

Abstract
Multivalency has an important but poorly understood role in molecular self-organization. We present the noncovalent synthesis of a multicomponent supramolecular polymer in which chemically distinct monomers spontaneously coassemble into a dynamic, functional structure. We show that a multivalent recruiter is able to bind selectively to one subset of monomers (receptors) and trigger their clustering along the self-assembled polymer, behavior that mimics raft formation in cell membranes. This phenomenon is reversible and affords spatiotemporal control over the monomer distribution inside the supramolecular polymer by superselective binding of single-strand DNA to positively charged receptors. Our findings reveal the pivotal role of multivalency in enabling structural order and nonlinear recognition in water-soluble supramolecular polymers, and it offers a design principle for functional, structurally defined supramolecular architectures.
Keywords: self-assembly, simulations, energy transfer
One of the most fascinating features of living matter is the precise control over biological activity in space and time. The cell membrane provides a remarkable example of such high-fidelity spatiotemporal control in a complex biological setting, wherein thousands of different components, namely lipids and proteins, self-assemble into a 2D fluid mosaic (1). To perform the functions that the cell requires, lipids and proteins are heterogeneously distributed and specific biomolecules are segregated in active nanometer-sized domains often referred to as rafts (2). This distribution is highly dynamic, such that these platforms can be rapidly assembled and disassembled (3). The principles that underlie control over the molecular composition of the cellular microenvironment in space and time are the subject of great scientific debate as they are of crucial importance for cell functioning, signaling, growth, and division (4).
One of the main goals of supramolecular chemistry is the noncovalent synthesis of functional molecular architectures through weak and reversible interactions (5). In this framework, a key challenge is the design of molecular building blocks that are able to self-organize hierarchically and in a cooperative fashion (6), thus mimicking the dynamic and structural complexity of living systems as well as their functionality. Various modular multicomponent systems have been successfully developed (7), but the spatiotemporal control of the localization of distinct components within synthetic supramolecular assemblies has yet to be realized. Mastering the spatial distribution of assembled molecules in a noncovalent synthesis is as crucial for their functionality as regio-selectivity impacts the molecular properties of organic molecules synthesized in a classical covalent manner. An interesting supramolecular polymer, where multiple components coassemble cooperatively in water, is based on 1,3,5-benzenetricarboxamide derivatives (BTAs) (8). Of particular relevance for the present work, is the fact that reversible interactions between monomers confer a dynamic behavior to BTA supramolecular polymers, wherein monomers are constantly being exchanged with the bulk (8). Most of these studies were in organic solvent, but we recently describe how pegylated BTAs self-assemble in water through a combination of hydrogen bonding and hydrophobic effects to create long supramolecular polymers of ∼0.1–10 μm in length (9).
In the biology and chemistry communities, there is intense interest in the role of multivalency as a tool to encode structure and function into soft materials. Several fascinating papers reported how nature exploits multivalency to exert control over spatial segregation and phase separation inside living cells (10). Segregation of proteins and sphingolipids in lipid raft domains (11) is a remarkable example of this phenomenon. Likewise, the chemistry community is keen to use the efficacy of multivalent interactions as a “chemical organization and action principle” (12). Interfacing chemistry with biology, two recent innovative studies demonstrate the use of synthetic multivalent binders to drive receptor clustering and raft formation on the cell membrane (13) (14). Collectively, these reports convincingly show how multivalency can be used to create well-defined structures and elicit a biological response. Moreover, a recent study has proposed an analytical model describing the nature of superselective binding as a consequence of multivalency (15). Can we now profit from the same organization principle to induce order into synthetic supramolecular structures?
Inspired by natural systems and the formation of rafts in the cell membrane in particular, we present a study that demonstrates control of the spatiotemporal distribution of assembled BTAs through dynamic and multivalent interactions in water. We find that both the dynamic nature of the molecular (dis)assembly and multivalency are key requirements to establish such control at the molecular level. Finally, we demonstrate the emergence of a strongly nonlinear effect, namely superselective recognition, as a direct consequence of the multivalent nature of synthetic supramolecular structures. The observation of these phenomena has important ramifications for the understanding and application of supramolecular materials in water. The control over the spatiotemporal distribution of distinct components in a supramolecular aggregate is an important step toward the design of switchable functional systems. Moreover, the adaptive behavior and the superselective binding, reported in this work, shows that BTA-based supramolecular polymers are versatile tools for molecular recognition in a broad range of applications including biomedical materials.
Results and Discussion
Multicomponent supramolecular polymers based on BTAs were prepared via coassembly of different types of pegylated BTAs (14) as displayed in Fig. 1A (see SI Text for details on the BTA synthesis). The three different building blocks reported in this work present the same hydrogen-bonding motif in the hydrophobic core but vary in the chemical composition of the peripheral side chains (Fig. 1A). Neutral BTA monomers have been coassembled together with cationic species, indicated as BTA3+, into multicomponent one-dimensional structures (9). BTA3+ are asymmetric monomers bearing at the PEG termini two primary amines, positively charged at neutral pH, and a cyanine dye (Cy3 or Cy5, both bearing one positive charge) as a fluorescent label. In our design, the neutral monomers serve as a scaffold; i.e., they represent the synthetic analog of the liquid phase of the plasma membrane. The cationic monomers are incorporated in small amounts ranging from 0.1% to 8% and act as receptors capable of electrostatic binding to oppositely charged ligands. The coassembly process results in a supramolecular analog of random copolymers with a stochastic distribution of monomers along the polymer chain (Fig. 1B, Left). The ligand-mediated rearrangement, similar to an isomerization, represents a subsequent step in the noncovalent synthesis and results in nonrandom, segmented monomer sequences (Fig. 1B, Right). Here, we exploit single-stranded DNA (ssDNA) as the multivalent, polyanionic tool necessary to recruit the cationic BTA3+ receptors and induce their clustering.
Fig. 1.

Multivalency-induced order in multicomponent supramolecular polymers. Supramolecular polymers are prepared by coassembling neutral BTA monomers (A, Left) with positively charged derivatives (A, Center). The neutral unit is the predominant component and represents the “liquid phase” of the assembly. Charged monomers (incorporated at 0.1–8% molar ratio) are capable of electrostatic binding and act as receptors. ssDNA (A, Right), due to its multiple negative charges, is able to bind and recruit receptors within the assembly. B shows a schematic representation of the reversible clustering of receptors along the supramolecular polymer. (Please note that the supramolecular polymers are represented in the cartoon as single stacks for simplification. We acknowledge that the chemical nature of the side chains can induce modifications of the structure from single stacks to double helixes.) Owing to recruiter multivalency and to the dynamic behavior of supramolecular polymers, DNA can reversibly induce a rearrangement in the monomer distribution along the polymer.
The characterization of the chemical composition (i.e., the monomer sequence) of multicomponent supramolecular polymers is complicated by the fact that their structure is dynamic and will change with a variation of the experimental conditions, such as monomer concentration, temperature, and the presence of ligands. In fact, the presence of spatial inhomogeneities in the dynamic environment of the plasma membrane remained controversial for decades, precisely because of the lack of suitable experimental techniques to probe such a complex system with sufficient temporal and spatial resolution. In recent years, progress has been made as various assays, mostly based on fluorescence, have been developed to probe the formation and breakup of solid-like domains within the otherwise liquid-like cell membrane (16). Similarly, the present study relies on sensitive fluorescence assays to probe the reversible assembly and disassembly of clusters of charged BTAs in a neutral BTA scaffold. For this purpose, we labeled BTA3+ monomers with a pair of dyes well suited for Förster resonance energy transfer (FRET). It is one of the most powerful biophysical tools used to demonstrate clustering of receptors inside the plasma membrane (17). We selected the FRET pair Cy3/Cy5, with a Förster radius of 50 Å (18), to probe monomer–monomer proximity on the molecular scale.
Exploiting this fluorescent tool, we first evaluated the dynamics of the multicomponent supramolecular polymers as it is a prerequisite for spatiotemporal control over the monomer sequence. Obviously, BTA receptors can only cluster upon binding to a multivalent ligand if they are able to exchange position within the supramolecular chain. To investigate whether water-soluble supramolecular polymers are able to reconfigure on a molecular level, we separately assembled supramolecular polymers incorporating either Cy3 or Cy5 BTAs as shown in Fig. 2A. After equilibration, the two polymer solutions were mixed and the FRET ratio monitored in time. If the structures are unable to exchange monomers, no change in fluorescence should be observed after mixing. However, if the polymers are endowed with a dynamic nature, mixed fibers containing both Cy3 and Cy5 should be obtained resulting in an increase of FRET due to proximity.
Fig. 2.

Dynamics of supramolecular polymers in water. A shows a schematic representation of the mixing experiment. Preequilibrated BTA assemblies incorporating either BTA-Cy3 (green) or Cy5 (red) (2% of labeled monomers) are mixed in water and the monomer exchange is monitored by FRET. The evolution of FRET ratio over time after mixing (B) reveals an increase of the FRET ratio (defined as FCy5/FCy3 upon Cy3 excitation) in time, indicating an increase in proximity between labeled BTAs due to monomer exchange. No significant dependence of the exchange kinetics on the BTA concentration was observed (B) while the exchange became significantly faster upon increasing the temperature (at constant total BTA concentration of 50 µM) (C), suggesting that monomer release and attachment is the rate-determining step.
Fig. 2B shows the results of the mixing experiments. As can be observed, FRET increases in time after mixing and reaches a plateau in ∼3 h. This indicates that the monomer sequence is not frozen like in a covalent polymer but molecules can dynamically exchange position. This exchange is remarkably slow in comparison with what has been observed for supramolecular polymers in organic solvent, where the exchange occurs on a second timescale (19). This difference can be attributed to the stronger association in water owing to the hydrophobic effect, which results in an enhanced BTA aggregation that slows down the depolymerization of the aggregates. To gain insight into the rearrangement process, we measured the exchange kinetics at different concentrations (Fig. 2B) and temperatures (Fig. 2C). Concentration does not significantly influence the rate of the reorganization over one order of magnitude. In contrast, the exchange rate is significantly faster at higher temperatures. These results resemble observations on self-assembled polymeric micelles, in which monomer expulsion and insertion is the rate-determining step of system dynamics (20, 21). Moreover, monomer polymerization and depolymerization are reported to be the main exchange mechanism of supramolecular polymers in organic solvents (22). The limited effect of concentration on the exchange rate indicates that stack–stack interactions do not play a pivotal role while free monomers seem to be the mediators of the polymer chain rearrangement. The information obtained about the dynamic behavior and the timescales for monomer exchange are of great importance for the study of spatiotemporal control proposed in this work.
Having verified that it is possible to rearrange the monomer sequence within and between supramolecular polymers, we continued to study the change in BTA distribution upon ssDNA addition. ssDNA is particularly appealing as recruiter as its multivalency (i.e., the number of charged groups) can be precisely tuned through solid-phase DNA synthesis. Fig. 3A shows the plot of FRET versus time upon DNA treatment.
Fig. 3.
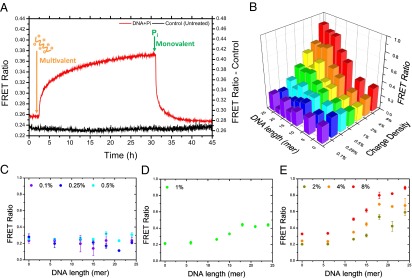
Nonlinearity in ssDNA-induced clustering. A strong increase in FRET upon ssDNA (18-mer) addition reveals multivalency-driven clustering of labeled monomers (A). The process is reversible; the addition of an excess of monovalent competitor (50 mM phosphate ions, Pi) reverts the trend in FRET. B shows steady-state FRET values for different densities of charged monomers and different DNA lengths. The highest values are observed for densities >1% and DNA length of >12-mer. The dependence of the FRET ratio on the receptor density and DNA length are highlighted in C–E. Both trends are strongly nonlinear. No effect upon DNA addition is observed for low densities (C) while clustering appears only above a critical value (1% of receptors; D). At higher densities, a strong clustering is observed (E). A nonlinear dependence of the FRET ratio on DNA length is observed at a critical value around 10–12 bases.
The FRET signal displays a dramatic increase upon ssDNA addition, as a result of the interactions of the polyanion with the labeled charged receptors. The change in FRET is correlated to the significant reduction in the average distance between charged monomers that occurs when the random distribution is converted into a more segmented one. This finding supports the idea that the DNA is able to bind the charged monomers and, due to its multivalent character, force their clustering along the stack into 1D domains. Interestingly, when a large excess of a monovalent anionic competitor, phosphate ion, is added, the trend is completely reverted (Fig. 3A). This result has two major implications. First, it confirms that the binding is dominated by electrostatic interactions, as an excess of phosphate ions disrupts the clustering. It also reveals the necessity of multivalency for receptor clustering: the FRET ratio measured in the presence of monovalent phosphate ions indicates that the monomer distribution is comparable to the stochastic one.
The clustering kinetic is remarkably slow: more than 24 h are needed to reach a plateau in FRET efficiency. However, this time frame is in full agreement with the time needed for monomer exchange. Interestingly, BTAs retain their dynamic behavior even when bound to ssDNA, owing to the reversible nature of electrostatic interactions. Indeed, when ssDNA is added to the individual polymers with only Cy3 or Cy5 BTAs first, followed by mixing, monomers can still exchange between different clusters as shown in SI Text. This interesting feature further highlights the fundamental difference between the exertions of multiple weak interactions compared with the use of permanent covalent crosslinking. Noncovalent interactions afford control over the monomer distribution without irreversibly “freezing” the polymer structure.
To clarify the role of multivalency in monomer clustering, we performed a series of FRET experiments varying recruiter length and receptor density (Fig. 3B). As expected, the highest efficiencies are obtained for the combinations of high density and long DNA strands (top right corner of the 3D plot in Fig. 3B). However, a marked nonlinear dependence on both receptor density and DNA length is observed, as highlighted in Fig. 3 C–E. At lower densities (from 0.1% to 0.5%), no clustering occurs regardless of the length of the DNA (Fig. 3C). At a critical threshold around 1% (Fig. 3D), an increase of FRET is observed, which becomes stronger upon an increase in the receptor density (Fig. 3E). Moreover, it is evident from Fig. 3E that the clustering has a nonlinear dependence on the length of the DNA; no effect is observed for short DNA strands and clustering starts to occur for strands longer than 12–15 DNA bases.
To fully understand the observed experimental results, we performed μVT lattice Monte Carlo (MC) simulations (23) of a coarse-grained model. DNA chains are represented as beads on a lattice connected through bonds, with each bead representing a DNA base. We simulated only a single BTA columnar stack, by prealigning BTA molecules (represented as single lattice sites) along the z direction of the simulation box. When a bead from a DNA chain is in nearest-neighbor contact with a charged BTA molecule, the system lowers its energy by an amount equal to ε. In this work, ε was varied between 0 and 10 kT, where k is Boltzmann’s constant and T is the absolute temperature. To simulate the dynamical behavior of the molecules in the polymer, the positions of charged and uncharged molecules are exchanged by means of standard Metropolis MC moves (23). A detailed description of the implementation of the simulations is reported in SI Text.
A typical snapshot of such simulations is shown in Fig. 4A and shows that DNA binds selectively to the charged monomers forcing their clustering along the BTA chain. To compare the computational results with the FRET measurements reported, we analyzed the simulations and gathered information about clustering and selectivity (Fig. 4 B–D). As FRET between red and green dyes is effective only within a cutoff distance of a few nanometers, we tracked in the simulations the number of nearest-neighbor red–green pairs as an indication of the FRET signal intensity and plotted it as a function of DNA length (Fig. 4B). Note that taking contributions beyond nearest neighbors into account was shown to have an insignificant effect on the observed results (SI Text). We find that, for DNA–BTA3+ energy of interaction ε = 4.50 kT, the simulation results (Fig. 4B) are remarkably similar to the experimental results presented in Fig. 3, whereas a variety of qualitatively different behaviors are found for other values of ε (SI Text). For low fractions of charged BTA (<1%), no clustering is observed and the number of green–red dye pairs is not dependent on DNA length. In contrast, for higher receptor densities (>1%), we observed a distinctive nonlinear behavior: no signal is observed below a critical DNA length (∼12-mer), whereas a rapid increase of clustering is observed above that threshold.
Fig. 4.

Superselectivity in supramolecular polymers. (A) Snapshot of μVT lattice MC simulations. Three types of beads represent three types of BTA molecules: neutral (gray), charged (red), and charged (green). The aquamarine beads represent the DNA chain. ssDNA binds charged monomers and forces their clustering along the BTA stack. (B) Average number of nearest-neighbor green–red pairs as a function of the length of the DNA in solution for different percentage of charged BTA molecules. Results are normalized to one in the absence of DNA. A good match with FRET experiments is observed with ε = 4.50 kT. (C) The calculated value of α for various DNA lengths at ε = 4.50 kT indicates that the supramolecular polymer behaves superselectively toward DNA binding. (D) Distribution of the number of BTA3+ monomers within a “macrolattice” for different values of ε and for a 20-mer DNA chain. Bimodal distributions indicate clustering. Lines are just a guide to the eye.
The excellent agreement between the experiments and simulations is remarkable given the simplicity and coarse-grained nature of our model, suggesting that the principles behind multivalency and recruitment are general and system independent. Indeed, the nonlinearity of our results can be understood in the light of multivalency and superselectivity. We previously reported an analytical model in which is demonstrated that, under specific conditions, multivalent particles are capable of superselective binding to receptors, i.e., that the fraction of bound particles varies nonlinearly with the receptor density (15). Although this model was originally developed for the case in which the receptors are “fixed” to the surface (i.e., nondynamic behavior), we have extended it for the case in which the receptors are mobile (SI Text). In this case, the model predicts that the mobility of the receptors allows for an even sharper dependence of the number of bound particles on receptor density, increasing the superselective behavior of the system at the appropriate conditions (SI Text). In fact, our MC simulations show that the number of bound ssDNA molecules depends sharply on the receptor (i.e., BTA3+) concentration. Hence, for a given DNA length, there should be a critical charge density in the polymer above which DNA binds significantly to the supramolecular polymer and receptors can be clustered. At low receptor concentration, few chains are bound and little clustering occurs. However, as the percentage of receptors increases and we approach the threshold concentration, the number of bound chains starts to increase dramatically resulting in receptor clustering. Because for dilute solutions this transition becomes more abrupt—in a nonlinear way—more clustering occurs for longer chains. Thus, at the conditions studied we can expect that for chains with lengths above the 12-mer the threshold receptor concentration occurs slightly above 1% of charged BTA, whereas for shorter chains the threshold concentration is higher than the concentrations studied (i.e., 8%) and therefore is not observed.
As discussed in ref. 15, the threshold concentration corresponds to the receptor concentration at which the quantity α reaches a maximum value. The quantity α is defined as follows:
 |
where Nb is the number of DNA chains bound to the supramolecular polymer and nR is the charge density. Because locally Nb ∼ nRα, the value of α indicates how fast the number of bound particles changes with receptor concentration. Hence, at the point where α is maximum, the number of bound particles changes the fastest with nR, therefore indicating the onset of DNA binding and clustering. To test this hypothesis, we computed α from the simulations for various DNA lengths (Fig. 4C). DNA chains with lengths of 4- and 10-mers do not reach a peak in α for nR < 8%, indicating that shorter chains do not reach the threshold concentration and cannot induce significant clustering. Conversely, longer chains (e.g., 20-mers) present a peak in the value of α around 1–2% consistent with the experimental results. Thus, our simulations strongly suggest that, in the experiments, our supramolecular system is behaving superselectively. Hence, the phenomenon of superselectivity, which has only been described for biological systems, can explain the behavior of fully synthetic architecture as well.
To further understand the change in receptor distribution along the polymer, we turned to a quantification of BTA3+ clustering. We divided the supramolecular polymer into arbitrary “macrolattices” of a large enough size (20 beads in this work) and we computed the frequency of a given number of charged monomers within these macrolattices. Fig. 4D displays such distributions for a 20-mer chain, with 8% charged monomers, for different values of ε. When there is no attractive energy (i.e., ε = 0), the charged beads distribute randomly and the distribution approaches a binomial distribution. However, as the attractive interaction becomes stronger, the distribution becomes more bimodal, indicating the existence of regions of the polymer with many charged monomers (i.e., clusters) and regions of no charged monomers, while keeping the total charge fixed. The appearance of clustering under conditions of multivalency, high values of ε, and low bulk concentration of DNA, is consistent with the prediction of the simple analytical model presented in SI Text, where we have extended the multivalency model proposed in ref. 15 to take the mobility of the receptors into account.
The computational analysis provides important insights into the roles of multivalency and dynamics in our supramolecular system. From a kinetic point of view, a critical number of initial binding interactions is required to stably anchor DNA onto the BTA fiber after which recruitment of more charged monomers can occur. The initial number of anchoring points depends both on DNA multivalency and receptor density, which is consistent with the observed nonlinear dependence on these two quantities. According to the model proposed by Hlavacek and coworkers for multiple receptor–ligand interaction (24), the number of initial binding sites determines the time that the DNA is bound to the BTA stack before detachment. If the residence time is comparable to the timescale of monomers rearrangement, which is about 2–3 h (Fig. 2), monomer exchange can provide additional receptors for DNA binding. The resultant increase in the number of binding sites further strengthens the DNA–polymer interaction and augments the residence time of the DNA onto the BTA stack. This synergy creates a positive-feedback loop in which multivalency and dynamics, if synchronized, act in a cooperative fashion to induce order in self-assembled systems.
Conclusions
In conclusion, we present a supramolecular system in which multiple components coassemble in a dynamic fashion. We investigated the role of multivalency and dynamics and show how they drive clustering of molecules within a synthetic self-assembled structure. These phenomena have two main implications. First, these findings demonstrate how a multivalent binder can induce order into a supramolecular polymer, allowing control over the spatiotemporal distribution of the monomers. This is of great interest to design well-defined functional supramolecular systems through noncovalent synthesis. Second, we have shown how the dynamic behavior of noncovalent architectures makes them adaptive, and affords superselective recognition of specific biomolecules. This responsive behavior is of great interest for biological recognition and makes functional supramolecular polymers a promising scaffold for several applications in the biological environment.
Methods
Chemicals were purchased from Sigma-Aldrich and used without further purifications. Cy3-NHS and Cy5-NHS esters were purchased from Lumiprobe. DNA oligos with different length were obtained from Eurofins. Dialysis membranes were obtained from Spectrum Laboratories. 1H-NMR and 13C-NMR spectra were recorded on a Varian Mercury Vx 400 MHz NMR spectrometer. Matrix-assisted laser desorption/ionization mass spectra were obtained on a PerSeptive Biosystems Voyager DE-PRO spectrometer or a Bruker autoflex speed spectrometer using α-cyano-4-hydroxycinnamic acid (CHCA) and 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) as matrices. Infrared spectra were recorded on a Perkin-Elmer Spectrum One 1600 FT-IR spectrometer or a Perkin-Elmer Spectrum Two FT-IR spectrometer, equipped with a Perkin-Elmer Universal ATR Sampler Accessory.
The synthetic scheme for the synthesis of cationic BTA3+ is reported in Fig. S1. The detailed procedures for the synthesis are reported in SI Text. The synthesis of neutral pegylated BTAs was previously reported (9). For BTA polymers assembly procedure stock solution of BTA (10 mM) and labeled BTA3+ (1 mM) in MeOH were prepared. The organic solvent solutions were mixed at the desired ratio to control the molar ratio of BTA3+ on BTA, injected in filtered Milli-Q water (total concentration BTA, 50 μM) and equilibrated for 24 h before experiment. Samples at different concentrations were obtained by serial dilution with Milli-Q water of the 50 μM stock. For mixing kinetic experiments, solution containing either 2% of BTA-Cy3 or 2% of BTA-Cy5 were mixed 1:1 in a 500-μL glass cuvette and immediately measured at the fluorimeter. For clustering experiments, different DNA (stock solutions in Milli-Q water at a concentration of 25 μM) was added to the BTA solutions to reach a final concentration of 500 nM.
To achieve FRET ratio, samples were excited at 540 nm (Cy3 excitation) and fluorescence emission measured at 570 nm (Cy3 emission) and 670 nm (Cy5 emission). Temperature was kept at 20 °C with the in-built peltier of the fluorimeter. The array of FRET measurements reported in Fig. 3B was obtained by means of a plate reader. Samples with different BTA3+ densities were prepared (total volume, 50 μL) and incubated with different DNA strands for 48 h in a 96-well plate at 20 °C. Three samples were independently prepared for every BTA3+–BTA combination. After 48 h, fluorescence emission at 570 and 670 nm was measured for every sample and the FRET ratio ± SD (n = 3) calculated.
The details of the MC simulation and the analytical model for the binding of multivalent recruiter to dynamic receptors are reported in SI Text.
Supplementary Material
Acknowledgments
We thank Dr. Anja Palmans for useful discussions, Bas de Waal for his support during the synthetic procedures, and Dr. Koen Pieterse and the Institute for Complex Molecular Systems animation studio for the 3D modeling. The Netherlands Institute for Regenerative Medicine is gratefully acknowledged for financial support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303109110/-/DCSupplemental.
References
- 1.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175(4023):720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 2.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327(5961):46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 3. Simons K, Sampaio JL (2011) Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol 3(10):a004697. Available at http://cshperspectives.cshlp.org/content/3/10/a004697. Accessed December 3, 2012.
- 4.Leslie M. Mysteries of the cell. Do lipid rafts exist? Science. 2011;334(6059):1046–1047. doi: 10.1126/science.334.6059.1046-b. [DOI] [PubMed] [Google Scholar]
- 5.Lehn J-M. Supramolecular Chemistry. 1st Ed. Weinheim, Germany: Wiley-VCH; 1995. [Google Scholar]
- 6.Aida T, Meijer EW, Stupp SI. Functional supramolecular polymers. Science. 2012;335(6070):813–817. doi: 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petkau-Milroy K, Sonntag MH, van Onzen AHAM, Brunsveld L. Supramolecular polymers as dynamic multicomponent cellular uptake carriers. J Am Chem Soc. 2012;134(19):8086–8089. doi: 10.1021/ja3029075. [DOI] [PubMed] [Google Scholar]
- 8.Cantekin S, de Greef TFA, Palmans ARA. Benzene-1,3,5-tricarboxamide: A versatile ordering moiety for supramolecular chemistry. Chem Soc Rev. 2012;41(18):6125–6137. doi: 10.1039/c2cs35156k. [DOI] [PubMed] [Google Scholar]
- 9.Leenders CMA, et al. Supramolecular polymerization in water harnessing both hydrophobic effects and hydrogen bond formation. Chem Commun (Camb) 2013;49(19):1963–1965. doi: 10.1039/c3cc38949a. [DOI] [PubMed] [Google Scholar]
- 10.Hyman AA, Simons K. Cell biology. Beyond oil and water—phase transitions in cells. Science. 2012;337(6098):1047–1049. doi: 10.1126/science.1223728. [DOI] [PubMed] [Google Scholar]
- 11.Coskun Ü, Simons K. Cell membranes: The lipid perspective. Structure. 2011;19(11):1543–1548. doi: 10.1016/j.str.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Fasting C, et al. Multivalency as a chemical organization and action principle. Angew Chem Int Ed Engl. 2012;51(42):10472–10498. doi: 10.1002/anie.201201114. [DOI] [PubMed] [Google Scholar]
- 13.Courtney AH, Puffer EB, Pontrello JK, Yang Z-Q, Kiessling LL. Sialylated multivalent antigens engage CD22 in trans and inhibit B cell activation. Proc Natl Acad Sci USA. 2009;106(8):2500–2505. doi: 10.1073/pnas.0807207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O’Reilly MK, Paulson JC (2010) Multivalent ligands for Siglecs. Methods in Enzymology, ed Fukuda M (Academic, San Diego), pp 343–363. Available at www.sciencedirect.com/science/article/pii/S0076687910780174. Accessed December 3, 2012.
- 15.Martinez-Veracoechea FJ, Frenkel D. Designing super selectivity in multivalent nano-particle binding. Proc Natl Acad Sci USA. 2011;108(27):10963–10968. doi: 10.1073/pnas.1105351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simons K, Gerl MJ. Revitalizing membrane rafts: New tools and insights. Nat Rev Mol Cell Biol. 2010;11(10):688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- 17.Sharma P, et al. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 2004;116(4):577–589. doi: 10.1016/s0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- 18.Bastiaens PI, Jovin TM. Microspectroscopic imaging tracks the intracellular processing of a signal transduction protein: Fluorescent-labeled protein kinase C beta I. Proc Natl Acad Sci USA. 1996;93(16):8407–8412. doi: 10.1073/pnas.93.16.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smulders MMJ, Schenning APHJ, Meijer EW. Insight into the mechanisms of cooperative self-assembly: The “sergeants-and-soldiers” principle of chiral and achiral C3-symmetrical discotic triamides. J Am Chem Soc. 2008;130(2):606–611. doi: 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- 20.Lund R, Willner L, Richter D, Dormidontova EE. Equilibrium chain exchange kinetics of diblock copolymer micelles: Tuning and logarithmic relaxation. Macromolecules. 2006;39(13):4566–4575. [Google Scholar]
- 21.Zinn T, Willner L, Lund R, Pipich V, Richter D. Equilibrium exchange kinetics in n-alkyl–PEO polymeric micelles: Single exponential relaxation and chain length dependence. Soft Matter. 2011;8:623–626. [Google Scholar]
- 22.De Greef TFA, et al. Supramolecular polymerization. Chem Rev. 2009;109(11):5687–5754. doi: 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- 23. Frenkel D, Smit B (2001) Understanding Molecular Simulation: From Algorithms to Applications (Academic, San Diego), 2nd Ed.
- 24.Goldstein B, Faeder JR, Hlavacek WS. Mathematical and computational models of immune-receptor signalling. Nat Rev Immunol. 2004;4(6):445–456. doi: 10.1038/nri1374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.