

Abstract
Guided by features of molecular, cellular, and circuit dysfunction affecting the prefrontal cortex in clinical investigations, we targeted prefrontal cortex in studies of a model for neuropsychiatric illness using transgenic mice expressing a putative dominant-negative disrupted in schizophrenia 1 (DN-DISC1). We detected marked augmentation of GAPDH–seven in absentia homolog Siah protein binding in the DISC1 mice, a major hallmark of a nuclear GAPDH cascade that is activated in response to oxidative stress. Furthermore, deficits were observed in well-defined tests for the cognitive control of adaptive behavior using reversal learning and reinforcer devaluation paradigms. These deficits occurred even though DN-DISC1 mice showed intact performance in simple associative learning and normal responses in consumption of reward. In an additional series of assessments, motivational functions also were impoverished in DN-DISC1 mice, including tests of the dynamic modulation of reward value by effortful action, progressive ratio performance, and social behavior. Augmentation of an oxidative stress-associated cascade (e.g., a nuclear GAPDH cascade) points to an underlying condition that may contribute to the profile of cognitive and motivational impairments in DN-DISC1 mice by affecting the functional integrity of the prefrontal cortex and dysfunction within its connected networks. As such, this model should be useful for further preclinical research and drug discovery efforts relevant to the burden of prefrontal dysfunction in neuropsychiatric illness.
Keywords: cognition, depression, orbitofrontal cortex
It has long been noted that the cognitive impairments in patients with major mental illness, such as schizophrenia and mood disorders, point to dysfunction of the frontal lobe (1–3). More recent translational studies based on cognitive and affective neuroscience approaches suggest the associated deficits in neuropsychiatric populations reflect abnormal functioning of prefrontal–subcortical circuitry responsible for cognitive and emotional control (4–7). By examining disorders of brain circuitry, these studies offer the potential to advance therapeutic treatment (7, 8).
The impaired integrity affecting prefrontal circuitry in schizophrenia and mood disorders in part reflects neuropathology associated with decreased parvalbumin immunoreactivity (9–11), a marker for fast-spiking inhibitory interneurons, which are critical for cognitive function (9, 10). One possible cause of impaired integrity affecting that interneuron population comes from evidence that decreased parvalbumin immunoreactivity is elicited by oxidative stress in mouse models relevant to neuropsychiatric illness (12–15). Oxidative stress can be driven by a deficiency in antioxidant defenses, which leads to increased oxidative signaling resulting in damage to DNA, proteins, and fatty acids (16). Central nervous system cells are particularly vulnerable to these effects because of reduced antioxidant levels and elevated metabolic rate (13).
Genetic studies also are contributing to our understanding of the molecular basis of schizophrenia and mood disorders, giving clues about the inherited risk for these conditions. Although no gene thus far has been found to cause major mental illness per se, and disrupted in schizophrenia 1 (DISC1) is not a direct susceptibility gene for disease, its variations are likely to play a key role in neurodevelopmental processes important for the formation of cortical neural networks and their function (17, 18). In at least one pedigree in Scotland, the disruption of this gene leads to major mental illnesses, including major depressive disorder and schizophrenia, with very high penetrance (19). Consistent with that evidence, many roles for DISC1 in neurodevelopment and synapse formation have been reported (17). Furthermore, mouse models that genetically modulate DISC1 by various methods demonstrate a decrease in parvalbumin immunoreactivity, consistent with the observations in human brains of patients as well as behavioral phenotypes relevant to psychiatric disease (20–23).
In the current series of experiments, we studied the phenotype of a genetically engineered model for DISC1. We previously reported on a transgenic model expressing a putative dominant-negative DISC1 (DN-DISC1) under control of the alpha calcium/calmodulin-dependent kinase II (αCaMKII) promoter in a heterozygous status, which showed a significant decrease in parvalbumin immunoreactivity in the frontal cortex, but in that model behavioral changes were subtle in the assessments used (21). We generated the present homozygous model by systematic cross-breeding to augment the pathology associated with abnormal DISC1 (24).
Here we aim to illuminate the status of functions at the interface of cognition and affect in guiding actions in homozygous DN-DISC1 mice in parallel with molecular changes in the prefrontal cortex, providing potential targets for experimental testing of interventions in future research. We found deficits in this model in tests of flexible behavioral control (reversal learning and reinforcer devaluation) and motivational status (reward hedonics regulated by differing levels of effort and tests of responding under a progressive ratio). In addition to strong evidence for behavioral impairment in prefrontal-dependent tasks, we also observed clear evidence of activation in the nuclear GAPDH cascade in the frontal cortex, but not in the striatum, of DN-DISC1 mice. The nuclear GAPDH cascade is an important downstream indicator of oxidative stress, and we have developed specific inhibitors against this cascade.
Results
A hallmark of prefrontal dysfunction in primates and rodents is an inability to adjust behavior based on changing contingencies in the environment. Homologous prefrontal circuitry involving the orbitofrontal cortex (OFC) plays a critical role in simple reversal learning across species (25–28). Here we examined the performance of DN-DISC1 mice in a reversal learning task in which mice first acquired a simple discrimination and then adapted to changes in the reward contingencies.
DN-DISC1 mice showed no difficulty learning a simple nose-poke discrimination (e.g., left nose-poke → sucrose; right nose-poke → no reward; SI Materials and Methods). Control and DN-DISC1 mice took a similar number of sessions to reach criterion (mean ± SEM: control, 12.75 ± 0.83; DN-DISC1, 12.56 ± 0.45; P = 0.85) and performed at a similar level of accuracy immediately before reversal training (correct responses ± SEM: control, 99 ± 0.14%%; DN-DISC1, 99 ± 0.26; P = 0.52). In the reversal phase, the contingencies in the discrimination task were switched so that the previously incorrect nose-poke became the rewarded response.
Control mice adapted to the new contingencies during reversal training more readily than DN-DISC1 mice. The groups differed in the initial reversal session (Fig. 1 Left), as indicated by the Group × Block interaction [F(4,96) = 3.13, P = 0.01], with the control mice performing better in the final block [F(1,24) = 8.67, P < 0.01]. The nature of the deficit in DN-DISC1 mice reflected response perseveration, because DN-DISC1 mice continued to respond with the incorrect (formerly rewarded) nose-poke (incorrect responses, 37.84 ± 13.04; correct responses, 3.68 ± 2.03). Although the overall number of responses did not differ between groups, a significant interaction reflected persistence of incorrect responding [F(1,23) = 4.76, P < 0.05]. By the second training session performance was equivalent in both groups (no effect of group or of interaction among variables; F < 1; P > 0.65), suggesting a transient impairment in reversal learning (Fig. 1 Right). The perseverative deficits seen in the early stages of reversal training in DN-DISC1 mice are consistent with damage to OFC in rodents (27).
Fig. 1.

Deficits in behavioral flexibility following reversal of reward contingencies. DN-DISC1 (n = 14) mice were impaired in their ability to alter their responses in a flexible manner following a shift in the response contingencies (Reversal training 1). The performance of DN-DISC1 mice improved to the same level as controls (n = 12) with further training (Reversal training 2). *P = 0.01, Group × Block interaction. Error bars indicate SEM.
Outcome Expectancy and Goal-Directed Behavior.
A deficit in outcome expectancy, i.e., in the use of information associated with predictive cues and actions, could contribute to reversal learning impairment. Such encoding is a feature of neurons in the OFC that are critical for guiding goal-directed behavior. We behaviorally assessed this function in DN-DISC1 mice using procedures that have been used in research with rodents and primates (29, 30). The reinforcer devaluation paradigm assesses the ability to guide responses based on changes in the value of an anticipated outcome. After training in which responses for obtaining two different rewarding foods are established, the value of one food is reduced by prefeeding to satiety. Normally this prefeeding spontaneously reduces actions directed toward obtaining the devalued (satiated) reward.
Both DN-DISC1 mice and control mice readily acquired lever-responding behavior during the training stage (Fig. 2A) when responses on two separate levers resulted in the delivery of different food rewards. After consuming one of the food rewards to satiation, the control mice showed the expected effect on their performance, responding less on the lever for obtaining the devalued food (Fig. 2B). It should be noted that responses on each lever during the test stage were not rewarded with food to ensure that any decrease in lever-responding would be mediated only by expectancy of the outcome, not the reinforcer itself. Unlike the control mice, DN-DISC1 mice continued to respond equally on both levers after satiation (Fig. 2C). Two-way ANOVA (Response × Group) conducted on the overall data (Fig. 2D) revealed a significant interaction [F(1,24) = 6.21, P = 0.01] caused by a reinforcer devaluation effect in control mice [F(1,24) = 8.11, P < 0.01] but not in DN-DISC1 mice (F < 1) with devalued [F(1,24) = 6.02, P = 0.02] but not maintained [F(1,24) = 1.31, P = 0.26] responses differing between the groups.
Fig. 2.

Impaired responses in a reinforcer devaluation task in DN-DISC1 mice. (A) DN-DISC1 mice showed normal acquisition of lever-responding behavior for the delivery of food rewards (e.g., lever 1 → food 1; lever 2 → food 2; data averaged across the two levers). After prefeeding with one of the two foods to satiety, mice were placed into the chambers with access to the two levers. (B) Control mice (n = 16) decreased responses on the lever associated with the devalued outcome. (C) In contrast, DN-DISC1 (n = 10) mice did not adjust their lever-responding behavior but instead had similar rates of response on both levers. (D) Overall lever response rates at test differed as a function of group (*P = 0.01, Response × Group interaction). Error bars indicate SEM.
The failure of DN-DISC1 mice to suppress performance on the devalued lever occurred even though these mice reduced consumption of the devalued food itself in a manner similar to the control mice in choice tests given after satiation (consumption in milliliters ± SEM: control/devalued, 1.59 ± 0.14; control/maintained, 2.12 ± 0.13; DN-DISC1/devalued, 1.3 ± 0.20; DN-DISC1/maintained, 1.7 ± 0.17); there was no effect of group or interaction among the variables (P > 0.05). Thus, although prefeeding was equally effective in promoting satiety in all mice, DN-DISC1 mice were unable to use an expectation of reduced reward value to guide responses during the lever test. This failure is consistent with disruption of prefrontal encoding of reward expectancy (31).
Modulation of the Hedonics of Reward Value by Effort.
Recently we have reported that effort can have relatively durable effects on the acquired value of cues that are associated with effortful action and on the hedonics of reward obtained with greater effort (32). We assessed the effects of effort on such processes in DN-DISC1 mice.
DN-DISC1 mice acquired lever-responding behavior in a task that required different amounts of effort to obtain reward, with 15× more responses required on a high-effort lever than on a low-effort lever (SI Materials and Methods). Performance of this task using operant training procedures did not differ significantly for the groups (high-effort lever, total responses ± SEM: control, 343 ± 13.51; DN-DISC1, 284 ± 36.33; low-effort lever: control, 25.05 ± 0.03; DN-DISC1, 24.05 ± 0.62; P > 0.05). Although training performance was roughly comparable, DN-DISC1 mice failed to show an acquired acceptance for rewards associated with high effort in later consumption tests (Fig. 3A). A significant Group × Testing Condition interaction [F(1,33) = 5.26, P < 0.05] was revealed by the acceptance exhibited by control mice tested for the high-effort reward compared with all other groups [smallest F-value: control high-effort reward vs. control low-effort reward, F(1,33) = 8.61, P < 0.01]. At test there were no interactions with these effects and the type of reinforcers (polycose or sucrose) used (Table S1).
Fig. 3.

Modulation of hedonic value following high-effort training. (A) Only control mice showed an increase in the consumption of the food reward associated with high effort (n = 12) relative to low effort (n = 10). Control mice also showed licking behaviors consistent in part with a greater hedonic value of that food. (B) Burst size. (C) Bursts initiated. DN-DISC1 mice showed comparable intake and measures of licking microstructure across high-effort (n = 7) and low-effort (n = 8) conditions. Error bars indicate SEM.
A refined analysis of licking microstructure previously showed an increase in the hedonic properties of the food that had been obtained with greater effort in normal mice (32). Here control mice tested with the high-effort food reward showed a similar profile, but DN-DISC1 mice did not (Fig. 3 B and C). Control mice tested with the high-effort food reward exhibited a larger burst size relative to all other groups [significant Group × Testing Condition ANOVA, F(1,33) = 4.48, P < 0.05] (Fig. 3B). In addition, DN-DISC1 mice failed to show greater numbers of licking bursts for the high-effort reward as exhibited by control mice [Group × Testing Condition interaction, F(1,33) = 7.54, P < 0.01] (Fig. 3C). As with the previous analyses, this result reflected differences between control mice tested with the high-effort food relative to all other groups [smallest F-value, control high-effort reward vs. control low-effort reward, F(1,33) = 11.71, P < 0.01].
Progressive Ratio Performance.
Actions not only are guided by information about outcomes but also reflect motivational and volitional processes in behavioral control. The willingness to engage and maintain goal-directed performance under conditions of increasing effort has been studied classically using progressive ratio procedures (33–35). Here motivation is assessed by systematically escalating the number of responses required to obtain the reward. The progressive ratio schedule refers to the number of responses required before reward delivery, which is increased over the course of a test session.
Before testing on a progressive ratio schedule, both groups of mice were shown to respond similarly in consumption tests for the sucrose food reward under free-access conditions (consumption in milliliters ± SEM: control, 1.49 ± 0.13; DN-, 1.50 ± 0.14; P = 0.89). In contrast, when mice were required to make an increasing number of licks to an empty well to obtain sucrose, consumption in the DN-DISC1 mice was markedly reduced compared with the control mice (Fig. 4A; P < 0.01). Here the ratio of responses was increased from 20 per reward at the beginning of the session to as many as 300 responses per reward at the end. The DN-DISC1 mice were less willing to respond for the food reward (licks at the sucrose delivery well; Fig. 4B) and ceased responding earlier in the session as the requirement for responses increased on the progressive ratio schedule (Fig. 4C) (analyses of the survival functions for mice detected a significant group effect; χ2 = 8.41, P < 0.01). Thus, despite normal sucrose intake during free-access conditions, DN-DISC1 mice displayed a marked impairment in their willingness to respond for sucrose as the effort required for its delivery increased within a test session.
Fig. 4.

Reduced operant motivation in DN-DISC1 mice. (A and B) DN-DISC1 mice (n = 8) showed a reduction in sucrose intake under progressive ratio test conditions (A) and displayed a corresponding attenuation in the number of licks they were willing to perform during this test relative to controls (n = 7) (B). (C) Dramatic differences in the rate of attrition were revealed between the groups. Main effect of group, *P < 0.05. Error bars indicate SEM.
Three-Chamber Social Interaction Test.
Next we used a three-chamber apparatus to assess sociability and preference for social novelty, which may be relevant to negative symptoms of schizophrenia and possibly to some features of depression (36). In this task a stranger mouse is introduced into the enclosure of one side of the chamber (37). Control mice preferably sniffed the enclosure with the stranger mouse (see SI Materials and Methods for sniffing counts), but DN-DISC1 mice showed no preference for either side (Fig. 5A). Group × Chamber ANOVA detected a significant interaction [F(1,20) =\11.26, P ≤ 0.001] resulting from a preference for the enclosure containing the stranger mouse in control [F(1,20) = 11.00, P < 0.01] but not in DN-DISC1 mice [F(1,20) = 1.58, P = 0.22]. In the social novelty phase a different stranger was inserted into the previously empty enclosure. The control mice preferred the unfamiliar stranger, but the DN-DISC1 mice preferred the by-then familiar mouse (Fig. 5B). Group × Chamber ANOVA detected a significant interaction [F(1,20) = 12.56, P < 0.001] because of a preference for the novel chamber in control mice [F(1,20) = 8.07, P = 0.01] and a preference for the familiar chamber in DN-DISC1 mice [F(1,20) = 4.52, P = 0.05]. Finally, to confirm that the deficit in DN-DISC1 mice did not reflect impairment in the detection of olfactory social cues, we adapted a procedure used to assess odor detection in rodents (38). A separate group of control [F(1,33) = 27.96, P < 0.01] and DN-DISC1 [F(1,33) = 18.30, P < 0.001] mice showed a preference in exploring a cube with familiar cage bedding rather than an empty odor cube, confirming intact olfactory detection in all mice (Fig. 5C). Collectively, these data add to the profile of impoverished motivational functions in DN-DISC1 mice.
Fig. 5.
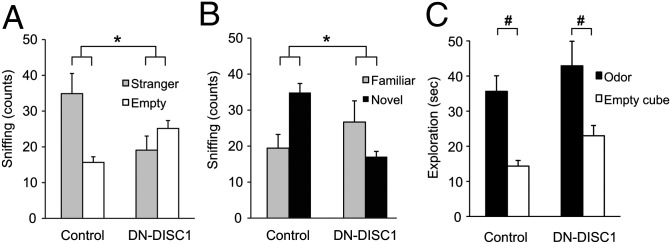
Three-chamber social interaction test. (A) Sociability: Although wild-type control mice (n = 9) preferred the enclosure with the stranger mouse, DN-DISC1 mice (n = 13) showed no preference. (B) Social novelty: Although control mice preferred the stranger, DN-DISC1 mice preferred the familiar mouse. (C) Social odor detection: There were no differences in initial exploratory activity to the open field as reflected by distance traveled (in centimeters ± SEM) in DN-DISC1 mice (853 ± 67.7, n = 15) and control mice (995 ± 68.4, n = 20). Importantly, both groups of mice showed a preference for the cube containing familiar cage bedding relative to an empty odor cube. *P < 0.05, Group × Chamber interaction.). #P < 0.01, odor detection effect. Error bars indicate SEM.
Together, these experimental paradigms have provided evidence that behavioral changes in homozygous DN-DISC1 are likely to be elicited by the DN-DISC1 transgene. However, homozygous DN-DISC1 mice in all the above experiments were maintained by inbreeding and as such do not have littermate controls with identical background, raising the risk that random mutations accumulating in the inbred homozygous DN-DISC1 line might contribute to the observed abnormalities. To address this issue, we included heterozygous DN-DISC1 mice (littermates of the wild-type mice) in the social interaction test (Fig. S1). Heterozygous mice showed an intermediate phenotype both in the sociability and in the social novelty phases of the test and also displayed intact olfactory detection, suggesting that the behavioral abnormalities are indeed caused by the DN-DISC1 transgene. Furthermore, the control nonlittermate wild-type mice and C57BL/6N mice show similar behavior patterns, indicating no major behavioral differences among different strains of mice in the control group (Table S2).
Oxidative Stress-Associated Pathology in the Prefrontal Cortex.
The previous behavioral findings suggest dysfunction of prefrontal circuitry and are consistent with decreased immunoreactivity of parvalbumin in interneurons of the medial prefrontal cortex (mPFC) in DN-DISC1 mice (21). To extend our assessment of such pathology, we used methods to examine the presence of oxidative stress affecting prefrontal networks in DN-DISC1 mice (SI Materials and Methods). We first examined a gross measurement using a protein carbonyl assay in which irreversible oxidative modification of proteins was quantified. Here we observed a trend in the increase in oxidative modification in DN-DISC1 mice compared with wild-type controls in a global analysis of prefrontal tissue (P = 0.07) (Fig. 6A). In an anatomically defined analysis of 8-oxo-dG staining (a marker for oxidative damage to the DNA), we observed a significant increase for this marker in the OFC but not in the mPFC in DN-DISC1 mice as compared with controls (Fig. 6 E and F). Those results suggest subregional variation in effects of oxidative stress within the prefrontal cortex of the DN-DISC1 model as defined by the 8-oxo-dG marker.
Fig. 6.

Oxidative stress-associated pathology in the prefrontal cortex. (A) A trend of increased oxidative stress in the prefrontal cortex of adult DN-DISC1 mice (n = 13) compared with C57BL/6N control mice (n = 11) as shown by the carbonyl assay. #P = 0.065. (B) Activation of the GAPDH–Siah cascade, assayed by GAPDH–Siah protein interaction by coimmunoprecipitation, reflects oxidative stress. Augmented activation of this cascade was observed in DN-DISC1 mice (n = 7) compared with wild-type controls (n = 8); *P < 0.005. (C and D) Increased oxidative stress was not detected in the striatum by either the carbonyl assay (C) or GAPDH–Siah interaction (D). (E and F) 8-Oxo-dG staining to assess oxidative damage to the DNA in subregions of prefrontal cortex in mice. (E) High-power photomicrograph showing 8-oxo-dG–positive (red) cells and the total number of cells (DAPI) in the OFC. (F) A significant (*P < 0.001) increase in the percentage of 8-oxo-dG–positive cells was revealed in the OFC but not in the mPFC in DN-DISC1 mice (n = 4 mice, 35–36 counts), compared with wild-type controls (n = 4 mice, 28–36 counts). Error bars indicate SEM.
To address possible downstream signaling elicited by oxidative stress, we focused on GAPDH. Under exposure to excess oxidative stress, GAPDH is posttranslationally modified at a specific cysteine residue, C150, which enables GAPDH to bind with Siah, which has a strong nuclear localization signal. The GAPDH–Siah complex then translocates to the nucleus where it can affect epigenetic and transcriptional machinery (39, 40). Thus, the levels of GAPDH–Siah protein interaction can be used to monitor the activation of the nuclear GAPDH signaling cascade elicited by oxidative stress. We observed an almost 10-fold up-regulation of GAPDH–Siah binding in DN-DISC1 mice compared with controls (P < 0.01) (Fig. 6B). Notably, this remarkable oxidative modification and GAPDH–Siah binding localized to the prefrontal cortex and was not observed in the striatum (Fig. 6 C and D).
Discussion
In this study we addressed prefrontal cortex-associated behavioral changes in the DN-DISC1 mice. We also provided an example of promising molecular changes with translational potential in the model. In the presence of pronounced signatures for oxidative stress in the prefrontal cortex, DN-DISC1 mice exhibited a behavioral profile consistent with prefrontal dysfunction. DN-DISC1 mice had selective deficits in the flexible use of information to guide goal-directed behavior, as reported here for reversal learning and a test of outcome expectancy using reinforcer devaluation procedures. We also assessed the DN-DISC1 mice for motivational and affective functions to model endophenotypes that may underlie some symptoms of major psychiatric illness, including schizophrenia and depression. We found pronounced deficits in such assessments, including tests for the role of effort in incentive learning, progressive ratio performance, and social interaction. Together these results reveal a phenotypic profile encompassing both cognitive and motivational deficits that are dependent on prefrontal cortex or are subserved by prefrontal connections with limbic and striatal circuitry. The localization of pronounced oxidative stress in prefrontal cortex, in particular the OFC, points to prefrontal dysfunction, underscoring a potential role of oxidative stress in the pathophysiology associated with neuropsychiatric illnesses (41, 42). At the same time, it should be noted that the absence of increased oxidative stress in the striatum does not exclude a striatal contribution to deficits in DN-DISC1 mice.
It is important to note the behavioral functions that were unaffected in the DN-DISC1 mice in considering the basis for their deficits. The DN-DISC1 mice exhibited unimpaired acquisition of learning in operant tasks, including operant licking and lever-pressing behavior. DN-DISC1 mice also exhibited normal consumption when given free access to reward and reduced reward consumption normally under satiated conditions. Thus, no generalized deficits in sensorimotor performance and in simple learning capacity account for the DN-DISC1 behavioral profile. Rather deficits were apparent in settings that depend on anticipation and decision making in the context of reward valuation. In addition, a dynamic modulation of hedonics by effortful action was impaired in these mice, possibly accounting, at least in part, for the impaired progressive ratio performance in DN-DISC1 mice.
The behavioral studies of DN-DISC1 mice used assessments that have become well characterized in cognitive and affective neuroscience research. Such research has revealed the encoding properties of neurons in prefrontal circuits that support adaptive and flexible behavioral control. For example, neurons in OFC that acquire associative responding to cues that predict reward rapidly shift their encoding when reward contingencies are reversed or the predicted value of an outcome is changed (43). Consistent with these encoding properties, interference with the normal function of OFC leads to impaired adaptive control, with deficits in reversal learning and devaluation protocols similar to those reported here for DN-DISC1 mice. Importantly, the properties of such prefrontal networks in studies of both rodents and primates reveal consistent core neurobehavioral functions across species, and neuroimaging studies suggest a similar role for this circuitry in the human brain (27, 44). Therefore the condition of prefrontal dysfunction in the DN-DISC1 model should be useful for translational purposes to discover the mechanisms leading to impairment and to test therapeutics in disease models.
Significant dysfunction in the prefrontal cortex could be expected to affect the range of cognitive and motivational processes found in the DN-DISC1 model. The functions of prefrontal cortex are integrated across prefrontal systems and in broader networks connecting cortical and subcortical systems (45). Critical limbic interconnections, for example, are required for the associative encoding of outcome expectancy by OFC neurons. Connectivity between the frontal cortex and striatum, via a series of parallel cortico–striatal and pallidal–thalamo–cortical loops (46, 47), is important in other behavioral assessments we studied in DN-DISC1 mice, integrating motivational functions in decision making and the behavioral control of action. Within this neurobehavioral framework, significant oxidative stress in the prefrontal cortex reported here points to a mediating condition that could contribute to both cognitive and motivational impairments in the DN-DISC1 model by causing pathology in the prefrontal cortex and dysfunction of its connected networks. Thus, the possibility that a broad range of untreated symptoms could benefit from a common therapy could be tested experimentally in this model by targeting oxidative stress.
As summarized in recent review articles, DISC1 is not a risk factor for any single disease but contributes to the susceptibility to a wide range of mental illnesses, including schizophrenia and major depression (17, 48). Thus, the behavioral changes observed in this study have implications for a wider range of mental illnesses rather than for specific diagnoses defined in the Diagnostic and Statistical Manual of Mental Disorders or in International Classification of Diseases criteria. Our approach with the present mouse model is analogous to that described in the National Institute of Mental Health Research Domain Criteria, which focuses on behavioral domains (e.g., cognition impairment) associated with dysfunction in specific neural circuitry (e.g., prefrontal cortex) rather than on a specific disease diagnosis (8). Given that DISC1 has multiple functions, including a role in mitochondrial function, regulation of cAMP signaling, and NMDA receptor dynamics (17, 48), it is crucial to consider that mutant DN-DISC1 may contribute to the behavioral abnormalities in this DISC1 model via multiple pathways.
A crucial role for oxidative stress in the neuropathology of major mental illness is supported by studies showing that interneuron deficits most affected in brains of patients with schizophrenia and mood disorders can be reproduced by exposure to oxidative stress in animal models. Moreover, oxidative stress can affect cellular processes (e.g., mitochondrial signaling and neuronal excitability), which could adversely affect neuronal phenotypes in addition to parvalbumin interneuronal deficits, contributing to the pathology of schizophrenia and mood disorders (49). Here we demonstrate that behavioral deficits reflecting prefrontal dysfunction occur in the setting of signs of oxidative stress in a mouse model that also exhibits loss of prefrontal parvalbumin interneuron integrity (21). In this context, the augmented GAPDH–Siah binding, as an index of marked activation of the nuclear GAPDH cascade, in the cortex of DN-DISC1 mice could be a specific target for testing the contribution of oxidative stress to prefrontal pathology and to functional impairment. Preclinical assessment of such a treatment could be used experimentally to test whether augmented oxidative stress causes impaired behavioral performance in the DN-DISC1 model. In addition to behavioral assessments that are relevant to untreated symptoms of mental illnesses, such preclinical studies also could assess the potential link between oxidative stress and the specific neuropathology affecting prefrontal cortex, such as loss of interneuron integrity. Regardless of the efficacy of this posited experimental treatment, the current findings suggest that DN-DISC1 mice offer an integrative model that captures key features of prefrontal dysfunction contributing to psychiatric endophenotypes that currently are untreated by existing therapies.
Materials and Methods
Detailed methods are described in SI Materials and Methods.
Mice.
Behavioral testing was conducted with male age-matched homozygous DN-DISC1 mice and control mice including both nonlittermate wild types and control C57BL/6N mice.
Nose-Poke Discrimination and Reversal Training.
After food cup shaping with sucrose, mice received daily nose-poke training sessions, during which correct nose-poke responses led to delivery of sucrose, and incorrect nose-poke responses were nonrewarded. Mice were trained to meet a pretest criterion (>85% correct responses for two consecutive sessions and zero trial omissions). During the reversal training stage, mice received two reversal sessions separated by a 24-h interval. Each response to the formerly incorrect nonrewarded nose-poke was rewarded (i.e., now was correct), whereas responses to the formerly correct nose-poke were nonrewarded (i.e., now was incorrect).
Reinforcer Devaluation.
Food cup shaping occurred separately with both sucrose and polycose (Abbott Laboratories) solutions followed by lever training (28), in which responses to a particular lever led to delivery of a particular reward. Mice received a total of 10 sessions of lever training. On each test day mice received satiety treatment by prefeeding with one of the two outcomes for a 2-h period followed by placement in the chamber for a 10-min extinction test with access to both levers. Then 24 h after the completion of these tests the effectiveness of the prefeeding devaluation treatment in altering reward preference via satiation was assessed (choice test).
Reward Hedonics and Effort.
Consumption tests and lever training were similar to those previously described (31). Mice received lever training in which a single response on one lever led to the delivery of reward, whereas 15 responses on a second lever led to the delivery of a different reward. On completion of training, mice received a single-access consumption test session in the consummatory chamber, with access to either the low-effort or high-effort reinforcers.
Progressive Ratio Testing.
After four consumption sessions with sucrose in the consummatory chambers, mice underwent progressive ratio testing. At this stage, mice were required to lick at an empty well to receive sucrose delivery; after each delivery of sucrose, the number of licks required for the next delivery increased under a progressive ratio schedule. This process was repeated until the 3-h session was complete or until the period between individual licks was longer than 5 min [i.e., breakpoint (32)].
Dissection of the Prefrontal Cortex.
Dissection of mouse brain was modified based on procedures described by Glowinski and Iversen (50). The anterior block of brain at the level of the anterior commissure, achieved by a transverse section at the optic chiasm, was dissected to yield striatum and prefrontal cortex samples, the latter including prelimbic and infralimbic tissue, OFC, and most of the cingulate cortex together with other anterior cortical tissue in that tissue block.
Supplementary Material
Acknowledgments
We thank Ms. Yukiko Lema for figure preparation. This work was supported by National Institute of Mental Health Grants MH-084018, MH-94268 Silvo O. Conte Center, MH-069853, MH-085226, MH-088753, and MH-092443 (all to A.S.) and DK-084415 (to A.W.J.) and by grants to A.S. from Stanley, S-R, RUSK, The John Hopkins University Brain Science Institute, and the Maryland Stem Cell Research Fund and to A.S. and H.J.-P. from the Brain and Behavior Research Foundation (formerly the National Alliance for Research in Schizophrenia and Affective Disorders).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307925110/-/DCSupplemental.
References
- 1.Baaré WF, et al. Volumetric analysis of frontal lobe regions in schizophrenia: Relation to cognitive function and symptomatology. Biol Psychiatry. 1999;45(12):1597–1605. doi: 10.1016/s0006-3223(98)00266-2. [DOI] [PubMed] [Google Scholar]
- 2.Hoff AL, Riordan H, O’Donnell DW, Morris L, DeLisi LE. Neuropsychological functioning of first-episode schizophreniform patients. Am J Psychiatry. 1992;149(7):898–903. doi: 10.1176/ajp.149.7.898. [DOI] [PubMed] [Google Scholar]
- 3.Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213(1-2):93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316–338. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murrough JW, Iacoviello B, Neumeister A, Charney DS, Iosifescu DV. Cognitive dysfunction in depression: Neurocircuitry and new therapeutic strategies. Neurobiol Learn Mem. 2011;96(4):553–563. doi: 10.1016/j.nlm.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35(1):192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treadway MT, Zald DH. Reconsidering anhedonia in depression: Lessons from translational neuroscience. Neurosci Biobehav Rev. 2011;35(3):537–555. doi: 10.1016/j.neubiorev.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Insel T, et al. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 9.Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52(7):708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- 10.Fung SJ, et al. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry. 2010;167(12):1479–1488. doi: 10.1176/appi.ajp.2010.09060784. [DOI] [PubMed] [Google Scholar]
- 11.Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol. 2011;14(6):721–734. doi: 10.1017/S1461145710001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behrens MM, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318(5856):1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- 13.Maes M, Galecki P, Chang YS, Berk M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(3):676–692. doi: 10.1016/j.pnpbp.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Steullet P, et al. Redox dysregulation affects the ventral but not dorsal hippocampus: Impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci. 2010;30(7):2547–2558. doi: 10.1523/JNEUROSCI.3857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao JK, Keshavan MS. Antioxidants, redox signaling, and pathophysiology in schizophrenia: An integrative view. Antioxid Redox Signal. 2011;15(7):2011–2035. doi: 10.1089/ars.2010.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maes M, et al. Increased 8-hydroxy-deoxyguanosine, a marker of oxidative damage to DNA, in major depression and myalgic encephalomyelitis / chronic fatigue syndrome. Neuroendocrinol Lett. 2009;30(6):715–722. [PubMed] [Google Scholar]
- 17.Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011;12(12):707–722. doi: 10.1038/nrn3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaaro-Peled H, et al. Neurodevelopmental mechanisms of schizophrenia: Understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32(9):485–495. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blackwood DH, et al. Schizophrenia and affective disorders—cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: Clinical and P300 findings in a family. Am J Hum Genet. 2001;69(2):428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen S, et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28(43):10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hikida T, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA. 2007;104(36):14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaaro-Peled H, Ayhan Y, Pletnikov MV, Sawa A. Review of pathological hallmarks of schizophrenia: Comparison of genetic models with patients and nongenetic models. Schizophr Bull. 2010;36(2):301–313. doi: 10.1093/schbul/sbp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayhan Y, et al. Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: Evidence for neurodevelopmental origin of major psychiatric disorders. Mol Psychiatry. 2011;16(3):293–306. doi: 10.1038/mp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaaro-Peled H, et al. Subcortical dopaminergic deficits in a DISC1 mutant model: A study in direct reference to human molecular brain imaging. Hum Mol Genet. 2013;22(8):1574–1580. doi: 10.1093/hmg/ddt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dias R, Robbins TW, Roberts AC. Primate analogue of the Wisconsin Card Sorting Test: Effects of excitotoxic lesions of the prefrontal cortex in the marmoset. Behav Neurosci. 1996;110(5):872–886. doi: 10.1037//0735-7044.110.5.872. [DOI] [PubMed] [Google Scholar]
- 26.Rygula R, Walker SC, Clarke HF, Robbins TW, Roberts AC. Differential contributions of the primate ventrolateral prefrontal and orbitofrontal cortex to serial reversal learning. J Neurosci. 2010;30(43):14552–14559. doi: 10.1523/JNEUROSCI.2631-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Doherty J, Critchley H, Deichmann R, Dolan RJ. Dissociating valence of outcome from behavioral control in human orbital and ventral prefrontal cortices. J Neurosci. 2003;23(21):7931–7939. doi: 10.1523/JNEUROSCI.23-21-07931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boulougouris V, Dalley JW, Robbins TW. Effects of orbitofrontal, infralimbic and prelimbic cortical lesions on serial spatial reversal learning in the rat. Behav Brain Res. 2007;179(2):219–228. doi: 10.1016/j.bbr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Johnson AW, et al. Localized disruption of Narp in medial prefrontal cortex blocks reinforcer devaluation performance. Learn Mem. 2010;17(12):620–626. doi: 10.1101/lm.1937210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izquierdo A, Suda RK, Murray EA. Bilateral orbital prefrontal cortex lesions in rhesus monkeys disrupt choices guided by both reward value and reward contingency. J Neurosci. 2004;24(34):7540–7548. doi: 10.1523/JNEUROSCI.1921-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holland PC, Gallagher M. Amygdala-frontal interactions and reward expectancy. Curr Opin Neurobiol. 2004;14(2):148–155. doi: 10.1016/j.conb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Johnson AW, Gallagher M. Greater effort boosts the affective taste properties of food. Proc Biol Sci. 2011;278(1711):1450–1456. doi: 10.1098/rspb.2010.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodos W. Progressive ratio as a measure of reward strength. Science. 1961;134(3483):943–944. doi: 10.1126/science.134.3483.943. [DOI] [PubMed] [Google Scholar]
- 34.Sclafani A. Sucrose motivation in sweet “sensitive” (C57BL/6J) and “subsensitive” (129P3/J) mice measured by progressive ratio licking. Physiol Behav. 2006;87(4):734–744. doi: 10.1016/j.physbeh.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 35.Ward RD, Simpson EH, Kandel ER, Balsam PD. Modeling motivational deficits in mouse models of schizophrenia: Behavior analysis as a guide for neuroscience. Behav Processes. 2011;87(1):149–156. doi: 10.1016/j.beproc.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett JR. Assessment of social interaction behaviors. J Vis Exp. 2011;48:2473–2477. doi: 10.3791/2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moy SS, et al. Sociability and preference for social novelty in five inbred strains: An approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3(5):287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 38.Smith DR, Burruss DR, Johnson AW. An assessment of olfaction and responses to novelty in three strains of mice. Behav Brain Res. 2009;201(1):22–28. doi: 10.1016/j.bbr.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 39.Hara MR, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7(7):665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 40.Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell Signal. 2011;23(2):317–323. doi: 10.1016/j.cellsig.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnsten AF. Prefrontal cortical network connections: Key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. 2011;29(3):215–223. doi: 10.1016/j.ijdevneu.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: Advancing toward rational pharmacological therapies. J Clin Invest. 2009;119(4):706–716. doi: 10.1172/JCI37335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallis JD. Orbitofrontal cortex and its contribution to decision-making. Annu Rev Neurosci. 2007;30:31–56. doi: 10.1146/annurev.neuro.30.051606.094334. [DOI] [PubMed] [Google Scholar]
- 44.Valentin VV, Dickinson A, O’Doherty JP. Determining the neural substrates of goal-directed learning in the human brain. J Neurosci. 2007;27(15):4019–4026. doi: 10.1523/JNEUROSCI.0564-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kouneiher F, Charron S, Koechlin E. Motivation and cognitive control in the human prefrontal cortex. Nat Neurosci. 2009;12(7):939–945. doi: 10.1038/nn.2321. [DOI] [PubMed] [Google Scholar]
- 46.Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20(6):2369–2382. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: Microcircuitry. Neuropsychopharmacology. 2010;35(1):27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porteous DJ, Millar JK, Brandon NJ, Sawa A. DISC1 at 10: Connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699–706. doi: 10.1016/j.molmed.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19(2):220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 50.Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem. 1966;13(8):655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.