring and attached to an N-methyl-guanidine tail (Fig. 1A). BlaS has long been known to be a potent inhibitor of protein synthesis in bacteria and eukaryotes (6, 7). A crystal structure of the 50S subunit from Haloarcula marismortui bound to BlaS in the absence of tRNA revealed that BlaS occupies the P site of the large subunit (5). Two molecules of BlaS interact with the P loop and form base pairs with the universally conserved G2251 and G2252 of 23S ribosomal RNA (Escherichia coli 70S ribosome numbering is used throughout this article). These base pairing interactions closely mimic those of the two cytosine residues of the conserved CCA 3′ terminus of tRNA bound in the P site. Based on the finding that two molecules of BlaS mimic the cytosine residues of the CCA 3′ end of the P-site tRNA, BlaS was proposed to inhibit protein synthesis by competing with tRNA binding to the P site (5). However, early biochemical studies demonstrated that BlaS enhances P-site binding of aminoacyl-CACCA, which mimics the 3′ end of aminoacylated tRNA (8) and modestly inhibits the binding of aminoacyl-CACCA to the A site (8, 9). Moreover, BlaS competes with A-site binding antibiotics, such as sparsomycin (10) and puromycin (11). Mammalian cells resistant to BlaS were shown to be also resistant to sparsomycin and puromycin, further suggesting that the inhibitory mode of action of BlaS may involve the binding of the drug to the A site (12). Thus, the mechanism of BlaS action remains a conundrum: how can BlaS, whose binding site overlaps with that for P-site tRNA, stabilize the binding of the pentanucleotide mimic of the 3′ end of tRNA to the P site on the 50S subunit (8) and inhibit binding of tRNA analogs and antibiotics to the A site?
Fig. 1.

Effect of BlaS on fMet-tRNAfMet binding to the P site of the 70S ribosome. (A) Chemical structure of BlaS. (B) Binding of [35S]-fMet-tRNAfMet to the ribosome in the presence of mRNA and various concentrations of BlaS measured by a filter-binding assay. Bars show the P-site occupancy by [35S]-fMet-tRNAfMet relative to that in the ribosomes not treated with the antibiotic (the occupancy in the absence of the antibiotic is set to 1.0). Error bars show SDs calculated from triplicate measurements.
In this work, we elucidate the mechanism of BlaS action by X-ray crystallography, single-molecule Förster resonance energy transfer (smFRET) and biochemical assays. We show that BlaS inhibits protein synthesis through a unique mechanism by bending the CCA end of P-site tRNA toward the A site of the large subunit. Stabilization of this unusual, deformed conformation of P-site tRNA by BlaS results in inhibition of peptide bond formation and peptidyl–tRNA hydrolysis by release factors.
Results and Discussion
BlaS Stabilizes tRNA in the P Site of the Large Ribosomal Subunit.
Early biochemical experiments showed that BlaS stabilizes binding of tRNA analogs to the 50S P site (8), which appears to conflict with crystallographic data showing that the binding sites of BlaS and the CCA end of P-site tRNA overlap (5). To reexamine the effect of BlaS on P-site binding, we used a filter-binding assay to monitor the binding of N-formyl-methionyl-tRNAfMet (fMet-tRNAfMet) to the P site of E. coli 70S ribosomes preincubated with various concentrations of BlaS in the presence of defined mRNA. Importantly, in contrast to previous biochemical experiments (8, 9), we used full-length initiator [35S]-fMet-tRNAfMet instead of short tRNA analogs, such as CACCA. No inhibition of tRNA binding was observed at concentrations of the antibiotic ≤1 mM (Fig. 1B). BlaS interfered with [35S]-fMet-tRNAfMet binding to the ribosome only at millimolar antibiotic concentrations (IC50 ∼20 mM), i.e., concentrations that exceed the inhibition constant of BlaS for peptidyl transfer (200–400 nM; see 13, 14) by nearly five orders of magnitude. Notably, these concentrations exceed by up to four orders of magnitude the concentration of BlaS sufficient to abolish growth of bacterial and eukaryotic cells (7, 15–17). The destabilizing effect of large amounts of the antibiotic on tRNA binding is likely due to BlaS binding to a second, low-affinity site on the ribosome observed in the structure of the large ribosomal subunit from H. marismortui crystallized in the presence of 1–10 mM BlaS (5). In sum, the antibiotic does not hinder tRNA binding to the P site of the ribosome at concentrations that are inhibitory for protein synthesis and cell growth.
To further explore the effects of BlaS on tRNA affinity to the 50S P site, we asked whether BlaS affects the dynamics of ribosome-bound tRNAs, whose translocation through the ribosome is essential for protein synthesis. tRNA translocation, which is catalyzed by elongation factor G [EF-G (EF-2 in eukaryotes)], occurs in two steps. The acceptor ends of tRNAs first move relative to the large ribosomal subunit, from the classical A/A and P/P states into hybrid A/P and P/E states (Fig. 2A). The next step is the movement of anticodon stem loops of tRNAs on the small ribosomal subunit into posttranslocation P/P and E/E states. Whereas tRNA translocation on the small subunit requires EF-G and GTP, the movement of acceptor stems of peptidyl- and deacylated tRNAs on the large subunit occurs spontaneously, fluctuating between the classical (A/A and P/P) and hybrid (A/P and P/E) states (18–20). These spontaneous fluctuations of tRNAs are coupled to a rotation of the small ribosomal subunit relative to the large subunit between the nonrotated and rotated conformations, respectively (21–24). We previously developed a method to follow the intersubunit dynamics of the ribosome by measuring smFRET between fluorophores attached to protein S6 on the platform of the small ribosomal subunit and protein L9 on the large subunit (23). Using FRET in combination with total internal reflection microscopy, we have reported spontaneous fluctuations in single pretranslocation ribosomes between ∼0.6 and ∼0.4 FRET values, which correspond to the classical (nonrotated) and hybrid (rotated) states, respectively (22).
Fig. 2.

Blasticidin S inhibits counterclockwise rotation of the 30S subunit coupled to the movement of deacylated tRNA into the hybrid P/E state. (A) Schematic depiction of intersubunit rotation, which is measured by smFRET, and tRNA movement during ribosomal translocation. (B and C) Histograms showing distributions of FRET values in ribosomes containing deacylated tRNAPhe in the P site in the absence (B) or presence (C) of BlaS. N, number of single-molecule FRET traces compiled.
We examined how BlaS affects the affinity of tRNA to the 50S P site by monitoring the dynamics of single-tRNA fluctuations between the classical P/P and hybrid P/E states. To this end, we added tRNAPhe to the P site of S6-Cy5/L9-Cy3-labeled ribosomes in the presence of defined mRNA and then flowed 1 mM BlaS into a sample cell. In the absence of BlaS, ribosomes underwent frequent intersubunit rotation and spent ∼70% of the time in the rotated, hybrid-state conformation (Fig. 2B), consistent with prior observations (18–20). Addition of BlaS dramatically increased the fraction of nonrotated ribosomes, from ∼30% to ∼80% (Fig. 2C and Table S1). The rate of clockwise rotation of the 30S subunit coupled to tRNA transition from the hybrid P/E to the classical P/P state was unaffected by BlaS. By contrast, BlaS addition resulted in a fivefold reduction of the rate of counterclockwise rotation coupled with transition of tRNA from the classical P/P to the hybrid P/E state (Table S1). Ribosomes containing a single elongator tRNAMet or initiator tRNAfMet instead of tRNAPhe behaved similarly (Fig. S1), underscoring that the effect of BlaS does not depend on tRNA identity. Inhibition of counterclockwise intersubunit rotation was recently observed in the presence of another peptidyl-transferase inhibitor, sparsomycin, which is also known to enhance tRNA binding to the 50S P site (25). We conclude that BlaS inhibits the transition of deacylated tRNA into the hybrid P/E state (Fig. 2C) via stabilization of the acceptor end of tRNA in the 50S P site, consistent with previous biochemical data (8, 9).
Effects of BlaS on the Dynamics of Pretranslocation Ribosomes and mRNA Translocation.
We next tested the effect of BlaS on spontaneous intersubunit dynamics of authentic pretranslocation ribosomes containing tRNAs in both A and P sites (Fig. 2A). Similarly to ribosomes containing a single tRNA in the P site, the majority (∼70%) of pretranslocation ribosomes containing the peptidyl-tRNA analog (18, 19, 26) N-acetyl-Phe-tRNAPhe in the A site and deacylated elongator tRNAMet in the P site adopt the rotated, hybrid-state conformation (Fig. 3A). In the presence of 1 mM BlaS, the majority of pretranslocation ribosomes (∼60%) remained in the rotated state. This was surprising because BlaS dramatically shifted the distribution of ribosomes containing a single deacylated tRNAMet toward the nonrotated, classical-state conformation (Fig. S1). However, kinetic analysis of single-molecule traces revealed that BlaS reduced the rates of both clockwise and counterclockwise rotation approximately sixfold, leaving the equilibrium constant (the ratio of rates for counterclockwise and clockwise intersubunit rotation) largely unaffected (Fig. S2 and Table S1). Inhibition of counterclockwise rotation of the 30S subunit coupled to transition of tRNA into hybrid states is likely due to stabilization of the acceptor end of deacylated tRNA in the 50S P site by BlaS, which fixes P-site tRNA in the P/P classical state (Fig. 2A). This echoes our observation for ribosomes containing a single deacylated tRNA in the P site (Fig. 2B). By contrast, inhibition of clockwise rotation of the 30S evidently occurs only in the presence of an A-site tRNA and is likely due to stabilization of the acceptor end of peptidyl-tRNA in the 50S P site by BlaS, which fixes A-site tRNA in the hybrid A/P state (Fig. 2A). Thus, in pretranslocation ribosomes containing tRNAs in both A and P sites, BlaS inhibits both the forward and reverse transitions between the classical and hybrid states of tRNA binding.
Fig. 3.

Effects of BlaS on spontaneous intersubunit rotation in pretranslocation ribosomes and kinetics of EF-G-catalyzed mRNA translocation. (A and B) smFRET distribution histograms for pretranslocation ribosomes containing tRNAMet and N-acetyl-Phe-tRNAPhe in the P and A sites, respectively, in the absence (A) or presence (B) of BlaS. (C) Kinetics of EF-G-catalyzed translocation of mRNA in ribosomes preincubated with BlaS (red) or not treated with the antibiotic (blue). Kinetic measurements were performed in presteady-state stopped-flow kinetic experiments by the quenching of fluorescein attached to the mRNA. Pretranslocation ribosomes containing deacylated elongator tRNAMet in the P site and N-acetyl-Phe-tRNAPhe in the A site were mixed with GTP and EF-G. Double-exponential fits for fluorescein quenching are black curves.
Because BlaS significantly slows down tRNA and intersubunit dynamics in pretranslocation ribosomes in the absence of EF-G, we next asked whether BlaS also inhibits EF-G-catalyzed translocation of mRNA and tRNA. EF-G⋅GTP transiently stabilizes the rotated, hybrid-state conformation (27) and induces translocation of the anticodon stem loops of tRNA and cognate codons of mRNA from A and P to P and E sites, respectively (26). Here, we followed the kinetics of mRNA translocation in the presence of EF-G⋅GTP and BlaS by measuring the fluorescence quenching of a fluorescein dye attached to the 3′ end of the mRNA as it moves within the ribosome (28). Pretranslocation ribosomes assembled with deacylated tRNAMet in the P site, N-acetyl-Phe-tRNAPhe in the A site, and fluorescein-labeled mRNA were mixed with EF-G⋅GTP in a stopped-flow apparatus. mRNA translocation was detected by rapid (3.7 ± 0.6 s−1) quenching of the fluorescein dye (Fig. 3C and Table S2). When pretranslocation ribosomes were preincubated with BlaS, the apparent rate of translocation was only slightly reduced (2.6 ± 0.4 s−1) (Fig. 3C and Table S2). Therefore, despite significant inhibition of spontaneous ribosome and tRNA dynamics by BlaS, EF-G-catalyzed translocation is almost unaffected by the presence of the antibiotic. Notably, perturbations of the P site of the large subunit by mutations in the P loop of 23S ribosomal RNA (G2252C and G2251C) were also shown to considerably affect the frequency of spontaneous tRNA fluctuations between classical and hybrid states (29) without dramatic effects on the rate of EF-G-induced translocation (30). Together, these observations suggest that formation of the hybrid state intermediate is accelerated by EF-G and is not a rate-limiting step of translocation.
The Crystal Structure of the 70S⋅tRNA Complex Bound with BlaS Reveals That BlaS Induces a Deformed Conformation of P-site tRNA.
A previous crystal structure of BlaS bound to the vacant 50S subunit of the H. marismortui ribosome revealed that the binding site for two molecules of BlaS overlaps with the binding site for P-tRNA (5). How can the enhancement of tRNA binding to the 50S P site observed in our smFRET experiments be achieved by an antibiotic that appears to compete with tRNA? To resolve this paradox, we determined a 3.4 Å crystal structure of BlaS bound to the 70S Thermus thermophilus ribosome in the presence of deacylated tRNAfMet molecules in the P and E sites (Fig. 4A and Table S3). In previously reported structures of 70S elongation (31, 32) or termination (33–35) complexes, the overall conformations of deacylated initiator tRNAfMet are nearly identical to those of elongator tRNAs and peptidyl-tRNA analogs. Together with the finding that the action of BlaS is unlikely to depend on the identity of tRNA (Fig. S1), this suggests that our crystal structure reflects the structural mechanism of action of BlaS on initiating and elongating ribosomes.
Fig. 4.

Crystal structure of T. thermophilus 70S ribosome bound with BlaS in the presence of tRNAfMet in the P and E sites. (A) Positions of BlaS (blue), P-site (orange), and E-site (red) tRNAs; 30S subunit is in cyan and 50S subunit in gray. (B) Unbiased Fobs-Fcalc density corresponding to BlaS and the CCA tail of the P-site tRNA. (C) Interactions of BlaS with the P site of the 50S subunit. (D) Superposition of tRNA in the 70S⋅tRNA⋅BlaS complex (orange, this work) with that from the antibiotic-free 70S·tRNA complex (green, ref. 31), demonstrating the BlaS-induced shift of the 3′ end toward the A site.
Unlike in the crystal structure of the vacant 50S subunit bound with two molecules of BlaS, only one molecule is found in the 70S⋅tRNA complex (Fig. 4B). The conformation of BlaS is similar to that of molecule 1 in the 50S crystal structure (5); the all-atom rms difference between them is 0.57 Å, similar to the coordinate error at 3.4 Å resolution. The N-methyl-guanidine moiety of BlaS is stacked on the base of A2439 of 23S ribosomal RNA and is stabilized by interaction with the negatively charged pocket formed by the phosphate groups of A2439, A2600, and C2601 (Fig. 4C). A single binding site for BlaS next to G2251 agrees with chemical probing and mutational data. In particular, interaction of the N-methyl-guanidine tail of BlaS with A2439 explains protection of A2439 from chemical modification in the presence of BlaS (36, 37). Mutation of U2438 of 23S ribosomal RNA confers BlaS resistance in the archaeon Halobacterium halobium (38). This can be explained by disruption of the U2438:A2071 base pair, which likely results in displacement of the adjacent A2439 and disruption of the interaction between A2439 and BlaS.
In the 70S structure, BlaS forms a base pair with G2251 of the 23S ribosomal RNA and intercalates between C74 and A76 of the CCA end of P-site tRNA. This interaction replaces the base pairing between G2251 and C75 observed in antibiotic-free 70S⋅tRNA complexes (31, 32, 39), resulting in displacement of the acceptor arm and the CCA end of the P-tRNA toward the A site (Fig. 4D). The ribose-phosphate backbone of C75 is shifted by more than 7 Å, whereas those of the adjacent C74 and A76 are displaced by ∼4 and ∼2 Å, respectively. This large local rearrangement results in a ∼4o rotation of the acceptor arm of the tRNA toward the 50S A site. The orientation of the rest of the P-site tRNA is nearly identical to that of the tRNA bound in the absence of BlaS (Fig. 4D). The movement of the CCA backbone caused by BlaS suggests that BlaS competition with A-site binding antibiotics (10, 11) is induced allosterically.
Whereas C74 is rearranged with respect to the 50S P site, it remains paired with G2252, whose base is moved by more than 1 Å codirectionally with the base of C74 (Fig. 4B). The ribose-phosphate backbone of the P loop is not affected by BlaS. The base of the terminal nucleotide A76, which is stacked on BlaS, remains in a position similar to that in the absence of BlaS (31, 32, 39). Intercalation of the cytosine moiety of BlaS between the bases of C74 and A76 likely compensates for the loss of stacking interaction of these nucleotides with C75. In addition, the 2′-hydroxyl group of C75 is positioned to hydrogen bond with the N4 position of the cytosine ring of the antibiotic, providing additional stabilization to the distorted conformation of tRNA. In sum, the stabilizing interactions that BlaS forms with ribosomal RNA and P-site tRNA explain the observed stabilization of the acceptor arm in the P site (Fig. 2B).
Our crystal structure provides insights into the mechanism of inhibition of peptidyl transfer by BlaS. Although the adenine moiety of A76 of the P-site tRNA is positioned to form an A-minor interaction with the A2450–C2063 bp of 23S ribosomal RNA (as in antibiotic-free complexes; see refs. 31, 32, 39), rearrangement of the CCA backbone results in detachment of the ribose of A76 from C2063. Rearrangement of the ribose moiety of A76 may result in suboptimal positioning of the peptidyl-tRNA and aminoacyl-tRNA substrates for nucleophilic attack, leading to inhibition of peptidyl transfer by BlaS.
BlaS Strongly Inhibits Peptide Release Mediated by RF1.
The nucleophilic substitution reaction of peptide-bond formation is mechanistically similar to peptidyl-tRNA hydrolysis mediated by release factors 1 and 2 (RF1 and RF2), which bind to the ribosomal A site in response to stop codons and catalyze peptidyl-tRNA hydrolysis (40–43). Release factors are directly involved in peptidyl-tRNA hydrolysis by providing a catalytic group located in the universally conserved GGQ motif (40–43). We reasoned that in addition to its peptidyl-transfer inhibitory activity, BlaS may be a translation termination inhibitor. Superimposition of the BlaS-bound 70S structure with structures of 70S termination complexes (34, 35, 44) revealed that the distorted CCA end of the P-site tRNA in the BlaS-bound ribosomal complex substantially overlaps with the binding site for the GGQ-containing region of domain 3 of the release factors (Fig. 5A). This suggests that distortion of the P-site tRNA by BlaS may interfere with binding of release factors to the ribosome and/or prevent proper positioning of the GGQ motif for peptidyl-tRNA hydrolysis.
Fig. 5.
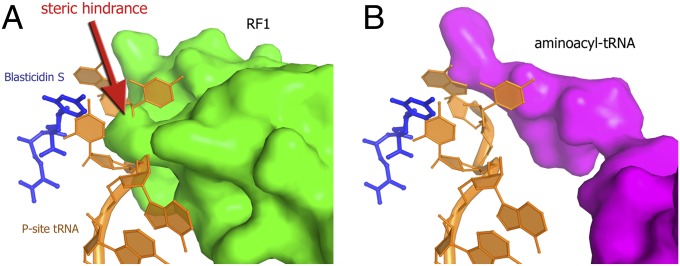
Superposition of the 70S⋅BlaS structure (this work) with structures of 70S complexes whose A site is occupied by RF1 (35) or aminoacyl-tRNA analog (32). The superposition demonstrates that steric hindrance in the presence of BlaS is more pronounced in the case of release factor (A, green) than in the case of aminoacyl-tRNA (B, magenta).
To test our hypothesis, we monitored the effect of BlaS on RF1-mediated peptide release from 70S ribosomes. We found that BlaS dramatically inhibits RF1-mediated [35S]-fMet release from [35S]-fMet-tRNAfMet bound to the P site of the ribosome in the presence of mRNA containing a UAA stop codon adjacent to an AUG codon. Specifically, BlaS nearly abolishes RF1-mediated release (Fig. 6A) at concentrations that were only modestly inhibitory to peptidyl transfer of [35S]-fMet to puromycin (Fig. 6B), which is an A-site antibiotic that mimics the 3′ end of an aminoacyl-tRNA. The apparent inhibition constant (Ki) of 182 ± 39 nM for BlaS-dependent inhibition of the puromycin reaction determined from our measurements is consistent with previous estimates of a Ki between 200 nM (13) and 380 nM (14). Remarkably, the apparent Ki for the RF1-mediated release (32 ± 18 nM) is almost one order of magnitude lower than the Ki for the puromycin reaction. It is noteworthy that the sensitivity of the termination assay does not permit measurements of peptide release at concentrations of the pretermination complex below 30 nM. Thus, the actual Ki for BlaS inhibition of termination may be even lower than the apparent Ki (∼32 nM) determined from our experiments. Hence, our biochemical experiments demonstrate that BlaS is a more efficient inhibitor of translation termination than of peptidyl transfer (Fig. 6 C and D).
Fig. 6.

BlaS is a potent inhibitor of RF1-mediated peptide release. (A and B) Time progress curves for RF1-mediated release (A) and peptidyl transfer to puromycin (B), respectively, in the absence (blue) or presence (red) of 800 nM BlaS. Pretermination complex was formed by incubating ribosomes with [35S]-fMet-tRNAfMet and mRNA containing the UAA termination codon next to the AUG codon. Then, pretermination ribosomes were incubated with either RF1 (A) or puromycin (B) and amounts of [35S]-labeled release product were determined at several time points. Error bars represent SDs obtained from duplicate experiments. (C and D) Dependence of catalytic rates (kobs) for RF1-mediated release (C) and puromycin reaction (D) on BlaS concentration. Ki for the peptide release and puromycin reaction was determined by hyperbola fitting (red) of kobs values obtained at different concentrations of BlaS. Error bars represent residuals of single-exponential fitting of kobs values to time progress curves.
Conclusions
Our findings demonstrate that BlaS inhibits translation via a unique molecular mechanism that has not been previously documented for any antibiotic. The crystal structure of the BlaS-bound 70S ribosome complex reveals that unlike most peptidyl-transfer inhibitors, which bind in the vicinity of the A site of the 50S subunit, BlaS binds to the P site of the 50S subunit and bends the CCA end of the P-site tRNA toward the A site. The dramatic (>7 Å) shift of the ribose-phosphate backbone of C75 of the P-site tRNA underscores the conformational flexibility of tRNA. This observation expands our knowledge about the conformational space that tRNAs can sample during translation. More importantly, the distortion of the P-site tRNA explains the puzzling, previously reported ability of BlaS to interfere with binding of ribosomal ligands to the A site (8–11).
Our smFRET measurements show that BlaS slows down the intersubunit rotation in pretranslocation ribosomes that is coupled to spontaneous movement of tRNAs on the large subunit (Fig. 3A). This observation is consistent with early biochemical data demonstrating that BlaS increases tRNA affinity toward the 50S P site (8). Despite the inhibitory effect on tRNA fluctuations in a pretranslocation complex, EF-G-catalyzed translocation is affected only modestly (Fig. 3B). Our data therefore rule out the possibility that inhibition of translocation is a mode of BlaS action in vivo. Instead, our biochemical experiments suggest that BlaS may specifically target the termination step of protein synthesis.
We found that BlaS efficiently inhibits peptide release mediated by release factor RF1 during termination of protein synthesis and, to a lesser extent, peptidyl transfer during translation elongation. Inhibition of these steps of translation likely originates from a combination of two effects. First, the shift of the distorted CCA end of the P-site tRNA may occlude access of aminoacyl-tRNA and release factors into the A site on the 50S subunit (Figs. 4 and 5). The stronger inhibition of RF1-mediated peptide release by BlaS may result from a more pronounced steric interference of the distorted CCA end with the GGQ-containing region of release factors than with the A-site aminoacyl-tRNA (Fig. 5). Second, the terminal adenosine residue of the P-site tRNA is shifted from the position it occupies in the absence of the antibiotic (Fig. 4D). This rearrangement likely results in poor positioning of the peptidyl-tRNA substrate for nucleophilic attack by aminoacyl-tRNA during peptidyl transfer or by a water molecule during translation termination.
The strong inhibition of the translation termination step by BlaS is noteworthy because no specific inhibitors of translation termination have been identified so far (2, 45–47). Some inhibitors of peptide bond formation have been shown to also inhibit peptide release (2), but none has been quantitatively demonstrated to inhibit the latter step to a greater degree than peptidyl transfer (48). A large number of inherited and acquired diseases are attributed to the occurrence of a premature termination codon within the normal open reading frame, which precludes synthesis of full-length protein (49, 50). Because BlaS predominantly targets peptide release and also inhibits protein synthesis in eukaryotes (15, 17), our findings may potentially serve as a platform for the development of therapeutics that specifically suppress translation termination in human diseases associated with premature termination codons.
Materials and Methods
Materials and methods are described in detail in SI Materials and Methods. Ribosomes, aminoacylated tRNAs, EF-G, and RF1 were prepared as previously described (23, 34, 44). Single-molecule FRET measurements were taken using a prism-type total internal reflection (TIR) microscope (22). Structure determination was performed using molecular replacement and yielded a structure with R/Rfree of 0.232/0.268.
Supplementary Material
Acknowledgments
We thank Rohini Madireddy for assistance with purification of RF1 and 70S ribosomes; staff members of beam lines 23ID-B and 23ID-D (Advanced Photon Source at Argonne National Laboratory) and X25 (National Synchrotron Light Source at Brookhaven National Laboratory, US Department of Energy, under Contract No. DE-AC02-98CH10886) for assistance with X-ray data collection; Jillian Dann and Enea Salsi for assistance with purification of ΔS6 and ΔL9 ribosomes and smFRET experiments, respectively; Peter Cornish for sharing MatLab scripts and advice on total internal reflection microscopy; Alexei V. Korennykh for stimulating discussions; and Harry F. Noller for comments on the manuscript. The study was supported by US National Institute of Health Grant GM-099719 (to D.N.E.) and Grant P30 GM092424 (to the Center for RNA Biology at University of Rochester). A.A.K. was supported by the Worcester Foundation for Biomedical Research and University of Massachusetts Medical School Center for AIDS Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4L6J, 4L6K, 4L6L, and 4L6M).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304922110/-/DCSupplemental.
References
- 1.Wimberly BT. The use of ribosomal crystal structures in antibiotic drug design. Curr Opin Investig Drugs. 2009;10(8):750–765. [PubMed] [Google Scholar]
- 2.Wilson DN. On the specificity of antibiotics targeting the large ribosomal subunit. Ann N Y Acad Sci. 2011;1241:1–16. doi: 10.1111/j.1749-6632.2011.06192.x. [DOI] [PubMed] [Google Scholar]
- 3.Yonath A. Antibiotics targeting ribosomes: Resistance, selectivity, synergism and cellular regulation. Annu Rev Biochem. 2005;74:649–679. doi: 10.1146/annurev.biochem.74.082803.133130. [DOI] [PubMed] [Google Scholar]
- 4.Blaha GM, Polikanov YS, Steitz TA. Elements of ribosomal drug resistance and specificity. Curr Opin Struct Biol. 2012;22(6):750–758. doi: 10.1016/j.sbi.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol. 2003;330(5):1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi H, Tanaka N. Inhibition of protein synthesis by blasticidin S. II. Studies on the site of action in E. coli polypeptide synthesizing systems. J Biochem. 1966;60(6):632–642. doi: 10.1093/oxfordjournals.jbchem.a128489. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi H, Yamamoto C, Tanaka N. Inhibition of protein synthesis by blasticidin S. I. Studies with cell-free systems from bacterial and mammalian cells. J Biochem. 1965;57(5):667–677. [PubMed] [Google Scholar]
- 8.Cerná J, Rychlík I, Lichtenthaler FW. The effect of the aminoacyl-4-aminohexosyl-cytosine group of antibiotics on ribosomal peptidyl transferase. FEBS Lett. 1973;30(2):147–150. doi: 10.1016/0014-5793(73)80639-8. [DOI] [PubMed] [Google Scholar]
- 9.Pestka S. The use of inhibitors in studies on protein synthesis. Methods Enzymol. 1974;30:261–282. doi: 10.1016/0076-6879(74)30030-4. [DOI] [PubMed] [Google Scholar]
- 10.Lazaro E, van den Broek LA, San Felix A, Ottenheijm HC, Ballesta JP. Biochemical and kinetic characteristics of the interaction of the antitumor antibiotic sparsomycin with prokaryotic and eukaryotic ribosomes. Biochemistry. 1991;30(40):9642–9648. doi: 10.1021/bi00104a011. [DOI] [PubMed] [Google Scholar]
- 11.Kalpaxis DL, Theocharis DA, Coutsogeorgopoulos C. Kinetic studies on ribosomal peptidyltransferase. The behaviour of the inhibitor blasticidin S. Eur J Biochem. 1986;154(2):267–271. doi: 10.1111/j.1432-1033.1986.tb09392.x. [DOI] [PubMed] [Google Scholar]
- 12.Kuwano M, Takenaka K, Ono M. The cross-resistance of mouse blasticidin S-resistant cell lines to puromycin and sparsomycin, inhibitors of ribosome function. Biochim Biophys Acta. 1979;563(2):479–489. doi: 10.1016/0005-2787(79)90066-2. [DOI] [PubMed] [Google Scholar]
- 13.Theocharis DA, Synetos D, Kalpaxis DL, Drainas D, Coutsogeorgopoulos C. Kinetics of inhibition of peptide bond formation on bacterial ribosomes. Arch Biochem Biophys. 1992;292(1):266–272. doi: 10.1016/0003-9861(92)90078-b. [DOI] [PubMed] [Google Scholar]
- 14.Petropoulos AD, Xaplanteri MA, Dinos GP, Wilson DN, Kalpaxis DL. Polyamines affect diversely the antibiotic potency: insight gained from kinetic studies of the blasticidin S AND spiramycin interactions with functional ribosomes. J Biol Chem. 2004;279(25):26518–26525. doi: 10.1074/jbc.M313634200. [DOI] [PubMed] [Google Scholar]
- 15.Fukunaga K, Misato T, Ishii I, Asakawa M. Blasticidin, a new anti-phytopathogenic fungal substance. Part I. Bull Agric Chem Soc Jpn. 1955;19:181–188. [Google Scholar]
- 16.Izumi M, et al. Blasticidin S-resistance gene (bsr): A novel selectable marker for mammalian cells. Exp Cell Res. 1991;197(2):229–233. doi: 10.1016/0014-4827(91)90427-v. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi S, Hirayama K, Ueda K, Sakai H, Yonehara H. Blasticidin S, a new antibiotic. J Antibiot (Tokyo) 1958;11(1):1–5. [PubMed] [Google Scholar]
- 18.Blanchard SC, Kim HD, Gonzalez RL, Jr, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc Natl Acad Sci USA. 2004;101(35):12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fei J, Kosuri P, MacDougall DD, Gonzalez RL., Jr Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol Cell. 2008;30(3):348–359. doi: 10.1016/j.molcel.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Munro JB, Altman RB, O’Connor N, Blanchard SC. Identification of two distinct hybrid state intermediates on the ribosome. Mol Cell. 2007;25(4):505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agirrezabala X, et al. Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol Cell. 2008;32(2):190–197. doi: 10.1016/j.molcel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornish PV, Ermolenko DN, Noller HF, Ha T. Spontaneous intersubunit rotation in single ribosomes. Mol Cell. 2008;30(5):578–588. doi: 10.1016/j.molcel.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ermolenko DN, et al. Observation of intersubunit movement of the ribosome in solution using FRET. J Mol Biol. 2007;370(3):530–540. doi: 10.1016/j.jmb.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 24.Julián P, et al. Structure of ratcheted ribosomes with tRNAs in hybrid states. Proc Natl Acad Sci USA. 2008;105(44):16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ermolenko DN, Cornish PV, Ha T, Noller HF. Antibiotics that bind to the A site of the large ribosomal subunit can induce mRNA translocation. RNA. 2013;19(2):158–166. doi: 10.1261/rna.035964.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ermolenko DN, Noller HF. mRNA translocation occurs during the second step of ribosomal intersubunit rotation. Nat Struct Mol Biol. 2011;18(4):457–462. doi: 10.1038/nsmb.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spiegel PC, Ermolenko DN, Noller HF. Elongation factor G stabilizes the hybrid-state conformation of the 70S ribosome. RNA. 2007;13(9):1473–1482. doi: 10.1261/rna.601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Studer SM, Feinberg JS, Joseph S. Rapid kinetic analysis of EF-G-dependent mRNA translocation in the ribosome. J Mol Biol. 2003;327(2):369–381. doi: 10.1016/s0022-2836(03)00146-3. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Altman RB, Blanchard SC. Insights into the molecular determinants of EF-G catalyzed translocation. RNA. 2011;17(12):2189–2200. doi: 10.1261/rna.029033.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorner S, Brunelle JL, Sharma D, Green R. The hybrid state of tRNA binding is an authentic translation elongation intermediate. Nat Struct Mol Biol. 2006;13(3):234–241. doi: 10.1038/nsmb1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313(5795):1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 32.Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol. 2009;16(5):528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weixlbaumer A, et al. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322(5903):953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korostelev A, et al. Crystal structure of a translation termination complex formed with release factor RF2. Proc Natl Acad Sci USA. 2008;105(50):19684–19689. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laurberg M, et al. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454(7206):852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Fonseca C, Amils R, Garrett RA. Fine structure of the peptidyl transferase centre on 23 S-like rRNAs deduced from chemical probing of antibiotic-ribosome complexes. J Mol Biol. 1995;247(2):224–235. doi: 10.1006/jmbi.1994.0135. [DOI] [PubMed] [Google Scholar]
- 37.Lichtenthaler FW, Trummlitz G. Structural basis for inhibition of protein synthesis by the aminoacyl-aminohexosyl-cytosine group of antibiotics. FEBS Lett. 1974;38(3):327–342. doi: 10.1016/0014-5793(74)80062-1. [DOI] [PubMed] [Google Scholar]
- 38.Porse BT, Rodriguez-Fonseca C, Leviev I, Garrett RA. Antibiotic inhibition of the movement of tRNA substrates through a peptidyl transferase cavity. Biochem Cell Biol. 1995;73(11-12):877–885. doi: 10.1139/o95-095. [DOI] [PubMed] [Google Scholar]
- 39.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126(6):1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Korostelev A, Lancaster L, Noller HF. Crystal structures of 70S ribosomes bound to release factors RF1, RF2 and RF3. Curr Opin Struct Biol. 2012;22(6):733–742. doi: 10.1016/j.sbi.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korostelev AA. Structural aspects of translation termination on the ribosome. RNA. 2011;17(8):1409–1421. doi: 10.1261/rna.2733411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loh PG, Song H. Structural and mechanistic insights into translation termination. Curr Opin Struct Biol. 2010;20(1):98–103. doi: 10.1016/j.sbi.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 43.Kisselev L, Ehrenberg M, Frolova L. Termination of translation: Interplay of mRNA, rRNAs and release factors? EMBO J. 2003;22(2):175–182. doi: 10.1093/emboj/cdg017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korostelev A, Zhu J, Asahara H, Noller HF. Recognition of the amber UAG stop codon by release factor RF1. EMBO J. 2010;29(15):2577–2585. doi: 10.1038/emboj.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nierhaus KH, Wilson DN (2004) Protein Synthesis and Ribosome Structure (Wiley-VCH, Weinheim)
- 46.Polacek N, et al. The critical role of the universally conserved A2602 of 23S ribosomal RNA in the release of the nascent peptide during translation termination. Mol Cell. 2003;11(1):103–112. doi: 10.1016/s1097-2765(02)00825-0. [DOI] [PubMed] [Google Scholar]
- 47.Caskey CT, Beaudet AL, Scolnick EM, Rosman M. Hydrolysis of fMet-tRNA by peptidyl transferase. Proc Natl Acad Sci USA. 1971;68(12):3163–3167. doi: 10.1073/pnas.68.12.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12(7):306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 49.Keeling KM, Bedwell DM. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip Rev RNA. 2011;2(6):837–852. doi: 10.1002/wrna.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med. 2013;64:407–425. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.