

Significance
Hepatitis B virus (HBV) is a major pathogen, yet no fully effective therapies exist. HBc is the multifunctional, capsid-forming protein essential for HBV replication. HBc structural plasticity is reportedly functionally important. We analyzed the folding mechanism of HBc using a multidisciplinary approach, including microscale thermophoresis and ion mobility spectrometry–mass spectrometry. HBc folds in a 3-state transition with a dimeric, helical intermediate. We found evidence of a strained native ensemble wherein the energy landscapes for folding, allostery, and capsid formation are linked. Mutations thermodynamically trapped HBc in conformations unable to form capsids, suggesting chemical chaperones could elicit similar, potentially antiviral, effects.
Keywords: capsid assembly, energy landscape, protein dynamics, thermodynamic coupling
Abstract
HBc, the capsid-forming “core protein” of human hepatitis B virus (HBV), is a multidomain, α-helical homodimer that aggressively forms human HBV capsids. Structural plasticity has been proposed to be important to the myriad functions HBc mediates during viral replication. Here, we report detailed thermodynamic analyses of the folding of the dimeric HBc protomer under conditions that prevented capsid formation. Central to our success was the use of ion mobility spectrometry–mass spectrometry and microscale thermophoresis, which allowed folding mechanisms to be characterized using just micrograms of protein. HBc folds in a three-state transition with a stable, dimeric, α-helical intermediate. Extensive protein engineering showed thermodynamic linkage between different structural domains. Unusual effects associated with mutating some residues suggest structural strain, arising from frustrated contacts, is present in the native dimer. We found evidence of structural gatekeepers that, when mutated, alleviated native strain and prevented (or significantly attenuated) capsid formation by tuning the population of alternative native conformations. This strain is likely an evolved feature that helps HBc access the different structures associated with its diverse essential functions. The subtle balance between native and strained contacts may provide the means to tune conformational properties of HBc by molecular interactions or mutations, thereby conferring allosteric regulation of structure and function. The ability to trap HBc conformers thermodynamically by mutation, and thereby ablate HBV capsid formation, provides proof of principle for designing antivirals that elicit similar effects.
The “protein-folding problem” describes how a polypeptide sequence contains all the information needed for it to adopt a specific 3D structure spontaneously (1). The chemistry and thermodynamic code that causes proteins to fold also underpins protein–protein interactions, allostery, and supramolecular assembly. An emerging trend has been the study of model proteins free from kinetic traps, aggregation, or metal binding, features that can confound experimental execution and data interpretation (2, 3). Consequently, model proteins are small (typically <130 residues), soluble monomers with few proline or cysteine residues and no prosthetic groups (2, 3).
Although model proteins have been instrumental in taking the field to its current zenith, there is a paucity of experimental insights into the conformational dynamics of larger, oligomeric proteins, especially those implicated in diseases (3). Such proteins usually have complex behavior refractory to detailed experimental studies. However, the connection between sequence, structure, dynamics, and allostery makes studies of larger proteins central to understanding biological function and aiding drug design (vide infra) (4). One such protein is HBc, the capsid-forming “core protein” of human hepatitis B virus (HBV), a major pathogen that kills 600,000 people annually (5). Although excellent vaccines exist, there are no effective cures for extant chronic infections (5, 6). In addition to capsid formation, HBc plays many essential roles in HBV replication (7–9), making it an attractive drug target (10–15).
WT HBc is a 183-residue polypeptide comprising a structured capsid-forming region (residues 1–149; Fig. 1A) and a basic, nucleic acid-binding domain (residues 150–183) (16–18). The structured N-terminal region (hereafter HBc1–149) spontaneously self-assembles in vitro and in vivo to form icosahedral capsid-like particles (CLPs) identical to nucleocapsids isolated from patient serum (19, 20). X-ray crystallography and cryo-EM have characterized the structure of HBc1–149 within the context of CLPs, virions, and hexamers (16, 19–23). HBc homodimers comprise two structural domains (Fig. 1A): Helices α3 and α4 from opposing monomers pack together and form a disulfide-linked, four-helix bundle dimerization interface (visible as protrusions on the capsid exterior; Fig. 1B), whereas α1, α2, and α5 pack together and around the base of the four-helix bundle to create the hydrophobic core of “contact” domains (19). Weak interdimer interactions between contact domains stabilize HBV capsids (19, 24) (Fig. 1B).
Fig. 1.

HBc1–149 dimer structure within HBV capsids. (A) Four-helix bundle dimerization interface (black) is flanked by contact domains (orange and red). Helices are numbered, and the N and C termini of one monomer are indicated. The disulfide link between C61 of each monomer is indicated (cyan). (B) Exterior surface of a T = 4 capsid HBc1–149 (PDB ID code 1QGT) (19). Dimers around the threefold and fivefold axes are indicated in blue/green and purple/orange, respectively. (Inset) Interacting quasiequivalent HBc1–149 dimers from the fivefold (purple and orange) and threefold (blue and green) axes are shown. Hydrophobic contacts between contact domains stabilize capsids. Residues that perturb capsid formation when mutated are indicated.
Multiple studies show clearly that HBc has a very malleable structure, with this structural plasticity argued to be functionally important (22, 23). This hypothesis accords well with antivirals that modulate HBc structure (11–15, 22, 23). Studies of HBV capsid assembly have inferred the existence of assembly-active (HBcAss) and assembly-incompetent (HBcInc) HBc conformations (12, 13, 21, 24, 25). However, there are few detailed insights on the thermodynamic origins of structure, allostery, and dynamics for the dimeric HBc1–149 protomer, where structural plasticity must originate. This arises from dimeric HBc1–149 being very challenging to study in vitro (compared with the model proteins described above) because it is a 298-residue disulfide-linked homodimer (containing 6 cysteine and 24 proline residues) that aggregates aggressively and forms capsids.
Here, we report detailed folding and stability studies of dimeric HBc1–149. These show HBc1–149 folds in a three-state transition with a populated, dimeric, α-helical intermediate. Of 29 “chemically conservative” mutants used to probe folding energetics (26), many had similar effects on the stability of the intermediate and native ensembles. The distribution of these mutations was consistent with the intermediate being stabilized by a significant native-like structure. However, some mutations destabilized the native state (N) much less than the intermediate state (I) relative to the denatured state (D), or significantly increased the free energy of unfolding (ΔGD–N) relative to WT HBc1–149. This suggests HBc1–149 contains structural strain arising from frustrated contacts (27, 28). We found evidence of HBc1–149 adopting multiple native conformers, where capsid assembly-competent conformers were less stable than those incapable of, or attenuated in, capsid formation. Frustrated regions likely contain structural gatekeepers that (28), when mutated, subtly tuned the folding energy landscape and altered capsid assembly. The presence of multiple native conformations and frustrated regions may explain the origins of allostery reported for HBc. Frustration is likely an evolved tradeoff that balances the conflicting requirements of HBc folding with allosteric regulation of native structure, capsid formation, and diverse functions of different conformers (29). The ability to trap HBc conformers thermodynamically by mutation and ablate capsid formation provides a proof of principle for designing antivirals that elicit similar effects.
Results
Equilibrium Chemical Denaturation of HBc1–149.
To trap HBc1–149 in a dimeric form and inhibit capsid formation, we studied freshly purified HBc1–149 at low protein concentrations and in alkaline buffers with low ionic strength (Materials and Methods). Reductants were used to prevent the tendency of HBc to form oligomers with nonnative disulfide links (30). We previously used 3D transverse relaxation-optimized spectroscopy (TROSY) NMR spectroscopy to demonstrate WT HBc1–149 was folded and dimeric under these conditions, with very similar ligand-binding properties and helical content as HBc1–149 within capsids (15).
Equilibrium guanidinium chloride (GdmCl) titrations probed by far-UV CD spectroscopy showed two well-resolved unfolding transitions (Fig. 2A), hereafter transition 1 and transition 2, with denaturation midpoints ([GdmCl]50%) of 1.97 ± 0.03 M and 5.32 ± 0.06 M, respectively. The m values for transitions 1 and 2 were similar (3.1 ± 0.1 and 3.6 ± 0.2 kcal/mol⋅M). Because m values correlate well with changes in accessible surface area on unfolding, transitions 1 and 2 involve denaturation of similar amounts of structure (31). The far-UV CD spectrum of N HBc1–149 mirrored reported spectra, whereas the D ensemble had a random coil spectrum (32) (Fig. 2B). The well-defined plateau separating transitions 1 and 2 was consistent with a populated I containing ∼50% of the helical content of N (Fig. 2B).
Fig. 2.

Equilibrium chemical denaturation of HBc1–149. (A) GdmCl denaturation of HBc1–149 measured by far-UV CD (▲) and fluorescence emission spectroscopy (○). Solid lines are fits to an equation describing a linear three-state transition with a dimeric intermediate (SI Materials and Methods). (B) Far-UV CD spectra of HBc1–149, corresponding to the main species populated in different GdmCl solutions: 0 M (black), 1.5 M (magenta), 4 M (green), and 7.5 M (cyan). The difference spectrum (Spectrum0M − Spectrum4M) is a dashed purple line. (C) FES spectra of the species populated in GdmCl titrations (using the same colors as in B). The spectra of assembled capsids (black dashed line) and HBc1–149 in 0.5 M GdmCl (dark blue line) are similar, consistent with salt-induced capsid formation (32, 36). (D) Raw MST data for chemical denaturation of HBc1–149. Fluorescence is measured for 5 s before and after applying a 30-s heating pulse (“Laser on/Laser off”), which creates a temperature gradient of 2–3 °C (33). The MST signal is typically determined from the normalized fluorescence intensity changes between any two points (here, A and B) after the temperature gradient has been established (34). Typically, point A is selected to be a time-point shortly after the laser is switched on, whereas point B is some time after significant thermophoresis has occurred. The measured properties for N (green), I (blue), and D (purple) are shown. (E) GdmCl titrations of HBc1–149 measured by far-UV CD (▲) and MST (◇). Solid lines are best fits to a three-state transition (as in Fig. 2A). (F) Urea denaturation of HBc1–149 in the presence (◇) or absence (▲) of 1 M NaCl. Solid circles indicate species with capsid-like spectra. A.U., arbitrary units.
Fluorescence emission spectroscopy (FES) experiments also showed two HBc1–149 transitions with similar [GdmCl]50% and m values as far-UV CD experiments (Fig. 2A). The FES spectrum of native HBc1–149 was highly quenched and blue-shifted but became increasingly red-shifted with each transition, consistent with fluorophores being displaced from hydrophobic environments (32) (Fig. 2C). The m values and [GdmCl]50% values were independent of excitation and emission wavelengths (Fig. S1), suggesting no additional intermediates were significantly populated.
Microscale Thermophoresis: A Fast, Efficient Way to Study Protein Stability.
To test for additional states invisible to FES and far-UV CD spectroscopy, we used microscale thermophoresis (MST) to probe HBc1–149 denaturation. Although MST was developed for measuring molecular interactions (33), we thought it would be an excellent probe of global structural changes during protein folding and unfolding because it reports on effects arising from changes in macromolecular shape and solvation shell (34).
MST measurements involve loading samples into a series of glass capillaries. An IR laser creates a local temperature gradient of 2–3 °C, along which macromolecules migrate to a new equilibrium distribution depending on their size, shape, and charge (33). This redistribution of proteins within the temperature gradient (thermophoresis) is measured using intrinsic fluorescence within a focal volume of just a few microns (34). Critically, it is the distribution of molecules along this gradient that is usually studied rather than bulk fluorescence (Fig. 2D) (33, 34).
Chemical denaturation of HBc1–149 probed by MST (Fig. 2E) showed two transitions, with [GdmCl]50% and m values consistent with those obtained using FES and CD (Fig. 2A). Although we cannot exclude a population of additional intermediates, we consider it unlikely, given the excellent concordance between techniques that probe polypeptide secondary (CD), tertiary (FES), and global (MST) structure. Our combined denaturation data support concerted folding of HBc1–149 in a three-state transition with a dimeric α-helical intermediate.
MST experiments use disposable glass capillaries and small sample volumes (∼4 μL) (33), thus eliminating the lengthy cuvette equilibration and cleaning steps inherent to most titration strategies. Our MST measurements used 40- to 200-fold less protein than conventional methods, did not require labeled protein, and took only 15–20 min (34) (Materials and Methods). Thus, a single operator can measure up to 20 denaturant titrations per day, an impossible feat using conventional instruments.
Ionic Strength and Capsid Assembly Do Not Affect the Stability of Dimeric HBc1–149.
A feature of GdmCl titrations probed by FES was a hump in the baseline preceding transition 1 (Fig. 2A). The FES spectrum of this species was identical to that of HBc1–149 CLPs (32) (Fig. 1C). Because HBc1–149 capsid formation is promoted by high salt concentrations, it seemed probable that GdmCl (an ionic chaotrope) induced capsid formation at low concentrations but ablated assembly at higher concentrations. To test this hypothesis, we measured HBc1–149 denaturation in urea (a nonionic denaturant) in the presence and absence of 1 M NaCl (Fig. 2F). Only one urea-induced transition was observed (Fig. 2F and Fig. S1) with the same spectroscopic properties and stability (ΔG of ∼6.2 kcal/mol) as transition 1 in GdmCl titrations. These data suggest the same unfolding event is observed using urea and GdmCl, with transition 1 only being observed with urea (which has half of the denaturant activity of GdmCl) (35).
In the absence of urea, 1 M NaCl induced the population of a species with an FES spectrum that matched HBc1–149 capsids (Fig. S1C), consistent with reports of salt-induced capsid formation (32, 36). However, even low concentrations of urea (e.g., 0.5 M) caused the spectrum to revert to that of native HBc1–149 (Fig. S1C). NaCl (1 M) had no effect on the stability of the urea-induced transition of HBc1–149 (Fig. 2F). These data showed that any capsid assembly induced by low GdmCl concentrations was minor and did not affect transition 1 and that the stability of native HBc1–149 was independent of ionic strength (in contrast to capsid formation) (24, 32).
Determining the Stoichiometry of the HBc1–149 Unfolding Mechanism.
Native HBc1–149 has an extensive (hydrophobic) dimerization interface with a disulfide link between monomers (19) (Fig. 1A). Because equilibrium denaturation of HBc1–149 follows an apparent three-state transition, three linear on-pathway schemes could describe HBc1–149 unfolding (off-pathway schemes are considered in Discussion):
To distinguish between these schemes, we measured the effects of protein concentration on HBc1–149 denaturation. A transition involving a change in oligomeric state will have a [GdmCl]50% value that depends on protein concentration (37). FES measurements were used to ensure a good signal-to-noise ratio over a range of protein concentrations (Fig. 3 and Fig. S2), which was not possible using CD spectroscopy. The hump in the native baseline caused by GdmCl-induced capsid formation was more apparent at higher HBc1–149 concentrations (Fig. 3A). However, these capsids did not intrude with transition 1 over the concentration range examined (because they were not populated at >1 M GdmCl). Capsid assembly was apparent only above 3 μM monomer, consistent with the reported pseudocritical concentration for capsid assembly (24, 38) (Fig. 3A, Inset).
Fig. 3.

Effects of protein concentration on chemical denaturation of HBc1–149. (A) GdmCl-induced denaturation over a 12.5-fold concentration range of HBc1–149. For clarity, these data are shown on a normalized scale (raw data are reported in Fig. S2). Each dataset is fitted to an equation (solid lines) describing a linear three-state transition with a dimeric intermediate (Materials and Methods and SI Materials and Methods). (Inset) Ratio of native baseline signal to the signal at 0.45 M GdmCl (where GdmCl-induced capsid formation was maximal). These data define a pseudocritical concentration of ∼3 μM (monomer) (24, 36, 38). (B) Transition 1 was independent of protein concentration (black circles), but transition 2 [GdmCl]50% values (red circles) were markedly dependent on HBc1–149 concentration. These data are consistent with a change in oligomeric state of HBc1–149 during transition 2.
Transition 1 thermodynamic parameters were independent of protein concentration. By contrast, the transition 2 [GdmCl]50% (but not its m value) increased significantly as a function of HBc1–149 concentration (Fig. 3B). These data support scheme 3 and are inconsistent with schemes 1 and 2. Further supporting scheme 3, removing the interdimer disulfide link (with the C61A mutation) destabilized transition 2 by ∼1 kcal/mol (Table 1), consistent with the destabilization expected for deleting a disulfide link (39). Fitting the WT GdmCl titration to an equation describing scheme 3 (SI Materials and Methods) defined values for ΔGI−N of ∼6.2 kcal/mol (transition 1) and for ΔGD−I of ∼26 kcal/mol (transition 2, where ΔGD−I = {−RT⋅ln([Protein]) + mD−I⋅[GdmCl]50%}, R is the gas constant, and T is the temperature in K) (37, 40). The free energy of denaturation for HBc1–149 (ΔGD−N of ∼32 kcal/mol) accords well with values reported for similarly sized oligomers (3, 40). Our data support the dimerization interface being at least partially intact in I (thus shielding the disulfide from reductants) but not in D.
Table 1.
Protein engineering studies of HBc1–149 stability
| Construct | Transition 1 [GdmCl]50%, M |
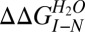 , kcal⋅mol−1 , kcal⋅mol−1
|
Transition 2 [GdmCl]50%, M |
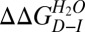 , kcal⋅mol−1 , kcal⋅mol−1
|
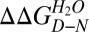 , kcal⋅mol−1 , kcal⋅mol−1
|
| Neither transition affected by mutation | |||||
| WT | 1.97 ± 0.03 | — | 5.32 ± 0.04 | — | — |
| S87G | 1.94 ± 0.04 | 0.1 ± 0.1 | 5.32 ± 0.05 | 0.0 ± 0.2 | 0.1 ± 0.3 |
| K96A | 1.92 ± 0.05 | 0.2 ± 0.2 | 5.40 ± 0.06 | −0.3 ± 0.3 | −0.1 ± 0.3 |
| Only transition 1 affected by mutation | |||||
| K7A | 2.22 ± 0.04 | −0.8 ± 0.2 | 5.31 ± 0.06 | 0.0 ± 0.3 | −0.7 ± 0.3 |
| C48A | 1.59 ± 0.06 | 1.2 ± 0.2 | 5.26 ± 0.07 | 0.2 ± 0.3 | 1.4 ± 0.4 |
| L60V | 2.74 ± 0.03 | −2.4 ± 0.2 | 5.26 ± 0.06 | 0.2 ± 0.3 | −2.2 ± 0.3 |
| S87D | 2.19 ± 0.04 | −0.7 ± 0.1 | 5.25 ± 0.06 | 0.3 ± 0.3 | −0.4 ± 0.3 |
| G111A | 1.30 ± 0.04 | 2.1 ± 0.2 | 5.21 ± 0.04 | 0.4 ± 0.2 | 2.5 ± 0.3 |
| Y132A | 3.42 ± 0.04 | −4.5 ± 0.2 | 5.33 ± 0.07 | 0.0 ± 0.3 | −4.6 ± 0.4 |
| Only transition 2 affected by mutation | |||||
| C61A | 1.94 ± 0.04 | 0.1 ± 0.1 | 5.10 ± 0.05 | 0.9 ± 0.3 | 0.9 ± 0.3 |
| Transitions 1 and 2 affected by mutation | |||||
| L16A | 1.29 ± 0.06 | 2.1 ± 0.2 | 4.97 ± 0.05 | 1.3 ± 0.3 | 3.4 ± 0.3 |
| F23A† | 2.60 ± 0.14 | −1.4 ± 0.6 | 4.69 ± 0.98 | 11.7 ± 2.3 | 10.3 ± 2.3 |
| L30A* | 1.48 ± 0.03 | −0.4 ± 0.2 | 5.11 ± 0.15 | 7.4 ± 0.4 | 7.0 ± 0.4 |
| L42A* | 2.96 ± 0.07 | −9.4 ± 0.4 | 4.95 ± 0.46 | 11.8 ± 0.7 | 2.4 ± 0.8 |
| L55A† | 1.22 ± 0.87 | 4.7 ± 1.1 | 5.13 ± 0.12 | 2.3 ± 1.8 | 7.0 ± 2.1 |
| L65A | 1.60 ± 0.03 | 1.2 ± 0.1 | 5.10 ± 0.05 | 0.8 ± 0.2 | 2.0 ± 0.3 |
| L68A | 1.37 ± 0.05 | 1.9 ± 0.2 | 5.01 ± 0.05 | 1.1 ± 0.2 | 3.0 ± 0.3 |
| W71A | 1.71 ± 0.06 | 0.8 ± 0.3 | 5.11 ± 0.07 | 0.8 ± 0.3 | 1.6 ± 0.4 |
| V72A | 2.16 ± 0.06 | −0.6 ± 0.2 | 5.10 ± 0.10 | 0.8 ± 0.4 | 0.2 ± 0.4 |
| F97A† | 0.65 ± 0.98 | 5.2 ± 1.4 | 4.69 ± 0.11 | 7.1 ± 1.7 | 12.3 ± 2.2 |
| F97L* | 1.89 ± 0.04 | −1.5 ± 0.4 | 5.12 ± 0.11 | 5.4 ± 1.7 | 3.9 ± 1.8 |
| L100A | 1.74 ± 0.04 | 0.7 ± 0.1 | 5.08 ± 0.04 | 0.9 ± 0.2 | 1.6 ± 0.3 |
| C107A* | 1.72 ± 0.04 | 0.6 ± 0.2 | 4.93 ± 0.05 | 1.8 ± 0.3 | 2.5 ± 0.3 |
| F122A | 1.18 ± 0.06 | 2.5 ± 0.2 | 5.00 ± 0.04 | 1.1 ± 0.2 | 3.6 ± 0.3 |
Most HBc1–149 mutants had essentially identical mI−N and mD−I values. Thus, unless otherwise stated, GdmCl titrations were fitted using the mean mI−N and mD−I values to improve precision (3.1 ± 0.1 and 3.6 ± 0.2 kcal/mol⋅M, respectively) (26). Errors on individual ΔGX−Y values typically ranged from 0.1 to 0.4 kcal/mol. To ensure conservative treatment of data, we assumed ΔΔG values were significant when they were >0.5 kcal/mol or the SE of fitting (whichever was greatest).
Denaturation curves for these proteins were fitted using a constrained mD−N value (determined from 17 independent measurements) to improve fitting precision (26).
These constructs had mI−N and mD−I values significantly different from most HBc1–149 mutants. Thus, thermodynamic parameters for these mutants were obtained using values obtained from free fits to individual titrations.
Gas Phase Unfolding of HBc1–149.
We, as well as others, have used multiangle laser light scattering (MALS) to measure the oligomeric state of native HBc1–149 dimers, capsids, and capsid assembly intermediates (15, 24). MALS experiments use refractive index measurements to determine the protein concentration. Unfortunately, the high refractive indices of concentrated GdmCl solutions and low protein concentrations used (to inhibit HBc1–149 aggregation and capsid assembly) made it impossible to use MALS to measure the oligomeric state of I and D.
Thus, we used ion mobility spectrometry (IMS) in combination with noncovalent electrospray ionization (ESI) MS (hereafter ESI-IMS-MS) to study gas phase unfolding of HBc1–149. ESI-IMS-MS allows the separation of gas phase ions by virtue of their rotationally averaged collision cross-section (CCS), which depends on molecular shape. This approach has been used to interrogate protein ligand interactions, gas phase stabilization of protein complexes, and conformation-dependent dissociation (41–45). Using ESI-IMS-MS, the internal energy of HBc1–149 ions was increased by collisions with an inert buffer gas. High internal energy caused noncovalent interactions to break, leading to gas phase unfolding of the protein ion and increases in CCS that were detected as delayed ion mobility arrival times.
We compared the gas phase unfolding of the 10+ charge state ions of HBc WT and C61A (Fig. 4 and Fig. S3). By applying Gaussian fits to the ion mobility arrival time distribution (a detailed procedural discussion is provided in SI Materials and Methods), we determined the number of components present at each collision voltage and the CCS of each ion (42, 46) (Fig. S4). Although the collision energy used to invoke gas phase unfolding does not directly correspond to the chemical denaturant concentration used in solution experiments, it provides a convenient parameter that allows the relative population of different species to be tuned and graphically represented (Fig. 4). We identified four dimeric states for WT HBc1–149 (N, I1, I2, and D) that each had distinct arrival times and became more expanded as the protein unfolded (Fig. 4A). As expected, the most compact state, N, dominated at low collision energies, with I1, and thereafter I2 and D2, becoming more populated as the collision energy was increased (with a concomitant decline in N; Fig. 4). No further unfolding of D was observed even at very high energies.
Fig. 4.

Gas phase unfolding profiles of HBc1–149. (A) Abundance of each state as a function of the center-of-mass collision energy (Materials and Methods and SI Materials and Methods). At low energies, N was the dominant species (black squares). As collision energy was increased, the population of N decreased and a partially unfolded state (I1) was populated (red circles). Further increases in energy induced the population of state I2 (blue triangles), which then populated the unfolded state D (green triangles) at higher energies. (B) C61A underwent a very similar unfolding process until high energies were reached; at that point, dissociation into monomeric D occurred. This illustrates gas phase dissociation of the unfolded state depends on the presence of the intermolecular disulfide link. Error bars show the SD from triplicate measurements.
Importantly, the monomer of WT HBc1–149 was not observed in these gas phase experiments (Fig. S4), suggesting the intermolecular C61-C61 disulfide link was intact in all species (consistent with our not using reductants in MS experiments). IMS-MS analysis of C61A, which eliminates this disulfide link, showed an unfolding profile similar to that of WT HBc1–149 but with an additional species present that had a CCS consistent with being a dissociated HBc1–149 monomer (Fig. 4B and Figs. S3 and S4). The C61A monomer was observed only once dimeric D was formed.
The “extra” intermediate observed in ESI-IMS-MS but not seen in GdmCl titrations is most likely a kinetic intermediate not observed in solution experiments, which arose from the different experimental designs used. In ESI-IMS-MS experiments, the time between HBc1–149 being injected into the mass spectrometer and different conformers being detected was milliseconds. By contrast, HBc1–149 was mixed with GdmCl solutions and equilibrated overnight before titrations. Thus, there is a greater likelihood of seeing metastable species in ESI-IMS-MS. We cannot exclude the extra intermediate arising from a gas phase artifact or being present in solution (but unresolved). However, these possibilities seem unlikely because HBc1–149 capsid assembly was studied in the gas phase and agreed well with solution data (47, 48) and no additional intermediates were detected using CD, FES, and MST (Fig. 2).
Mutational Analysis of HBc1–149.
Our data show HBc1–149 folds from an unstructured D to a dimeric, helical I and, thereafter, to a dimeric N. Our a priori hypothesis was HBc1–149 contained structured domains of different stability, with each transition corresponding to denaturation of only one domain. We generated 29 HBc1–149 mutants to test this hypothesis and determine which residues stabilized different populated species (Table 1). Conservative point mutations were used that should not significantly alter the structure of D (26). Thus, significant free energy changes most likely report on changes in the relative stability of N and I (26, 49).
We were unable to study the folding properties of six X→A mutants (Y6A, F9A, W102A, F103A, L119A, and W125A) that colocalized to the hydrophobic core of the HBc1–149 contact domain (19) (Fig. 5A). These mutants expressed but were aggregation-prone and refractory to biophysical study. Although we could not quantitatively determine their energetic contribution to folding, these residues are clearly essential for HBc1–149 folding (consistent with their conservation) (19).
Fig. 5.

Mutational analysis of HBc1–149. (A) Conserved residues that, when mutated, caused aggregation and were refractory to study colocalized to a conserved hydrophobic core between the four-helix bundle and contact domains (orange) (19). (B) Mutations that altered transition 1 stability only. (C) GdmCl titrations for representative HBc1–149 mutants: WT; C61A, the only mutant to affect transition 2 only; L16A, which destabilized both transitions; and L60V, which stabilized transition 1 only. (D) GdmCl titrations of WT HBc1–149 and capsid assembly-incompetent mutants (F23A, L42A, and Y132A). (E) Light-scattering analysis of HBc1–149 constructs that reduce (K7A, L30A, and V72A) or knock out capsid assembly (F23A and L42A) compared with WT. After dialysis into a buffer that promotes in vitro capsid formation, samples were run on an analytical Superose 6 column. HBc1–149 capsids eluted at ∼8 mL (molar mass of ∼4 × 106 Da), and dimers eluted at 16 mL (molar mass of ∼3.4 × 104 Da). For clarity, each capsid peak was normalized to an arbitrary value of 1 (15, 17). dRI, differential refractive index. (F) Location of residues discussed in E is shown. The binding site of an antiviral peptide is indicated with a red arrow (14, 15) (associated calorimetry data are provided in Fig. S6).
Only four X→A mutants affected only transition 1 (K7A, C48A, G111A, and Y132A; Table 1). Notably, these residues were not colocalized and had solvent-exposed side chains on native HBc1–149 (assuming the capsid structure accurately represents that of the free dimer) (19) (Fig. 5 A and B). C61A, which removes the intermonomer disulfide link, was the only mutation that affected only transition 2 (Fig. 5C and Table 1). All other X→A mutants affected both transitions 1 and 2 and decreased the overall stability of N compared with WT HBc1–149 (ΔΔGD−N > 0; Table 1). Most mutations in this category destabilized both transitions (L16A, L55A, L65A, L68A, W71A, F97A, L100A, C107A, and F122A; Fig. 5C and Table 1), consistent with them destabilizing both N and I. This is expected for conservative mutations that remove side-chain moieties, making partial, native-like contacts in I. However, a subset of mutants (F23A, L30A, L42A, and V72A) stabilized N relative to I (ΔΔGI−N < 0, making I less populated) while destabilizing I relative to D (ΔΔGD−I > 0; Fig. 5D and Table 1). Similarly, K7A and Y132A, which are solvent-exposed side chains in structures of dimeric HBc1–149 (19), stabilized N but did not affect the stability of I (ΔΔGD−I of ∼0; Fig. 5D). This unusual behavior is consistent with deletion of side-chain moieties at these loci, alleviating structural strain and differentially affecting the stability of N and I.
Surprisingly, F23A and L42A did not form capsids and purified as stable dimers, even in buffers that normally induce HBc1–149 capsid assembly (Fig. 5E and Table S1), a phenomenon reported for HBc1–149 Y132A (50). Although Y132 and F23 from opposing dimers interact directly within capsids, L42A is buried within the hydrophobic core of the HBc1–149 dimer and distant from capsid-stabilizing interdimer contacts (Figs. 1B and 5F). Intriguingly, N was stabilized relative to I for these assembly-incompetent mutants (Fig. 5D and Table 1). This suggests capsid assembly can be tuned by modulating the thermodynamics of the HBc1–149 dimer, including mutation of residues on the interdimer interface (F23 and Y132) or residues distant from it (L42; Discussion). Probing this trend further, equilibrium capsid assembly studies revealed other “unusual” mutants (e.g., K7A, L30A, V72A) with compromised capsid formation and increased dimer populations (Fig. 5F and Table S1). The result for V72A was surprising because this residue is located far from interdimer contacts that stabilize capsids (Fig. 5F). Our data suggest the presence of structural strain in N or I that, when modulated, affects capsid formation, ranging from attenuation (K7A, L30A, and V72A) to complete ablation (F23A, L42A, and Y132A).
Phenotypic Mutant Analysis.
We then analyzed HBc mutants that yield viruses with altered phenotypes in patients with chronic HBV infections to see if these phenotypes had origins in the thermodynamics of the native dimer (Table 1). F97L enhances HBV capsid assembly and causes premature virion secretion (25, 51–53). L60V causes low-level virion secretion in vivo (53, 54). S87G eliminates a phosphorylation site and is associated with lower levels of capsid formation than phosphorylated WT HBc (53, 55).
S87G had no effect on the stability of either transition (Table 1). Because the S87G phenotype could arise from loss of an important phosphorylation site, we generated S87D to mimic phosphoserine (18). However, S87D had only marginal effects on ΔΔGD−N. F97A significantly destabilized transitions 1 and 2 (Table 1), showing F97 makes significant contributions to stabilizing I and N (consistent with its buried locus in the dimerization interface). However, F97L significantly stabilized transition 1 and destabilized transition 2, showing this mutation destabilized I more than N. By contrast, L60V significantly stabilized N, although it had no effect on I (Fig. 5C and Table 1). It was surprising L60V increased ΔΔGD−N, given its solvent-exposed location on the native dimer (Fig. 5B). F97L slightly increased the extent of capsid assembly (25), but S87D and L60V were WT-like (Table S1).
Discussion
HBc1–149 Folds via a Structured, α-Helical Intermediate.
HBc1–149 is one of the largest, most complex proteins for which extensive protein engineering and stability studies have been performed (3). Despite its complexity and aggregation tendencies, there was excellent concordance in thermodynamic parameters obtained using techniques that probed its secondary (CD spectroscopy), tertiary (FES), and global (MST) structure (Fig. 2). Our data are consistent with a three-state folding mechanism involving a concerted loss of secondary and tertiary structure. Empirically derived relationships between m values and polypeptide length (31, 56) suggest dimeric HBc1–149 (containing two 149-residue monomers) should have an mD−N value of ∼7.1 kcal/mol⋅M, in excellent agreement with our experimental value (mD−N = 6.7 ± 0.2 kcal/mol⋅M for WT) (Fig. 2).
The transition 2 [GdmCl]50% value was dependent on protein concentration, suggesting dimer-to-monomer dissociation occurs in this transition (37, 40) (Fig. 3). C61A, which deletes the intermolecular disulfide link, was the sole mutation that affected only transition 2 (Fig. 5C). Thus, the four-helix bundle dimerization interface must be at least partially structured in the folding intermediate (with the disulfide intact but not needed for dimerization). However, dimerization is a prerequisite for folding, consistent with folded HBc1–149 monomers never being reported in solution. Thus, native HBc1–149 unfolds through at least one helical, dimeric intermediate that yields an unstructured ensemble on denaturation.
Structural Properties of the Intermediate.
Ultimately, a structural model of the HBc1–149 folding intermediate needs to reconcile the following: (i) residues from all regions of HBc1–149 could modulate the stability of transitions 1 and 2 (Table 1); (ii) the folding intermediate is an α-helical homodimer; (iii) mutation of some solvent-exposed residues increased native stability compared with WT HBc1–149 (ΔΔGD−N < 0; Table 1); and (iv) m values and far-UV CD spectra show that significant burial of hydrophobic surface area and helical structure formation occurs in each folding transition.
Our a priori expectation was HBc1–149 contains independent “foldons” (likely the dimeric four-helix bundle and contact domains; Fig. 1A), where each folds and unfolds independently (57), yielding a three-state transition. We define a foldon as a domain that folds independently as a discrete, stable entity. This is distinct from a structural domain, a coherent unit of structure recognized during inspection that does not fold independently. In this scenario, transitions 1 and 2 would report on the denaturation of only one foldon each and mutations within a foldon would affect only the associated transition. However, the mutations that affected only transition 1 did not colocalize to one domain (Fig. 5B), and the sole mutation that affected transition 2 only was C61A, which destabilized the dimer interface (Figs. 1A and 5C and Table 1). Thus, I is not composed of a single foldon, and our data exclude a mechanism where the four-helix bundle and contact domains fold independently (Fig. S5A). This is not surprising, given the extensive hydrophobic contacts between these domains. We can also exclude mechanisms where I loses tertiary packing but maintains secondary structure, or vice versa (Fig. S5B), because we found mutations that perturb transition 1 or 2 only, m values show a significant loss of hydrophobic packing during each transition, and I has ∼50% of the helicity of N.
It is more likely that I contains structured and unstructured regions, where the four-helix bundle and contact domains are partially formed (Fig. 6A) and contain largely native-like interactions. This explains why most mutations had similar effects on the stability of N and I (Table 1). However, superimposed on these native-like interactions is structural strain arising from frustrated contacts in N, explaining why some mutations stabilized N relative to I or increased the relative stability of N compared with WT HBc1–149 (Table 1 and Fig. 5 C and D). Because I is not a single foldon, its structure provides a thermodynamic linkage between the four-helix bundle and contact domains, which, together with strain in N, may facilitate the long-range structural rearrangements and allostery of HBc reported by many laboratories (discussed in detail below) (15, 21–23).
Fig. 6.

Three-state folding of HBc1–149. (A) Unfolded HBc1–149 monomers interact and form a partially structured, dimeric helical intermediate. This intermediate can thereafter fold to multiple native structures. One native conformer (HBcAss) can form capsids, whereas the other is assembly-incompetent (HBcInc). The occupancy of an alternative native structure can be tuned thermodynamically by point mutations, thereby modulating the extent of capsid formation. (B) Comparison of a previously reported crystal structure for Y132A (21) (cyan; PDB ID code 3KXS) with one determined by us under different conditions (pink; PDB ID code 4BMG). Arrows show structural variations between the dimers. These data demonstrate that native HBc1–149 dimers exhibit considerable structural variability, even within the same protein sequence. (C) Close-up of a Y132A dimer within the hexameric unit shown in B that highlights the structural differences between the same protein under different crystallization conditions.
Here, we assume I is on-pathway to the native state and a native-like structure accretes during folding (Fig. 6A). However, it is also possible that I could be off-pathway and N unfolds directly to D (and vice versa; scheme 4):
In the off-pathway scenario, I, rather than N, may have nonnative or strained regions, explaining why some mutants had more profound effects on the stability of I than N (Table 1), something that would not be expected if I contained only native-like interactions. An off-pathway I could, however, be a structural stepping-stone between alternative native conformers, where breathing from one N to the strained I provides access to another N within a different region of the energy landscape (29). This landscape could be tuned by HBc interacting with cognate partners encountered during viral replication, thus mediating diverse functions. We believe that two-state folding between D and N is rather unlikely for a large homodimer like HBc1–149. This, together with many mutations showing evidence of native-like interactions in I (on which strained, nonnative contacts are superimposed), leads us to believe the on-pathway folding scenario (scheme 3) is more likely.
Connections Between Folding, Allostery, and Capsid Assembly.
HBV has a very compact genome, and just four genes express seven viral proteins (9). HBV replicates efficiently because its few proteins (including HBc) have multiple functions. HBc not only forms capsids (and shuttles the viral genome within and between hosts) (18–20) but regulates viral replication in precapsid conformations (8) and communicates events occurring within capsids to the exterior (22), such that virion envelopment occurs at the correct cellular location (and only when genome replication is complete). Further, HBV also secretes an E antigen (HBe), a variant of HBc1–149 believed to confer immune tolerance during infections (58). Except for 10 extra residues at its N terminus, HBe has a sequence identical to HBc1–149. However, oxidized HBe is dimeric with a dimer interface that has its monomers rotated through ∼140° compared with HBc (58). Thus, structural plasticity appears inherent to HBc functions.
HBc has been described as a series of linked helical rods whose structure is modulated by interacting with other biomolecules: Interaction of its C terminus with viral RNA/DNA inside capsids induces profound structural changes within the four-helix bundle (and around its base) (22). Similarly, we previously used TROSY NMR spectra to show antiviral peptides that bind to the tip of the four-helix bundle induce long-range structural rearrangements within this bundle (and adjacent regions of contact domains) (15). Others have shown that inserting short linkers at the tip of the four-helix bundle mediates a 40° rotation of the contact domain relative to the bundle (within capsids) (23). Further, small molecules have been reported that bind to HBc and induce global structural rearrangements that impair HBV capsid formation (11–13, 50).
We initially thought F23A and Y132A abrogated capsid formation solely by destabilizing interdimer contacts (Fig. 1B). However, although this may contribute toward inhibiting capsid formation, other factors also play a role. For example, truncation of a solvent-exposed side chain would not normally be expected to stabilize native HBc1–149, yet this is what was observed for Y132A. Similarly, the F23A mutation destabilized I much more than N, suggesting the presence of nonnative interactions. These data suggest that F23 and Y132 not only stabilize interdimer contacts that support capsid formation but carry an energetic penalty for protein folding and stability. However, L42A also completely inhibited capsid formation (Fig. 5E), despite being buried in the hydrophobic core and distant to interdimer contacts that stabilize capsids (residues 128–143) (19) (Figs. 1B and 5F). Thus, the inability of L42A to form capsids cannot be attributed to the deletion of interdimer contacts. Like F23A, L42A destabilized N much less than I and affected the m values of both transitions compared with the WT, suggesting structural rearrangements of N and I (Fig. 5D and Table 1). We speculate that L42A, F23A, and Y132A elicit structural rearrangements of HBc1–149 that inhibit capsid formation, perhaps by promoting the population of HBcInc (the capsid assembly-incompetent conformer).
Evidence of structural plasticity of HBc1–149 is clearly seen in the previously reported Y132A crystal structure, which formed a trimer of dimers in an open triangle arrangement (effectively a single turn of the right-handed helix) (21). The structures of individual Y132A dimers were quite different, and also different from dimer structures in WT HBc1–149 capsids. We crystallized Y132A in a different form, a closed trimer of dimers resembling the threefold axes of HBV capsids [Protein Data Bank (PDB) ID code 4BMG; Fig. 6 B and C], where Y132A dimers have significant structural differences compared with Y132A dimers in the open triangle form. Although some structural diversity may arise from lattice contacts, the structural data support significant structural plasticity of HBc1–149 at multiple loci remote from these contacts (21) (Fig. 6 B and C).
Unfortunately, it has proved impossible to obtain diffraction-quality protein crystals for L42A, which prevented us from directly observing mutation-induced structural changes. Interestingly, L42A significantly reduced HBc1–149 affinity for antiviral peptides (Kd of ∼11 μM) compared with WT (15), F23A, and Y132A (Kd of ∼2 μM, ∼3 μM, and ∼3 μM, respectively; Fig. S6A), even though the peptide binds to the four-helix bundle tip (∼37 Å away from L42; Fig. 5F). Because L42A affects the interdimer contacts stabilizing capsids and also ligand binding, with both events occurring at loci remote from the mutational site, it seems reasonable that L42A elicits allosteric structural rearrangements in HBc1–149. Consistent with this hypothesis, L42A induced massive, long-range chemical shift perturbations (CSPs) in amide resonances in 2D TROSY spectra (relative to WT values) (15) (Fig. S6B). These CSPs mapped to the base of the four-helix bundle and also to the bundle itself (Fig. S6C).
Recent hydrogen/deuterium exchange MS experiments showed that the kinked helix containing L42 had the most significant breathing motions of any part of HBc1–149 (within capsids), with ligand binding at distant loci inducing more profound alterations in H/D exchange at this helix than at any other region (59). In addition, L42 lies close to a hydrophobic pocket that binds sulfanilamide derivatives that inhibit HBV capsid assembly (60). Thus, there is strong precedent for HBc1–149 being malleable in this region, and it is possible that mutations in this region elicit structural rearrangements that interfere with capsid assembly. The structural data support long-range structural arrangements within the four-helix bundle (Fig. S6) and evidence of alternative packing of contact domains at different angles relative to this bundle (11–15, 22, 23). Our data suggest native HBc1–149 is on a structural knife-edge, likely arising from structural strain, where small perturbations can “flip” the native structure into a different conformer with altered tertiary packing (and functions). This flipping may be achieved by the movement of proposed “subdomains” around glycine hinges, as seen for Y132A and the different WT conformations seen in the capsid (21). Truncation of the L42 side chain may affect the conformational freedom of the N terminus, which is highly dynamic in NMR spectroscopy, a motion that may be used in generating HBe (which has a grossly altered N-terminal structure) (15, 21, 22, 58).
We used these findings to predict other HBc1–149 mutations exhibiting reduced capsid assembly on the basis of their thermodynamic parameters. Whereas other mutants showed WT-like proportions of capsid and dimer in an equilibrium assembly study, we found that all X→A mutations that stabilized N relative to I (L30A, K7A, and V72A) had increased proportions of dimer (Fig. 5 E and F and Table S1). We note that K7 and L30, like L42, lie at the base of the four-helix bundle and, when mutated, may elicit allosteric effects in a similar manner to L42A (Fig. 5F and Fig. S6). However, the V72A result (while modest) was surprising, because this residue is located near the top of the four-helix bundle and remote from capsid-stabilizing interdimer contacts. This is consistent with our reported NMR studies showing peptide binding to the tip of the four-helix bundle induced long-range allosteric regulation of HBc1–149 structure within this bundle and around helix 2 (15).
Two of the mutants reported here (L60V and F97L) are reported phenotypic mutants (54). L60V stabilized N greatly but is capsid assembly-competent (Table S1), suggesting HBcAss and HBcInc may be stabilized by similar amounts. Thus, the low secretion phenotype associated with L60V may arise from making other functionally important conformers inaccessible or by compromising interactions with key-binding partners. F97L stabilized N relative to I but slightly increased the equilibrium proportion of capsids (Table S1), consistent with reports of enhanced in vitro capsid assembly and immature virion secretion (25, 51, 52). Because F97 is buried within the hydrophobic core of the four-helix bundle, a structurally diverse region reported to be allosteric (15), it may be that F97L simply accelerates capsid assembly by promoting occupancy of HBcAss (25).
Conclusion
It is clearly efficient for HBV to evolve structurally promiscuous proteins that adopt a range of structures to meet diverse functional demands. For HBc, this structural diversity is likely a thermodynamic balancing act, wherein the diverse structure–function requirements cause structural strain and frustrated contacts (29). The reported structural data for HBc1–149 point strongly toward its having a broad conformational ensemble whose properties can be tuned by solution conditions, ligand binding, domain boundaries (e.g., HBc vs. HBe), or mutations. It is likely that F23, L42, and Y132 are structural gatekeepers (28) that ensure WT HBc adopts a capsid assembly-active conformation (HBcAss). Mutation of these residues releases conformational restraints and promotes the adoption of a less strained native conformer (HBcInc) unable to form capsids or access conformations needed to function properly (Fig. 6A). Conversely, other residues (e.g., L60) may help HBc1–149 adopt other functional states.
The free energy change accompanying capsid assembly (∆Gcap) arises from the energetic tradeoff between making favorable contacts between HBc dimers (ΔGcontact) and unfavorable conformational rearrangements of HBc (∆Gconf) between assembly-inactive (HBcInc) and assembly-active (HBcAss) states (i.e., ∆Gcap = ∆Gcontact − ∆Gconf) (25, 36). This payoff makes dimer–dimer contacts relatively weak (Kd of ∼6 mM, equating to ΔGcap of −2.9 kcal/mol at 21 °C) yet yields stable capsids by summation of many weak contacts (21, 24).
Thus, only small energies are needed to regulate HBc structure and capsid assembly, presenting an “Achilles’ heel” that can be therapeutically targeted. It is likely that drugs to treat HBV could be developed that work by overstabilizing one HBc conformation, thereby throwing its regulatory system out of balance. Our findings provide thermodynamic proof of principle for such small molecules and the means to assay which species small molecules bind to. This could help identify new molecules targeting a specific HBc conformation that could be then tested for antiviral activity, helping the quest for much needed antivirals. Our work suggests HBc1–149, with its complex structural dynamics and therapeutic importance, is an excellent system for computational modeling of folding, allostery, and functional regulation of large oligomeric proteins.
Materials and Methods
Reagents.
A detailed list of reagents is provided in SI Materials and Methods.
Recombinant Protein Expression and Purification.
WT and capsid assembly-competent HBc1–149 variants were expressed in Escherichia coli and purified as described previously (15, 17, 32). This involved an ammonium sulfate precipitation, followed by sequential size-exclusion columns that resolved HBc1–149 capsids and dimers, respectively. Mutants defective at capsid assembly (F23A, L42A, and Y132A) were purified as dimers using a modified version of a reported protocol [involving ammonium sulfate precipitation, cation exchange, and size-exclusion chromatography (SEC); SI Materials and Methods] (50). The final purification step in all protocols was SEC in buffer A [30 mM sodium bicarbonate buffer (pH 9.5), 2 mM DTT], which yielded dimeric HBc1–149 at >95% purity (as determined by SDS/PAGE analysis). HBc protein concentrations are reported as monomer concentrations.
Equilibrium Chemical Denaturation.
Urea and GdmCl solutions were made gravimetrically in buffer A (but substituting 2 mM DTE instead of DTT for CD spectroscopy) and using HBc1–149 at a protein concentration of ∼7.5 μM (unless stated otherwise). Samples were equilibrated overnight at 20 °C before making measurements. Far-UV CD spectroscopy was performed using a JASCO J810 spectropolarimeter (Jasco) and 1-mm pathlength cuvettes. FES measurements were made using an Aminco SLM2 spectrometer (ThermoFisher) with excitation and emission wavelengths of 280 nm and 330 nm, respectively (unless stated otherwise).
MST.
Samples (32-μL sample size) of 7.5 μM HBc1–149 containing various GdmCl concentrations were made (as described above). Four microliters was loaded by capillarity into standard MST-grade capillaries, and MST experiments were performed using a Monolith.LabelFree instrument (NanoTemper, Inc.) (34). A thermal gradient of ∼2–3 °C was induced (33) over a period of 30 s using an IR laser (20% intensity setting). Intrinsic tryptophan and tyrosine fluorescence was excited at 280 nm (light-emitting diode set to 20% intensity). Emission at 360 nm was recorded for the entire heating period, and for an additional 5 s before and after heating, to resolve thermophoresis from intrinsic temperature-dependent changes in fluorescence emission (34) (Fig. 2D). Data from two sequential runs were concatenated, with 16 GdmCl concentrations measured per run (the maximum number held in a magazine). Multiple overlapping concentrations were used to ensure accurate data scaling between runs.
Data Analysis and Curve Fitting.
Equilibrium denaturant titrations were analyzed using SigmaPlot (Systat Software) and fitted to an equation describing scheme 3: a three-state transition where dimeric N unfolds to a dimeric I to monomeric D (more details are provided in SI Materials and Methods). Free energy values were calculated by the following equations.
For transition 1:
where 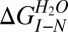 is the Gibbs free energy difference between I and N, mI−N is the dependence of the free energy on denaturant concentration, and [GdmCl]50% is the transition midpoint.
is the Gibbs free energy difference between I and N, mI−N is the dependence of the free energy on denaturant concentration, and [GdmCl]50% is the transition midpoint.
For transition 2 (including dimer-to-monomer dissociation):
where  is the free energy difference between D and I, R is the gas constant, and T is the temperature (in K). All mutants with WT-like m values were fitted to a common averaged value. For the few mutants that had different m values, and where using an average m value was inappropriate, data were free-fitted or fitted assuming the total mD−N value was the same as the mean mD−N value (but with mI−N and mD−I floating freely during fitting). More details are provided in Table 1 and SI Materials and Methods.
is the free energy difference between D and I, R is the gas constant, and T is the temperature (in K). All mutants with WT-like m values were fitted to a common averaged value. For the few mutants that had different m values, and where using an average m value was inappropriate, data were free-fitted or fitted assuming the total mD−N value was the same as the mean mD−N value (but with mI−N and mD−I floating freely during fitting). More details are provided in Table 1 and SI Materials and Methods.
Solution-State Molecular Mass Determination.
To promote capsid assembly, 100 μM stocks of HBc1–149 variants (or 400 μL for F23A and L42A) were dialyzed overnight at 20 °C in 0.1 M Hepes (pH 7.5; ionic strength adjusted to 250 mM with NaCl) (15). SEC combined with MALS measurements was performed at 20 °C using a Wyatt Heleos II light-scattering detector and Optilab refractometer (Wyatt) and an in-line Superose 6 10/300 GL SEC column (GE Healthcare). The molar mass of different species was determined using the manufacturer’s software.
ESI-IMS-MS.
All mass spectra and IMS-MS data were collected using a Synapt HDMS quadrupole-traveling wave TOF mass spectrometer (Micromass Ltd.). HBc1–149 solutions (8 μM) were electrosprayed from 20 mM ammonium acetate (pH 7.5). To construct gas phase unfolding profiles, the 10+ charge states for WT HBc1–149 and C61A were mass-selected and subjected to increasing collision voltages in the trap. Collision voltages of 6–54 V (in 2-V increments) were applied, and unfolding was probed by measuring changes in arrival time distribution of ions traversing the traveling-wave device. The contribution of all species populated at each collision energy was calculated by fitting the minimum number of Gaussian curves to the arrival time distribution. The components (unfolding states) were then correlated between collision energies, identified as N, I1, I2, or D, and plotted as a function of center-of-mass collision energy (SI Materials and Methods).
Protein Crystallization.
HBc1–149 Y132A was concentrated to ∼17 mg/mL in 20 mM Tris⋅HCl (pH 9.0) and 2 mM DTT, and was crystallized at 20 °C in 100 mM citrate (pH 5.0), 15% (vol/vol) isopropanol, and 1% (wt/vol) PEG 10,000. Crystals (200 μm) grew within 4 d. The crystal structure was solved by molecular replacement [using chains E and F from the PDB ID code 3KXS structure (21) as the initial search model] and standard procedures and software. The structure (PDB ID code 4BMG) was solved to a resolution of 3 Å with final Rwork and Rfree values of 25.6% and 31.7%, respectively (more details are provided in SI Materials and Methods). Rwork and Rfree were calculated from the following formalism: 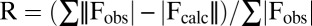 . 5% of the reflections were excluded from refinement and used to calculate Rfree, with the remaining 95% used to calculate Rwork.
. 5% of the reflections were excluded from refinement and used to calculate Rfree, with the remaining 95% used to calculate Rwork.
Supplementary Material
Acknowledgments
We thank Dr. L. Vogeley (Trinity College Dublin) for assistance with X-ray diffraction data collection and processing. Thanks also to NanoTemper Technologies GmbH (Munich, Germany) for technical assistance and providing access to microscale thermophoresis instrumentation. This work was supported by the Science Foundation Ireland (SFI) President of Ireland Young Researcher Award 09/YI/B1682 (to N.F.); SFI Stokes Lecturer Award 07/SK/B1224a (to N.F.); and Royal Irish Academy-Royal Society International Exchange Award IE111031 (to N.F. and A.E.A). D.A.S. was funded by an Engineering and Physical Sciences Research Council White Rose PhD Studentship. The Synapt mass spectrometer was purchased with Biotechnology and Biological Sciences Research Council funds (Grant BB/E012558/1).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4BMG).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1308846110/-/DCSupplemental.
References
- 1.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14(1):70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SE. How do small single-domain proteins fold? Fold Des. 1998;3(4):R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 3.Fulton KF, et al. PFD: A database for the investigation of protein folding kinetics and stability. Nucleic Acids Res. 2005;33(Database issue):D279–D283. doi: 10.1093/nar/gki016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teague SJ. Implications of protein flexibility for drug discovery. Nat Rev Drug Discov. 2003;2(7):527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization Publication Hepatitis B vaccines: WHO position paper—Recommendations. Vaccine. 2010;28(3):589–590. doi: 10.1016/j.vaccine.2009.10.110. [DOI] [PubMed] [Google Scholar]
- 6.Zoulim F. Are novel combination therapies needed for chronic hepatitis B? Antiviral Res. 2012;96(2):256–259. doi: 10.1016/j.antiviral.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Rabe B, Vlachou A, Panté N, Helenius A, Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci USA. 2003;100(17):9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewellyn EB, Loeb DD. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol. 2011;85(3):1298–1309. doi: 10.1128/JVI.01957-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponsel D, Bruss V. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J Virol. 2003;77(1):416–422. doi: 10.1128/JVI.77.1.416-422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katen SP, Chirapu SR, Finn MG, Zlotnick A. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol. 2010;5(12):1125–1136. doi: 10.1021/cb100275b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deres K, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299(5608):893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 12.Bourne CR, Finn MG, Zlotnick A. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J Virol. 2006;80(22):11055–11061. doi: 10.1128/JVI.00933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stray SJ, et al. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci USA. 2005;102(23):8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dyson MR, Murray K. Selection of peptide inhibitors of interactions involved in complex protein assemblies: Association of the core and surface antigens of hepatitis B virus. Proc Natl Acad Sci USA. 1995;92(6):2194–2198. doi: 10.1073/pnas.92.6.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freund SM, Johnson CM, Jaulent AM, Ferguson N. Moving towards high-resolution descriptions of the molecular interactions and structural rearrangements of the human hepatitis B core protein. J Mol Biol. 2008;384(5):1301–1313. doi: 10.1016/j.jmb.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 16.Zlotnick A, et al. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry. 1996;35(23):7412–7421. doi: 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- 17.Steven AC, et al. Structure, assembly, and antigenicity of hepatitis B virus capsid proteins. Adv Virus Res. 2005;64:125–164. doi: 10.1016/S0065-3527(05)64005-5. [DOI] [PubMed] [Google Scholar]
- 18.Wang JC, Dhason MS, Zlotnick A. Structural organization of pregenomic RNA and the carboxy-terminal domain of the capsid protein of hepatitis B virus. PLoS Pathog. 2012;8(9):e1002919. doi: 10.1371/journal.ppat.1002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3(6):771–780. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 20.Seitz S, Urban S, Antoni C, Böttcher B. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 2007;26(18):4160–4167. doi: 10.1038/sj.emboj.7601841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Packianathan C, Katen SP, Dann CE, 3rd, Zlotnick A. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J Virol. 2010;84(3):1607–1615. doi: 10.1128/JVI.02033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roseman AM, Berriman JA, Wynne SA, Butler PJ, Crowther RA. A structural model for maturation of the hepatitis B virus core. Proc Natl Acad Sci USA. 2005;102(44):15821–15826. doi: 10.1073/pnas.0504874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Böttcher B, Vogel M, Ploss M, Nassal M. High plasticity of the hepatitis B virus capsid revealed by conformational stress. J Mol Biol. 2006;356(3):812–822. doi: 10.1016/j.jmb.2005.11.053. [DOI] [PubMed] [Google Scholar]
- 24.Ceres P, Zlotnick A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry. 2002;41(39):11525–11531. doi: 10.1021/bi0261645. [DOI] [PubMed] [Google Scholar]
- 25.Ceres P, Stray SJ, Zlotnick A. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J Virol. 2004;78(17):9538–9543. doi: 10.1128/JVI.78.17.9538-9543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrano L, Kellis JT, Jr, Cann P, Matouschek A, Fersht AR. The folding of an enzyme. II. Substructure of barnase and the contribution of different interactions to protein stability. J Mol Biol. 1992;224(3):783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- 27.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins. 1995;21(3):167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 28.Otzen DE, Oliveberg M. Salt-induced detour through compact regions of the protein folding landscape. Proc Natl Acad Sci USA. 1999;96(21):11746–11751. doi: 10.1073/pnas.96.21.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. On the role of frustration in the energy landscapes of allosteric proteins. Proc Natl Acad Sci USA. 2011;108(9):3499–3503. doi: 10.1073/pnas.1018980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nassal M, Rieger A, Steinau O. Topological analysis of the hepatitis B virus core particle by cysteine-cysteine cross-linking. J Mol Biol. 1992;225(4):1013–1025. doi: 10.1016/0022-2836(92)90101-o. [DOI] [PubMed] [Google Scholar]
- 31.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4(10):2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wingfield PT, Stahl SJ, Williams RW, Steven AC. Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry. 1995;34(15):4919–4932. doi: 10.1021/bi00015a003. [DOI] [PubMed] [Google Scholar]
- 33.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol. 2011;9(4):342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidel SA, et al. Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding. Angew Chem Int Ed Engl. 2012;51(42):10656–10659. doi: 10.1002/anie.201204268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greene RF, Jr, Pace CN. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, alpha-chymotrypsin, and beta-lactoglobulin. J Biol Chem. 1974;249(17):5388–5393. [PubMed] [Google Scholar]
- 36.Zlotnick A, Johnson JM, Wingfield PW, Stahl SJ, Endres D. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry. 1999;38(44):14644–14652. doi: 10.1021/bi991611a. [DOI] [PubMed] [Google Scholar]
- 37.Galvagnion C, et al. Folding and association of thermophilic dimeric and trimeric DsrEFH proteins: Tm0979 and Mth1491. Biochemistry. 2009;48(13):2891–2906. doi: 10.1021/bi801784d. [DOI] [PubMed] [Google Scholar]
- 38.Katen S, Zlotnick A. The thermodynamics of virus capsid assembly. Methods Enzymol. 2009;455:395–417. doi: 10.1016/S0076-6879(08)04214-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betz SF. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993;2(10):1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mallam AL, Jackson SE. Folding studies on a knotted protein. J Mol Biol. 2005;346(5):1409–1421. doi: 10.1016/j.jmb.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 41.Hopper JT, et al. Evidence for the preservation of native inter- and intra-molecular hydrogen bonds in the desolvated FK-binding protein⋅FK506 complex produced by electrospray ionization. J Am Soc Mass Spectrom. 2012;23(10):1757–1767. doi: 10.1007/s13361-012-0430-y. [DOI] [PubMed] [Google Scholar]
- 42.Hopper JT, Oldham NJ. Collision induced unfolding of protein ions in the gas phase studied by ion mobility-mass spectrometry: The effect of ligand binding on conformational stability. J Am Soc Mass Spectrom. 2009;20(10):1851–1858. doi: 10.1016/j.jasms.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Han L, Hyung SJ, Mayers JJ, Ruotolo BT. Bound anions differentially stabilize multiprotein complexes in the absence of bulk solvent. J Am Chem Soc. 2011;133(29):11358–11367. doi: 10.1021/ja203527a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han L, Hyung SJ, Ruotolo BT. Bound cations significantly stabilize the structure of multiprotein complexes in the gas phase. Angew Chem Int Ed Engl. 2012;51(23):5692–5695. doi: 10.1002/anie.201109127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kükrer B, et al. Conformational isomers of calcineurin follow distinct dissociation pathways. J Am Soc Mass Spectrom. 2012;23(9):1534–1543. doi: 10.1007/s13361-012-0441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freeke J, Bush MF, Robinson CV, Ruotolo BT. Gas-phase protein assemblies: Unfolding landscapes and preserving native-like structures using noncovalent adducts. Chem Phys Lett. 2012;524:1–9. [Google Scholar]
- 47.Uetrecht C, Barbu IM, Shoemaker GK, van Duijn E, Heck AJ. Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat Chem. 2011;3(2):126–132. doi: 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- 48.Uetrecht C, et al. Subunit exchange rates in Hepatitis B virus capsids are geometry- and temperature-dependent. Phys Chem Chem Phys. 2010;12(41):13368–13371. doi: 10.1039/c0cp00692k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ferguson N, Fersht AR (2011) Phi-value analysis of protein folding transition states. Protein Engineering Handbook, eds Lutz S, Bornscheuer UT (Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany), pp 1–47.
- 50.Bourne CR, Katen SP, Fulz MR, Packianathan C, Zlotnick A. A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry. 2009;48(8):1736–1742. doi: 10.1021/bi801814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan TT, Tai PC, Shih C. Subtype-independent immature secretion and subtype-dependent replication deficiency of a highly frequent, naturally occurring mutation of human hepatitis B virus core antigen. J Virol. 1999;73(12):10122–10128. doi: 10.1128/jvi.73.12.10122-10128.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan TTT, Sahu GK, Whitehead WE, Greenberg R, Shih C. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J Virol. 1999;73(7):5731–5740. doi: 10.1128/jvi.73.7.5731-5740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akarca US, Lok ASF. Naturally occurring hepatitis B virus core gene mutations. Hepatology. 1995;22(1):50–60. [PubMed] [Google Scholar]
- 54.Le Pogam S, Yuan TT, Sahu GK, Chatterjee S, Shih C. Low-level secretion of human hepatitis B virus virions caused by two independent, naturally occurring mutations (P5T and L60V) in the capsid protein. J Virol. 2000;74(19):9099–9105. doi: 10.1128/jvi.74.19.9099-9105.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang HY, et al. Phosphorylation of hepatitis B virus Cp at Ser87 facilitates core assembly. Biochem J. 2006;398(2):311–317. doi: 10.1042/BJ20060347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neet KE, Timm DE. Conformational stability of dimeric proteins: Quantitative studies by equilibrium denaturation. Protein Sci. 1994;3(12):2167–2174. doi: 10.1002/pro.5560031202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panchenko AR, Luthey-Schulten Z, Cole R, Wolynes PG. The foldon universe: A survey of structural similarity and self-recognition of independently folding units. J Mol Biol. 1997;272(1):95–105. doi: 10.1006/jmbi.1997.1205. [DOI] [PubMed] [Google Scholar]
- 58.DiMattia MA, et al. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure. 2013;21(1):133–142. doi: 10.1016/j.str.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bereszczak JZ, et al. Epitope-distal effects accompany the binding of two distinct antibodies to hepatitis B virus capsids. J Am Chem Soc. 2013;135(17):6504–6512. doi: 10.1021/ja402023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cho MH, Song JS, Kim HJ, Park SG, Jung G. Structure-based design and biochemical evaluation of sulfanilamide derivatives as hepatitis B virus capsid assembly inhibitors. J Enzyme Inhib Med Chem. 2012 doi: 10.3109/14756366.2012.694879. 10.3109/14756366.2012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.