

Abstract
The abundance, genetic diversity, and crucial ecological and evolutionary roles of marine phages have prompted a large number of metagenomic studies. However, obtaining a thorough understanding of marine phages has been hampered by the low number of phage isolates infecting major bacterial groups other than cyanophages and pelagiphages. Therefore, there is an urgent requirement for the isolation of phages that infect abundant marine bacterial groups. In this study, we isolated and characterized HMO-2011, a phage infecting a bacterium of the SAR116 clade, one of the most abundant marine bacterial lineages. HMO-2011, which infects “Candidatus Puniceispirillum marinum” strain IMCC1322, has an ∼55-kb dsDNA genome that harbors many genes with novel features rarely found in cultured organisms, including genes encoding a DNA polymerase with a partial DnaJ central domain and an atypical methanesulfonate monooxygenase. Furthermore, homologs of nearly all HMO-2011 genes were predominantly found in marine metagenomes rather than cultured organisms, suggesting the novelty of HMO-2011 and the prevalence of this phage type in the oceans. A significant number of the viral metagenome sequences obtained from the ocean surface were best assigned to the HMO-2011 genome. The number of reads assigned to HMO-2011 accounted for 10.3%–25.3% of the total reads assigned to viruses in seven viromes from the Pacific and Indian Oceans, making the HMO-2011 genome the most or second-most frequently assigned viral genome. Given its ability to infect the abundant SAR116 clade and its widespread distribution, Puniceispirillum phage HMO-2011 could be an important resource for marine virus research.
Viruses are the most abundant biological entities in diverse marine environments, as revealed by electron microscopy, epifluorescence microscopy, and flow cytometry studies (1–3). The average number of virus-like particles in surface seawater is ∼107 per mL, and it typically exceeds the number of prokaryotes by an order of magnitude. Marine viruses play an important role in nutrient cycles by mediating a significant proportion of bacterial mortality. This so-called “viral shunt” diverts organic matter from particulate forms to dissolved forms, influencing the overall biogeochemistry of various elements (4). Viruses affect the community composition and genetic diversity of marine organisms by selectively infecting susceptible hosts (5, 6), which also increases the genetic diversity of the viruses themselves through mechanisms such as antagonistic coevolution (6, 7).
Recent metagenomic studies of the oceans have revealed the numerous novel genetic repertoires of marine viromes (8–14). However, most genome fragments in these viromes cannot be categorized into any known viral group (8, 9, 13, 14). Because most marine viruses are believed to be phages (15), a more thorough understanding of the ecological and evolutionary roles of marine viruses and a better interpretation of the rapidly increasing virome sequences require the isolation and genomic analysis of individual viruses, especially phages infecting diverse marine bacterioplankton.
The importance of phage isolation is exemplified by studies on marine cyanophages and pelagiphages. The isolation and characterization of cyanophages have shown that they have auxiliary metabolic genes (16), including photosynthesis-related genes that are expressed during infection and modulate host metabolism toward successful infection (17–21). Cyanophages are important in the diversification of hosts and horizontal gene transfer and are extensively used to interpret marine metagenome sequences (6, 22, 23). Very recently, four pelagiphages, viruses infecting the SAR11 clade, were isolated (24). Pelagiphage genome sequences have been found to be crucial for the interpretation of viromes from the Pacific Ocean (24). However, there are many abundant marine bacterial groups, including SAR86, SAR116, and Bacteroidetes, for which few phages have been isolated or characterized. Given the poor assignment of virome sequences into specific viral groups and the presence of difficult-to-cultivate or unculturable bacterial groups in the ocean, cultivating these bacteria and isolating the phages infecting them are prerequisites for understanding phage diversity.
The SAR116 clade is one of the most abundant groups of heterotrophic bacteria inhabiting the surface of the ocean. Since its initial discovery through the cloning of 16S rRNA genes from the Sargasso Sea (25), many culture-independent studies have shown that the SAR116 clade contributes significantly—more than 10% in some cases—to the bacterial assemblages of the marine euphotic zone (26–33). The genome sequences of two bacteria in this clade, HIMB100 and “Candidatus Puniceispirillum marinum” IMCC1322, have recently been reported (34, 35). Both strains have genes for proteorhodopsin-based photoheterotrophy, carbon monoxide dehydrogenase, and dimethylsulfoniopropionate demethylase, suggesting the diverse metabolic potential and biogeochemical importance of the SAR116 clade. Considering the widespread distribution and ecologically meaningful genome characteristics of this clade, their phages are likely to be abundant in marine environments and may provide useful references for functional and phylogenetic annotation of marine viromes.
Here, we report the isolation and genomic characterization of HMO-2011, a phage that infects strain IMCC1322 of the SAR116 clade. The HMO-2011 genome harbors many novel genes, such as a gene encoding a DNA polymerase with a DnaJ central domain. The genome of HMO-2011 was a notable resource for the identification of unknown marine viral gene fragments, as HMO-2011 accounted for 10.3%–25.3% of viral metagenome reads assigned to viruses.
Results and Discussion
Isolation and General Characterization.
A marine phage, designated HMO-2011, that can infect “Candidatus Puniceispirillum marinum” strain IMCC1322 of the SAR116 clade was isolated from a surface seawater sample from the East Sea (Sea of Japan), where the host strain had been previously cultivated (34). This bacteriophage is the first lytic phage isolate that infects the SAR116 clade. Transmission electron microscopy (TEM) images showed that HMO-2011 had an isometric head with an average diameter of ∼58 nm and a short tail (Fig. 1A). Although tails could often not be clearly identified, this morphological feature suggested that HMO-2011 belongs to the Podoviridae. A one-step growth curve indicated that HMO-2011 is lytic (Fig. 1B). The latent period was ∼6 h. The burst size was ∼500, which is similar to that of Silicibacter phage DSS3phi2 (36). The lytic ability of the phage was confirmed by coculture with strain IMCC1322. The titer of HMO-2011 increased until 3 d after inoculation and was maintained thereafter whereas the cell numbers of IMCC1322 decreased after 1 d (Fig. S1). In the host range experiment, HMO-2011 did not infect any type strain of the six species tested, which share greater than 90% 16S rRNA gene sequence similarity with strain IMCC1322 (SI Text).
Fig. 1.

Morphological features and one-step growth curve of HMO-2011. (A) Transmission electron microscopy images showing purified phage particles. A distal part of the tail was detached from the head in the lower image. (Scale bar, 20 nm.) (B) Increase in phage titers during one-step growth. The data shown are averages from triplicate experiments, and error bars indicate SDs.
Overall Genome Features.
The genome sequence of HMO-2011 was assembled as a 55,282-bp circular dsDNA, with a GC content of 43.1 mol%. Considering that the dsDNA genomes of all tailed phages are linear, the circular assembly of the HMO-2011 genome suggested circular permutation or terminal redundancy (37). Seventy-four ORFs were predicted in the genome sequence of HMO-2011 (Table S1). The HMO-2011 genome has a modular structure (Fig. 2) that is typical of phage genomes (38), and the genome contains modules for DNA metabolism and replication (6 ORFs), structure and assembly (19 ORFs), and packaging and lysis (3 ORFS), as well as two auxiliary metabolic genes and 44 ORFs predicted to encode hypothetical proteins. The gene content and order of the DNA metabolism/replication module were similar to those of marine podoviruses such as P-SSP7, Syn5, and SIO1, which supported the TEM image-based classification of HMO-2011 as a podovirus (Fig. 2; also see SI Text). Many ORFs in the HMO-2011 genome were highly similar to genes found only in metagenome sequences rather than those in isolated phages or bacteria. In the following sections, analyses of a few selected ORFs and comparison of the genome with viromes are discussed. ORFs for DNA replication and metabolism, including primase (Fig. S2A), helicase, endonuclease, and integrase; structure (Figs. S2B and S3), packaging, and lysis; and auxiliary metabolic genes are discussed in detail in SI Text.
Fig. 2.
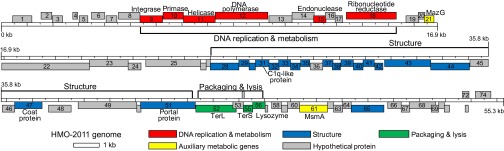
Genome map of HMO-2011 showing a modular structure. ORFs above the ruler are transcribed rightward and include genes for DNA replication and metabolism. ORFs below the ruler are transcribed leftward and include genes for structure, packaging, and lysis. The numbers in each box are the ORF numbers, which correspond to those used in the text and Table S1. TerL, Terminase, large subunit; TerS, Terminase, small subunit.
DNA Polymerase.
The DNA polymerase encoded by ORF12 has an unusual domain architecture. A part of the DnaJ central domain (Pfam protein families database ID PF00684), also called a zinc finger domain, was found between the 3′–5′ exonuclease domain (PF01612) and the DNA polymerase family A domain (PF00476) (Fig. 3A). The DnaJ central domain is a cysteine-rich domain that is usually found in type I DnaJ family proteins (molecular chaperones) and is characterized by four repeats of the CXXCXGXG motif involved in zinc binding (39, 40). ORF12 has two repeats of this motif, which is known to bind one zinc ion (Fig. 3B) (41). To the best of our knowledge, this is the first report of a DnaJ central domain found in a family A DNA polymerase of a cultured organism. In analyses using PfamAlyzer (42) and the Conserved Domain Architecture Retrieval Tool (43), no family A DNA polymerase in the Pfam database and nonredundant (nr) database of GenBank was found to have a DnaJ central domain (see SI Text for details). However, many protein sequences predicted from marine metagenomes had the same domain architecture as that of ORF12, indicating the wide distribution of this type of DNA polymerase in marine environments (Fig. 3B and Table S2; see also SI Text). Phylogenetic analysis of the polymerase domain of ORF12 supported the above findings; ORF12 formed a robust cluster with many metagenome-derived DNA polymerases, and this cluster was well separated from the DNA polymerases of other bacteria and phages (Fig. 3C). Among cultured organisms, the most closely related DNA polymerase was found in Vibrio phage VpV262, a member of the Podoviridae.
Fig. 3.
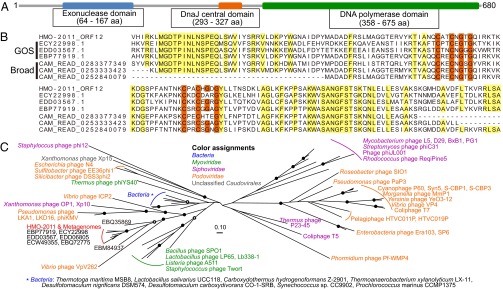
Analysis of the domain organization and phylogenetic position of the DNA polymerase (ORF12, 680 amino acids) of HMO-2011. (A) Domain organization of ORF12, showing the presence of the DnaJ central domain. Domains and their boundaries were predicted using Pfam. (B) Alignment of DNA polymerase sequences showing the existence of a partial DnaJ central domain in HMO-2011 and marine metagenome sequences. Amino acids corresponding to positions 228–386 of ORF12 are presented. Conserved residues in the CXXCXGXG motifs of the DnaJ central domain are in orange. Other conserved residues are in yellow. GOS indicates sequences obtained from the GOS project (478–682 amino acids). Broad indicates sequences from the BroadPhageMetagenomes database of the CAMERA (143–147 amino acids). Broad sequences are short because they were obtained through pyrosequencing. Alignment was generated using COBALT at the National Center for Biotechnology Information. (C) An unrooted maximum-likelihood tree of the polymerase domain (PF00476) of family A DNA polymerases. Filled circles indicate nodes with bootstrap values of ≥90. Empty circles indicate nodes with bootstrap values of ≥70. See SI Materials and Methods for more information about the procedure used for tree building.
Auxiliary Metabolic Genes.
Auxiliary metabolic genes (AMGs) are phage-encoded metabolic genes putatively involved in the regulation of host metabolism (15). HMO-2011 has two AMGs that are predicted to encode a MazG domain-containing protein and a hydroxylase alpha-subunit of methanesulfonate monooxygenase (MsmA).
A single MazG nucleotide pyrophosphohydrolase domain (PF03819) was found in ORF21. Many marine phages have proteins containing MazG domains, suggesting the importance of this domain in the proliferation of marine phages (44–46). In bacteria, MazG domain proteins have been shown to function in stress responses or removal of noncanonical nucleotides (47–49). It has been suggested that phage MazG proteins may contribute to phage propagation by helping sustain the metabolism of starved host bacteria (44, 46). However, the functions of the diverse MazG proteins in phages remain to be fully elucidated (45).
Another AMG (ORF61) was predicted to encode an MsmA protein. To the best of our knowledge, this is the first MsmA gene found in an isolated virus. Because methanesulfonate monooxygenase mediates the oxidation of methanesulfonic acid to formaldehyde and sulfite and because formaldehyde can be further metabolized by host strains (34, 50), ORF61 may assist in the C1 metabolism of hosts (see SI Text for further discussion). However, the Rieske domain of ORF61 lacked the four cysteine and histidine residues needed to bind the iron–sulfur cluster that is essential for electron transfer, making the functionality of this ORF unclear (51) (Fig. S2C). This sequence feature, found only in one of two putative MsmAs of IMCC1322 (SAR116_2109) among cultured organisms, was found in many global ocean sampling (GOS) metagenome sequences and marine viromes (Fig. S2C).
Distribution in Marine Environments.
The genome analyses of HMO-2011 showed that many ORFs shared a greater number of similar sequences in marine metagenomes than in the nr database of GenBank (Fig. 3 and Fig S2). In a more comprehensive analysis using all 74 ORFs as queries against the nr, environmental non-redundant (env_nr), GOS, and BroadPhageMetagenomes (BroadPhage) databases, sequences similar to the HMO-2011 genome were most frequently retrieved from the BroadPhage database of Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis (CAMERA) (52) (Fig. S4; also see SI Text). Considering that the BroadPhage database contains data for viromes from diverse marine habitats, this result suggested that HMO-2011 is a phage representing a previously unknown phage type prevalent in marine environments and that the HMO-2011 genome could be a good reference for taxonomic binning of marine viromes.
To test the usefulness of the HMO-2011 genome as a reference genome in metagenome analysis, seven marine viromes were selected and used for metagenomic binning analysis. These seven viromes, all from the ocean surface, included four from the Indian Ocean (13) and three from the Pacific Ocean (Table 1 and Table S3; see SI Materials and Methods for the selection process, brief description, and sequence processing). Each read from the selected viromes was used as a query in BLASTN analyses against a genome database that included HMO-2011 and 4,541 other viruses and was assigned to a single best-matching reference genome. The results showed that 6.7%–12.8% of the total reads were assigned to viruses and that 0.9%–2.0% of total reads were assigned to HMO-2011. HMO-2011 contributed 10.3%–25.3% of the reads assigned to viral genomes, being the most or second-most frequently assigned viral genome for all seven samples (Table 1, Fig. 4, and Table S3). In addition, more than 90% of the reads recruited by HMO-2011 were assigned to HMO-2011, suggesting that this phage does not share a significant fraction of viromes with other viruses (Table 1).
Table 1.
Binning of marine virome reads to reference viral genomes, including HMO-2011
| Sample* | Station† | Ocean | Depth, m | No. of sequence reads |
Percent, % |
Rank of HMO‖ | |||||
| Total (A) | Assigned to viruses‡ (B) | Assigned to HMO§ (C) | Recruited by HMO¶ | Virus proportion (B/A ×100) | HMO proportion (C/A × 100) | HMO contribution (C/B × 100) | |||||
| 108** | Cocos | Indian | 1.8 | 289129 | 23221 | 3205 | 3474 | 8.0 | 1.1 | 13.8 | 1 |
| 112** | Indian | Indian | 1.8 | 401817 | 33772 | 5185 | 5541 | 8.4 | 1.3 | 15.4 | 1 |
| 117** | St. Ann | Indian | 1.8 | 427102 | 52940 | 7291 | 7973 | 12.4 | 1.7 | 13.8 | 2 |
| 122** | Madagascar | Indian | 1.9 | 272553 | 18284 | 2637 | 2896 | 6.7 | 1.0 | 14.4 | 1 |
| 001011†† | NESAP | Pacific | 10 | 214886 | 16702 | 4227 | 4448 | 7.8 | 2.0 | 25.3 | 1 |
| 000990†† | SPOT | Pacific | 5–30 | 165223 | 21154 | 2812 | 3122 | 12.8 | 1.7 | 13.3 | 1 |
| 1336†† | Scripps | Pacific | 1 | 2574331 | 226400 | 23360 | 25870 | 8.8 | 0.9 | 10.3 | 1 |
HMO-2011 is abbreviated as HMO in this table.
See Table S3 and SI Materials and Methods for more information about the samples.
Location of stations: Cocos, 12.06S 96.53E (Cocos Islands, inside lagoon); Indian, 8.30S 80.23E; St. Ann, 4.39S 55.31E (St. Ann Island); Madagascar, 30.54S 40.25E (between Madagascar and South Africa); NESAP, 50N 145W (northeastern subarctic Pacific); SPOT, 33.55N 118.4W (Southern California Bight); Scripps, 32.87N 117.25W (Scripps Pier).
Total number of reads assigned to viral genomes.
Total number of reads assigned to the HMO-2011 genome.
Total number of reads recruited by the HMO-2011 genome.
Rank of HMO among cultured viruses was determined by comparing the number of reads assigned to each genome. See Fig. 4 and Table S3 for details, including other highly assigned viruses.
Sample names as used in ref. 13 (GSIOVIR).
Sample accession numbers as used on CAMERA website (CAM_SMPL_ or CAM_S_).
Fig. 4.
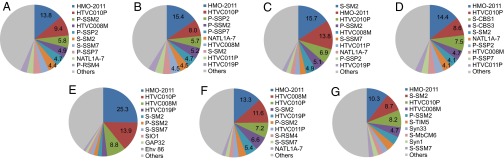
Highly assigned virus genomes in the binning of virome reads. The contribution of each virus (in %) was calculated using the same formula applied to HMO-2011 in Table 1 and is indicated when it exceeded 4.0%. Only the top 10 most highly assigned viruses are presented for each virome whereas other viruses are grouped as Others. (A) Cocos Islands, GSIOVIR108; (B) Indian Ocean, GSIOVIR112; (C) St. Ann Island, GSIOVIR117; (D) between Madagascar and South Africa, GSIOVIR122; (E) NESAP, CAM_SMPL_001011; (F) SPOT, CAM_SMPL_000990; (G) Scripps Pier, CAM_S_1336. Abbreviation for virus names: Ehv 86, Emiliania huxleyi virus 86; GAP32, Cronobacter phage vB_CsaM_GAP32; NATL1A-7, cyanophage NATL1A-7; SIO1, roseophage SIO1; Syn1, Prochlorococcus phage Syn1; Syn33, Prochlorococcus phage Syn33. All viruses starting with P- or S- are Prochlorococcus or Synechococcus phages, respectively. Four viruses starting with “HTVC” are pelagiphages.
HMO-2011 remained the most or second-most frequently assigned viral genome when virome reads were assigned to a single best-matching ORF in the reference viral genomes by BLASTX (Table S3). The proportion of sequences assigned to viral genomes by BLASTX increased 1.7- to 2.4-fold compared with those obtained by BLASTN analyses, ranging from 15.8% to 23.0%. HMO-2011 was assigned by 1.6%–3.5% of the total reads, contributing 9.1%–19.0% of the reads assigned to viral genomes (Table S3). Fragment recruitment plots of the virome reads assigned to HMO-2011 by BLASTX showed that many ORFs related to DNA replication/metabolism, packaging and lysis, and virion structure were highly assigned (Fig. S5). Some ORFs encoding hypothetical proteins, such as ORF4, ORF27, ORF57, ORF58, ORF59, ORF63, and ORF69, were also frequently assigned. Distribution patterns of percent identities, bitscores, and alignment lengths of the reads assigned to each of the highly assigned viruses showed that there were no consistent differences among viruses, suggesting that the effect of so-called “greedy recruitment” by viruses without close relatives in the search databases was not so significant (Fig. S6) (24). Taken together, the metagenomic binning results by BLASTN and BLASTX clearly showed that HMO-2011 is a phage that represents a group of phages widespread in the ocean surface and that the HMO-2011 genome is an important reference for binning of marine viromes.
Most highly assigned viruses other than HMO-2011 in our binning analyses were pelagiphages and cyanophages (Fig. 4 and Table S3), which coincide with the results obtained from the analyses of the Indian Ocean viromes (13) and the Pacific Ocean viromes (24). The binning analyses of the Indian Ocean viromes by Williamson et al. (13) in the absence of pelagiphages and HMO-2011 revealed that cyanophages were the most highly assigned viruses. When four pelagiphages were added for the binning of the Pacific Ocean viromes, the most abundant viruses were pelagiphages, followed by cyanophages (24). HTVC010P, a pelagipodovirus, has been found to be highly abundant in the Pacific Ocean viromes (24). When HMO-2011 was included in the present study, HMO-2011 became the most or second-most highly assigned virus whereas pelagiphages and cyanophages were still highly represented. HTVC010P was always in the top four viruses for all seven viromes in our analyses (Table S3). Our results confirm the abundance of pelagiphages and cyanophages shown in previous studies and establish SAR116 phages as a significant contributor in viral assemblages in the ocean surface.
The remarkable power of the HMO-2011 genome in the binning of metagenome reads was attributed to the high abundance of SAR116 bacteria in the ocean. The SAR116 clade is reported to be a significant contributor to bacterial communities in various ocean regions, including San Pedro Ocean Time-series (SPOT) site (26, 32), Scripps Pier (30), and the northeastern subarctic Pacific (33), from which three Pacific Ocean viromes listed in Table 1 were sampled. The proportion of the SAR116 clade at four stations where Indian Ocean viromes were reported ranged from 1.4% to 4.5% (53). The other highly assigned viruses were mostly pelagiphages and cyanophages that infect the SAR11 clade and Cyanobacteria, respectively. Considering the high abundance of the SAR11 clade and Cyanobacteria in surface seawater, this result implied that (i) a high occurrence of bacterial hosts leads to prevalence of the phages that infect them and (ii) genomes of phages infecting abundant groups of bacteria are essential for a thorough characterization of marine viromes (14, 24). An interesting finding of our study, especially considering previous studies that showed that the SAR11 clade is ∼10-fold more abundant than the SAR116 clade in the ocean surface (29, 53), was that the number of reads assigned to HMO-2011 was in a range comparable with the total number of reads assigned to four pelagiphages. Although the reason for this comparability cannot be clearly explained at present, we expect that it is related to the environmentally relevant host range of each phage and the degree of genome conservation among each group of pelagiphages or SAR116 phages.
The “Killing the Winner” (KtW) hypothesis has been proposed to explain phage–host dynamics (4, 54). The prevalence of SAR116 phages, together with the high abundance of SAR116 bacteria in the ocean, suggests that the KtW hypothesis may work on a relatively fine phylogenetic scale. Therefore, it can contribute to strain or genotype-level diversification without having significant effects on the community composition of hosts or phages on a coarse phylogenetic scale (6, 55, 56). In this regard, it is noteworthy that strain IMCC1322 has a CRISPR (clustered regularly interspaced short palindromic repeats) locus (http://crispr.u-psud.fr) (57). None of the 29 spacers in the CRISPR locus of IMCC1322 exactly matched the HMO-2011 genome, as was expected on the basis of the lytic ability of the phage. The presence of a CRISPR locus in IMCC1322 indicates that some SAR116 bacteria can acquire immunity to phages with a minimal fitness cost (58, 59), which may confer a competitive advantage in the marine euphotic zone, the main habitat of SAR116 bacteria.
Concluding Remarks.
Metagenomic research on marine viruses has yielded numerous novel sequences but has provided little insight into individual viruses. A few methods developed to characterize single viruses without culturing remain limited in the information they can provide about the host (60) or genomic content (61) of viruses. Our results of isolation and genome characterization of a SAR116 phage demonstrate the importance and strength of culture-based studies. HMO-2011 is a lytic phage infecting the SAR116 clade, one of major components of bacterial assemblages in the marine euphotic zone. The successful isolation of HMO-2011 was possible owing to the cultivation of a representative member of the abundant SAR116 clade, which emphasizes the importance of culturing presently underrepresented bacterial groups for phage study. The genome of HMO-2011 was found to have many new genes, including a DNA polymerase with a unique domain architecture, similar sequences of which are found mainly in marine metagenomes but not in other cultured organisms. This genomic uniqueness of HMO-2011 combined with the high abundance of the SAR116 clade contributed to the remarkable power of the HMO-2011 genome in the binning of marine viromes. The HMO-2011 genome explained a significant portion of viral metagenomes from the Indian and Pacific Oceans, demonstrating that this type of phage is widespread and among the most abundant phage groups in the global ocean. Conclusively, our study provides an example of the complementary nature of culture-dependent and culture-independent approaches, and HMO-2011 will be an indispensable resource and a valuable model system for marine virus research. To understand more comprehensively the abundance, distribution, and ecological roles of this phage type in the ocean, further studies may focus on comparative analyses with more viromes from various ocean regions, functional identification of novel genes in HMO-2011, and elucidation of infection network and host–phage dynamics using techniques such as viral tagging and phageFISH (62, 63).
Materials and Methods
Phage Isolation and Characterization.
A surface seawater sample collected at a station located in the East Sea (38°20′15″ N, 128°33′32″ E) of Korea was treated with chloroform, and 10-μL aliquots were placed into 96-well plates. Exponentially growing host bacteria (strain IMCC1322; 150-μL aliquots) were then added into each well. After 1 wk of incubation at 15 °C, the optical density of each well was compared with those of control wells inoculated with autoclaved seawater. Culture lysates were recovered from the wells with lower optical density and were used to purify putative phages. One of these lysates was purified three times via plaque assay, and a phage, designated HMO-2011, was established from a single plaque. See SI Materials and Methods for descriptions of host bacteria cultivation, plaque assays, the determination of growth kinetics, and morphological characterization using TEM.
Genome Sequencing.
Phage particles from a 100-mL culture lysate were filtered through a 0.2-μm filter and concentrated by ultracentrifugation (120,000 × g, 30 min; Beckman Coulter). DNA was extracted using a DNeasy Blood and Tissue kit (Qiagen). Library production (average insert size, ∼5 kb) and whole-genome shotgun sequencing using Big Dye chemistry was performed by Genotech Co. Ltd. Sequence reads were assembled using phred/phrap/consed, and the remaining gaps were closed using PCR and primer walking. The average fold coverage of the genome was 9.9.
Annotation of the Phage Genome.
Gene prediction was performed using Glimmer, based on an interpolated context model built with the IMCC1322 genome sequence. The GeneMark.hmm program, RAST server, and long-orfs program in Glimmer3 were used to check and edit the original predictions. Functional annotation of the ORFs was performed with BLASTP, PSI-BLAST, and DELTA-BLAST analyses against the nr database of GenBank. Domains were predicted using the Pfam database, InterProScan, and HHpred. SignalP 3.0 and TMHMM were used to predict localization and transmembrane helices.
Binning of Virome Reads.
A total of seven viromes were selected for metagenome binning analyses. Four viromes from the Indian Ocean were selected without any preliminary analyses because these represented the only viral metagenomes reported from the Indian Ocean (13). The remaining three viromes were selected from the metagenomes deposited in the CAMERA database, mainly based on the proportion of reads similar to the HMO-2011 genome (see SI Materials and Methods for details). Binning of virome reads was performed using BLASTN and BLASTX (version 2.2.25+). Genome or ORF sequences of HMO-2011 and four pelagiphages (HTVC010P, HTVC011P, HTVC019P, and HTVC008M) were added to the RefSeq Viral database (release 57) to construct search databases for binning. Each read of seven selected viromes was used as a query and assigned to a single best-matching viral genome (BLASTN) or viral ORF (BLASTX), only if the alignments satisfied the criteria of bitscore (≥40) and length (≥50 bp in BLASTN, ≥20 amino acids in BLASTX). All BLAST parameters were default values except for query filtering options. “dust” (BLASTN) and “seg” (BLASTX) were disabled. All reads assigned to viruses were checked against RefSeq microbial genomes or proteins at the CAMERA Web site, and reads satisfying the above criteria and showing higher bitscores for microbes were discarded. For BLAST searches at the CAMERA website, the low-complexity filter was disabled and the parameters were set as follows: match reward = 2, mismatch penalty = −3, gap open cost = 5, and gap extend cost = 2 for BLASTN; and gap open cost = 11 and gap extend cost = 1 for BLASTX. Recruitment of virome reads by HMO-2011 was also performed to determine the fraction of recruited reads that was assigned to HMO-2011. The HMO-2011 genome was used as a query for BLASTN against each of the seven viromes. All parameters and criteria were the same as those used in binning.
Supplementary Material
Acknowledgments
We thank Jed Fuhrman, Grieg Steward, and Matthew Sullivan for sharing metagenome information. This work was supported by a National Research Foundation of Korea grant funded by the Korean government (Grant 2010-0014604) and also by the Polar Academic Program (PD12010), Korea Polar Research Institute, 2012.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The genome sequence of HMO-2011 reported in this paper has been deposited in the GenBank database (accession no. GU557055).
See Commentary on page 12166.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219930110/-/DCSupplemental.
References
- 1.Bergh O, Børsheim KY, Bratbak G, Heldal M. High abundance of viruses found in aquatic environments. Nature. 1989;340(6233):467–468. doi: 10.1038/340467a0. [DOI] [PubMed] [Google Scholar]
- 2.Noble RT, Fuhrman JA. Use of SYBR Green I for rapid epifluorescence counts of marine viruses and bacteria. Aquat Microb Ecol. 1998;14:113–118. [Google Scholar]
- 3.Marie D, Brussaard CPD, Thyrhaug R, Bratbak G, Vaulot D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl Environ Microbiol. 1999;65(1):45–52. doi: 10.1128/aem.65.1.45-52.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suttle CA. Marine viruses—major players in the global ecosystem. Nat Rev Microbiol. 2007;5(10):801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 5.Bouvier T, del Giorgio PA. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environ Microbiol. 2007;9(2):287–297. doi: 10.1111/j.1462-2920.2006.01137.x. [DOI] [PubMed] [Google Scholar]
- 6.Marston MF, et al. Rapid diversification of coevolving marine Synechococcus and a virus. Proc Natl Acad Sci USA. 2012;109(12):4544–4549. doi: 10.1073/pnas.1120310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buckling A, Rainey PB. Antagonistic coevolution between a bacterium and a bacteriophage. Proc Biol Sci. 2002;269(1494):931–936. doi: 10.1098/rspb.2001.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breitbart M, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA. 2002;99(22):14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angly FE, et al. The marine viromes of four oceanic regions. PLoS Biol. 2006;4(11):e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bench SR, et al. Metagenomic characterization of Chesapeake Bay virioplankton. Appl Environ Microbiol. 2007;73(23):7629–7641. doi: 10.1128/AEM.00938-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dinsdale EA, et al. Functional metagenomic profiling of nine biomes. Nature. 2008;452(7187):629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- 12.Cassman N, et al. Oxygen minimum zones harbour novel viral communities with low diversity. Environ Microbiol. 2012;14(11):3043–3065. doi: 10.1111/j.1462-2920.2012.02891.x. [DOI] [PubMed] [Google Scholar]
- 13.Williamson SJ, et al. Metagenomic exploration of viruses throughout the Indian Ocean. PLoS ONE. 2012;7(10):e42047. doi: 10.1371/journal.pone.0042047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurwitz BL, Sullivan MB. The Pacific Ocean virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE. 2013;8(2):e57355. doi: 10.1371/journal.pone.0057355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breitbart M. Marine viruses: Truth or dare. Annu Rev Mar Sci. 2012;4:425–448. doi: 10.1146/annurev-marine-120709-142805. [DOI] [PubMed] [Google Scholar]
- 16.Breitbart M, Thompson LR, Suttle CA, Sullivan MB. Exploring the vast diversity of marine viruses. Oceanography (Wash DC) 2007;20:135–139. [Google Scholar]
- 17.Lindell D, Jaffe JD, Johnson ZI, Church GM, Chisholm SW. Photosynthesis genes in marine viruses yield proteins during host infection. Nature. 2005;438(7064):86–89. doi: 10.1038/nature04111. [DOI] [PubMed] [Google Scholar]
- 18.Clokie MRJ, et al. Transcription of a ‘photosynthetic’ T4-type phage during infection of a marine cyanobacterium. Environ Microbiol. 2006;8(5):827–835. doi: 10.1111/j.1462-2920.2005.00969.x. [DOI] [PubMed] [Google Scholar]
- 19.Lindell D, et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature. 2007;449(7158):83–86. doi: 10.1038/nature06130. [DOI] [PubMed] [Google Scholar]
- 20.Dammeyer T, Bagby SC, Sullivan MB, Chisholm SW, Frankenberg-Dinkel N. Efficient phage-mediated pigment biosynthesis in oceanic cyanobacteria. Curr Biol. 2008;18(6):442–448. doi: 10.1016/j.cub.2008.02.067. [DOI] [PubMed] [Google Scholar]
- 21.Thompson LR, et al. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc Natl Acad Sci USA. 2011;108(39):E757–E764. doi: 10.1073/pnas.1102164108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindell D, et al. Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc Natl Acad Sci USA. 2004;101(30):11013–11018. doi: 10.1073/pnas.0401526101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabehi G, et al. A novel lineage of myoviruses infecting cyanobacteria is widespread in the oceans. Proc Natl Acad Sci USA. 2012;109(6):2037–2042. doi: 10.1073/pnas.1115467109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Y, et al. Abundant SAR11 viruses in the ocean. Nature. 2013;494(7437):357–360. doi: 10.1038/nature11921. [DOI] [PubMed] [Google Scholar]
- 25.Mullins TD, Britschgi TB, Krest RL, Giovannoni SJ. Genetic comparisons reveal the same unknown bacterial lineages in Atlantic and Pacific bacterioplankton communities. Limnol Oceanogr. 1995;40:148–158. [Google Scholar]
- 26.Fuhrman JA, et al. Annually reoccurring bacterial communities are predictable from ocean conditions. Proc Natl Acad Sci USA. 2006;103(35):13104–13109. doi: 10.1073/pnas.0602399103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rusch DB, et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5(3):e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pham VD, Konstantinidis KT, Palden T, DeLong EF. Phylogenetic analyses of ribosomal DNA-containing bacterioplankton genome fragments from a 4000 m vertical profile in the North Pacific Subtropical Gyre. Environ Microbiol. 2008;10(9):2313–2330. doi: 10.1111/j.1462-2920.2008.01657.x. [DOI] [PubMed] [Google Scholar]
- 29.Biers EJ, Sun S, Howard EC. Prokaryotic genomes and diversity in surface ocean waters: Interrogating the global ocean sampling metagenome. Appl Environ Microbiol. 2009;75(7):2221–2229. doi: 10.1128/AEM.02118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayali X, Palenik B, Burton RS. Dynamics of marine bacterial and phytoplankton populations using multiplex liquid bead array technology. Environ Microbiol. 2010;12(4):975–989. doi: 10.1111/j.1462-2920.2004.02142.x. [DOI] [PubMed] [Google Scholar]
- 31.Swan BK, et al. Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science. 2011;333(6047):1296–1300. doi: 10.1126/science.1203690. [DOI] [PubMed] [Google Scholar]
- 32.Steele JA, et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011;5(9):1414–1425. doi: 10.1038/ismej.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright JJ, Konwar KM, Hallam SJ. Microbial ecology of expanding oxygen minimum zones. Nat Rev Microbiol. 2012;10(6):381–394. doi: 10.1038/nrmicro2778. [DOI] [PubMed] [Google Scholar]
- 34.Oh H-M, et al. Complete genome sequence of “Candidatus Puniceispirillum marinum” IMCC1322, a representative of the SAR116 clade in the Alphaproteobacteria. J Bacteriol. 2010;192(12):3240–3241. doi: 10.1128/JB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grote J, et al. Draft genome sequence of strain HIMB100, a cultured representative of the SAR116 clade of marine Alphaproteobacteria. Stand Genomic Sci. 2011;5(3):269–278. doi: 10.4056/sigs.1854551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Wang K, Jiao N, Chen F. Genome sequences of two novel phages infecting marine roseobacters. Environ Microbiol. 2009;11(8):2055–2064. doi: 10.1111/j.1462-2920.2009.01927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baudoux AC, et al. Genomic and functional analysis of Vibrio phage SIO-2 reveals novel insights into ecology and evolution of marine siphoviruses. Environ Microbiol. 2012;14(8):2071–2086. doi: 10.1111/j.1462-2920.2011.02685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol Mol Biol Rev. 2011;75(4):610–635. doi: 10.1128/MMBR.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheetham ME, Caplan AJ. Structure, function and evolution of DnaJ: Conservation and adaptation of chaperone function. Cell Stress Chaperones. 1998;3(1):28–36. doi: 10.1379/1466-1268(1998)003<0028:sfaeod>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linke K, Wolfram T, Bussemer J, Jakob U. The roles of the two zinc binding sites in DnaJ. J Biol Chem. 2003;278(45):44457–44466. doi: 10.1074/jbc.M307491200. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Yamout M, Legge GB, Zhang O, Wright PE, Dyson HJ. Solution structure of the cysteine-rich domain of the Escherichia coli chaperone protein DnaJ. J Mol Biol. 2000;300(4):805–818. doi: 10.1006/jmbi.2000.3923. [DOI] [PubMed] [Google Scholar]
- 42.Hollich V, Sonnhammer ELL. PfamAlyzer: Domain-centric homology search. Bioinformatics. 2007;23(24):3382–3383. doi: 10.1093/bioinformatics/btm521. [DOI] [PubMed] [Google Scholar]
- 43.Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: Protein homology by domain architecture. Genome Res. 2002;12(10):1619–1623. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bryan MJ, et al. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS ONE. 2008;3(4):e2048. doi: 10.1371/journal.pone.0002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sullivan MB, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12(11):3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duhaime MB, Wichels A, Waldmann J, Teeling H, Glöckner FO. Ecogenomics and genome landscapes of marine Pseudoalteromonas phage H105/1. ISME J. 2011;5(1):107–121. doi: 10.1038/ismej.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gross M, Marianovsky I, Glaser G. MazG — a regulator of programmed cell death in Escherichia coli. Mol Microbiol. 2006;59(2):590–601. doi: 10.1111/j.1365-2958.2005.04956.x. [DOI] [PubMed] [Google Scholar]
- 48.Lu LD, et al. Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response. J Biol Chem. 2010;285(36):28076–28085. doi: 10.1074/jbc.M109.088872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonçalves AMD, de Sanctis D, McSweeney SM. Structural and functional insights into DR2231 protein, the MazG-like nucleoside triphosphate pyrophosphohydrolase from Deinococcus radiodurans. J Biol Chem. 2011;286(35):30691–30705. doi: 10.1074/jbc.M111.247999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baxter NJ, Scanlan J, De Marco P, Wood AP, Murrell JC. Duplicate copies of genes encoding methanesulfonate monooxygenase in Marinosulfonomonas methylotropha strain TR3 and detection of methanesulfonate utilizers in the environment. Appl Environ Microbiol. 2002;68(1):289–296. doi: 10.1128/AEM.68.1.289-296.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt CL, Shaw L. A comprehensive phylogenetic analysis of Rieske and Rieske-type iron-sulfur proteins. J Bioenerg Biomembr. 2001;33(1):9–26. doi: 10.1023/a:1005616505962. [DOI] [PubMed] [Google Scholar]
- 52.Sun S, et al. Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: The CAMERA resource. Nucleic Acids Res. 2011;39(Database issue):D546–D551. doi: 10.1093/nar/gkq1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yilmaz P, et al. Ecological structuring of bacterial and archaeal taxa in surface ocean waters. FEMS Microbiol Ecol. 2012;81(2):373–385. doi: 10.1111/j.1574-6941.2012.01357.x. [DOI] [PubMed] [Google Scholar]
- 54.Winter C, Bouvier T, Weinbauer MG, Thingstad TF. Trade-offs between competition and defense specialists among unicellular planktonic organisms: The “killing the winner” hypothesis revisited. Microbiol Mol Biol Rev. 2010;74(1):42–57. doi: 10.1128/MMBR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Valera F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7(11):828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez-Brito B, et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010;4(6):739–751. doi: 10.1038/ismej.2010.1. [DOI] [PubMed] [Google Scholar]
- 57.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35(Web Server issue) suppl 2:W52-7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deveau H, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190(4):1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 60.Allen LZ, et al. Single virus genomics: A new tool for virus discovery. PLoS ONE. 2011;6(3):e17722. doi: 10.1371/journal.pone.0017722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tadmor AD, Ottesen EA, Leadbetter JR, Phillips R. Probing individual environmental bacteria for viruses by using microfluidic digital PCR. Science. 2011;333(6038):58–62. doi: 10.1126/science.1200758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deng L, et al. 2012. Contrasting life strategies of viruses that infect photo- and heterotrophic bacteria, as revealed by viral tagging. mBio 3(6):e00373-12.
- 63.Allers E, et al. Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environ Microbiol. 2013 doi: 10.1111/1462-2920.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.