

Abstract
In cancer research, as in all of medicine, it is important to classify patients into etiologically and therapeutically relevant subtypes to improve diagnosis and treatment. One way to do this is to use clustering methods to find subgroups of homogeneous individuals based on genetic profiles together with heuristic clinical analysis. A notable drawback of existing clustering methods is that they ignore the possibility that the variance of gene expression profile measurements can be heterogeneous across subgroups, and methods that do not consider heterogeneity of variance can lead to inaccurate subgroup prediction. Research has shown that hypervariability is a common feature among cancer subtypes. In this paper, we present a statistical approach that can capture both mean and variance structure in genetic data. We demonstrate the strength of our method in both synthetic data and in two cancer data sets. In particular, our method confirms the hypervariability of methylation level in cancer patients, and it detects clearer subgroup patterns in lung cancer data.
Clustering is an important type of unsupervised learning algorithm for data exploration. Successful examples include K-mean clustering and hierarchical clustering, both of which are widely used in biological research to find cancer subtypes and to stratify patients. These and other traditional clustering algorithms depend on the distances calculated using all of the features. For example, individuals can be clustered into homogeneous groups by minimizing the summation of within-clusters sum of squares (the Euclidean distances) of their gene expression profiles. Unfortunately, this strategy is ineffective when only a subset of features is informative. This phenomenon can be demonstrated by K-means clustering (1) results for a toy example using only the variables which determine the underlying true cluster compared with using all variables (which includes many uninformative variables). As can be seen in Fig. 1, clustering performance is poor when all variables are used in the clustering algorithm (2).
Fig. 1.
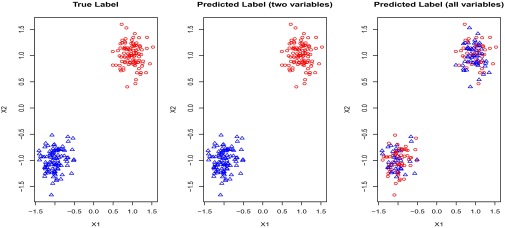
Data set contains two clusters determined by two variables 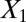 and
and 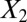 such that points around
such that points around 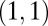 and
and 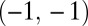 naturally form clusters. There are 200 observations (100 for each cluster) and 1,002 variables (
naturally form clusters. There are 200 observations (100 for each cluster) and 1,002 variables (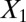 ,
, 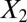 and 1,000 random noise variables). We plot the data in the 2D space of
and 1,000 random noise variables). We plot the data in the 2D space of 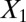 and
and 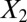 . Graphs with true cluster labels and predicted cluster labels obtained by clustering using only
. Graphs with true cluster labels and predicted cluster labels obtained by clustering using only 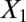 and
and 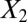 and clustering by using all variables are laid from left to right. The predicted labels are the same as the true labels only when
and clustering by using all variables are laid from left to right. The predicted labels are the same as the true labels only when 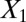 and
and 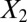 are used for clustering; however, the performance is much worse when all variables are used.
are used for clustering; however, the performance is much worse when all variables are used.
To solve this problem, sparse clustering methods have been proposed to allow clustering decisions to depend on only a subset of feature variables (the property of sparsity). Prominent sparse clustering methods include sparse principal component analysis (PCA) (3–5) and Sparse K-means (2), among others (6). However, sparse clustering still fails if the true sparsity is a local rather than a global phenomenon (6). More specifically, different subsets of features can be informative for some samples but not all samples, or, in other words, sparsity exists in both features and samples jointly. Biclustering methods are a potential solution to this problem, and further generalize the sparsity principle by considering samples and features as exchangeable concepts to handle local sparsity (6, 7). For example, gene expression data can be represented as a matrix with genes as columns, and subjects as rows (with various and possibly unknown diseases or tissue types). Traditional methods will either cluster the rows—as done, for example, in microarray research, where researchers want to find subpopulation structure among subjects to identify possible common disease status—or cluster the columns, as done, for example, in gene clustering research, where genes are of interest and the goal is to predict the biological function of novel genes from the function of other well-studied genes within the same clusters. In contrast, biclustering involves clustering rows and columns simultaneously to account for the interaction of row and column sparsity. This local sparsity perspective provides an intuition for using sparse singular value decomposition (SSVD) algorithms for biclustering (8–11). SSVD assumes that the signal in the data matrix can be represented by a low-rank matrix 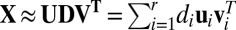 with
with 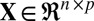 .
.  and
and  contain left and right sparse singular vectors and are orthonormal with only a few nonzero elements (corresponding to local sparsity).
contain left and right sparse singular vectors and are orthonormal with only a few nonzero elements (corresponding to local sparsity). 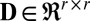 is diagonal (with diagonal elements
is diagonal (with diagonal elements 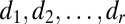 ) with
) with 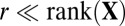 . The outer product of each pair of sparse singular vectors (
. The outer product of each pair of sparse singular vectors (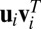 ,
, 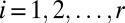 ) will designate two biclusters corresponding to positive and negative elements, respectively.
) will designate two biclusters corresponding to positive and negative elements, respectively.
A common assumption of existing SSVD biclustering methods is that the observed data can be decomposed into a signal matrix plus a fully exchangeable random noise matrix:
where X is the observed data, 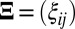 is an
is an 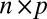 matrix representing the signal, and
matrix representing the signal, and 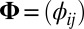 is an
is an 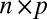 random noise/residual matrix with independent identically distributed (i.i.d.) entries (10, 12, 13). A method based on model 1 is proposed in ref. 9 which minimizes the sum of the Frobenius norm of
random noise/residual matrix with independent identically distributed (i.i.d.) entries (10, 12, 13). A method based on model 1 is proposed in ref. 9 which minimizes the sum of the Frobenius norm of 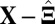 and a penalty function with variable selection, such as the
and a penalty function with variable selection, such as the 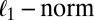 (14) or smoothly clipped absolute deviation (15). A similar loss plus penalty minimization approach can be seen in ref. 11. A different method for SSVD employs iterative thresholding QR decomposition to estimate
(14) or smoothly clipped absolute deviation (15). A similar loss plus penalty minimization approach can be seen in ref. 11. A different method for SSVD employs iterative thresholding QR decomposition to estimate 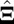 in ref. 10. We refer to ref. 9 as LSHM (for Lee, Shen, Huang, and Marron) and ref. 10 as fast iterative thresholding for SSVD (FIT-SSVD), and compare these approaches to our method. An alternative approach, which is more direct, is based on a mixture model (16, 17). For example, ref. 17 defines the bicluster as a submatrix with a large positive or negative mean. Although these approaches have proven successful in some settings, they are limited by their focus on only the mean signal approximation. In addition, the explicit homogeneous residual variance assumption is too restrictive in many applications.
in ref. 10. We refer to ref. 9 as LSHM (for Lee, Shen, Huang, and Marron) and ref. 10 as fast iterative thresholding for SSVD (FIT-SSVD), and compare these approaches to our method. An alternative approach, which is more direct, is based on a mixture model (16, 17). For example, ref. 17 defines the bicluster as a submatrix with a large positive or negative mean. Although these approaches have proven successful in some settings, they are limited by their focus on only the mean signal approximation. In addition, the explicit homogeneous residual variance assumption is too restrictive in many applications.
To our knowledge, the only extension of the traditional model given in [1] is the generalized PCA approach (18), which assumes that if the random noise matrix were stacked into a vector, 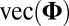 , it would have mean 0 and variance
, it would have mean 0 and variance  , where
, where 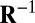 is the common covariance structure of the random variables within the same column, and
is the common covariance structure of the random variables within the same column, and 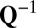 is the common covariance structure of the random variables within the same row. This approach is especially suited to denoising NMR data for which there is a natural covariance structure of the form given above (18). Drawbacks of the generalized PCA method, however, are that it remains focused on mean signal approximation and the structure of
is the common covariance structure of the random variables within the same row. This approach is especially suited to denoising NMR data for which there is a natural covariance structure of the form given above (18). Drawbacks of the generalized PCA method, however, are that it remains focused on mean signal approximation and the structure of 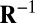 and
and 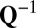 must be explicitly known in advance.
must be explicitly known in advance.
In this paper, we present a biclustering framework based on SSVD called heterogeneous sparse singular value decomposition (HSSVD). This method can detect both mean biclusters and variance biclusters in the presence of unknown heterogeneous residual variance. We also apply our method, as well as competing approaches, to two cancer data sets, one with methylation data and the other with gene expression data. Our method delivers more distinct genetic profile pattern detection and is able to confirm the biological findings originally made for each of the data sets. We also apply our method as well as other competing approaches on synthetic data to compare their performance quantitatively. We demonstrate that our proposed method is robust, location- and scale invariant, and computationally feasible.
Application to Cancer Data
Hypervariability of Methylation in Cancer.
We demonstrate the capability of variance bicluster detection with methylation data in cancer versus normal patients (19). The experiments were conducted by a custom nucleotide-specific Illumina bead array to increase the precision of DNA methylation measurements on previously identified cancer-specific differentially methylated regions (cDMRs) in colon cancer (20). The data set (GEO accession: GSE29505) consists of 290 samples including cancer samples (colon, breast, lung, thyroid, and Wilms’ tumor cancers) and matched normal samples. Each sample had 384 methylation probes which covered 151 cDMRs. The authors of the primary report concluded that cancer samples had hypervariability in these cDMRs across all cancer types (19).
First, we wish to verify that HSSVD can provide a good mean signal approximation of methylation. In this data set, all of the probes measuring the methylation are placed in the cDMRs identified in colon cancer patients. As a result, we would expect that mean methylation levels differ between colon cancer samples and the matched normal samples. Under this assumption, we require the biclustering methods to capture this mean structure before investigating the information gained from variance structure estimation. Note that the numerical range of methylation level is between 0 and 1. Hence, we applied the logit transformation on the original data for further biclustering analysis. We compare three methods, HSSVD, FIT-SSVD and LSHM, all based on SVD. Only colon cancer samples and their matched normal samples are used for this particular analysis. In Fig. 2, we can see from the hierarchical clustering analysis that the majority of colon cancer samples (labeled blue in the sidebar) are grouped together and most of the cDMRs are differentially expressed in colon tumor samples compared with normal samples. The conclusion is the same for all three methods compared, including our proposed HSSVD method.
Fig. 2.

Mean approximation of colon cancer and the normal matched samples. From left to right the methods are HSSVD, FIT-SSVD, and LSHM. Colon cancer samples are labeled in blue, and normal matched samples are labeled in pink in the sidebar. Genes and samples are ordered by hierarchical clustering. Colon cancer patients are clustered together, which indicates that the mean approximations for these three methods achieve the expected signal structure.
Second, our proposed HSSVD method confirms the most important finding in ref. 19 that cancer samples tended to have hypervariability in methylation level regardless of tumor subtype. We compared the mean approximation and variance approximation results of HSSVD. All samples were used in this analysis. The variance approximation of HSSVD (Fig. 3A) shows that nearly all normal samples have low variance compared with cancer samples, and this pattern is consistent across all cDMRs. Notably, our method provides additional information beyond the conclusion from ref. 19. Specifically, our variance approximation suggests that some cancer samples are not characterized by hypervariability in methylation level for certain cDMRs. More precisely, some cDMRs for a few cancer samples (surrounded by normal samples) are predicted to have low variance (lower left part of Fig. 3A). Our method also highlights cDMRs with the greatest contrast variance between cancer and normal samples. The corresponding cDMRs with high contrast variance (especially some of the first and middle columns of Fig. 3A) warrant further study for biological and clinical relevance. We also want to emphasize that the analysis in ref. 19 relies on the disease status information, whereas for HSSVD the disease status is only used for result interpretation. Note that most cancer patients cluster together by hierarchical clustering of the variance approximation from HSSVD. In contrast, clustering the mean approximation from HSSVD in Fig. 3B fails to reveal such a pattern. This indicates that most cancer samples may have hypervariability of methylation as a common feature whereas their mean-level methylation varies from sample to sample. Hence, identifying variance biclusters can provide potential new insight for cancer epigenesis.
Fig. 3.
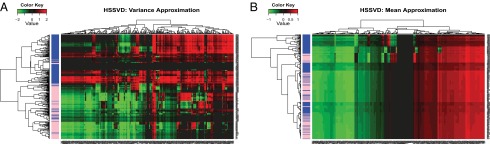
HSSVD approximation result for all samples. (A) Variance approximation; (B) mean approximation. Blue represents cancer samples, and pink represents normal samples in the sidebar. Genes and samples are ordered by hierarchical clustering. Red represents large values, and green represents small values. Only the variance approximation can discriminate between cancer and normal samples. More importantly, within the same gene, the heatmap for the variance approximation indicates that cancer patients have larger variance than normal individuals. This result matches the conclusion in ref. 19. In addition, the cDMRs with the greatest contrast variance across cancer and normal samples are highlighted by the variance approximation, whereas the original paper does not provide such information.
Gene Expression in Lung Cancer.
Some biological settings, in contrast with the methylation example above, do not express variance heterogeneity. Usually, the presence or absence of such heterogeneity is not known in advance for a given research data set. Thus, it is important to verify that the proposed approach remains effective in either case for discovering mean-only biclusters. We now demonstrate that even in settings without variance heterogeneity, HSSVD can better identify discriminative biclusters for different cancer subtypes than other methods, including FIT-SSVD (10), LSHM (9), and traditional SVD. We use a lung cancer data set which has been studied in the statistics literature (9, 10, 17). The samples are a subset of patients (21) having lung cancer with gene expression measured by the Affymetrix 95av2 GeneChip (22). The data set contains the expression levels of 12,625 genes for 56 patients, each having one of four disease subtypes: normal lung (20 samples), pulmonary carcinoid tumors (13 samples), colon metastases (17 samples), and small-cell carcinoma (6 samples).
The performance of different methods is evaluated based on the pattern difference of subtypes based on the mean approximations. For all methods, we set the rank of the mean signal matrix equal to 3 to maintain consistency with the ranks used in FIT-SSVD (10) and LSHM (9). Further, we use the measurement “support” to evaluate the sparsity of the estimated gene signal (10). Support is the cardinality of the nonzero elements in the right and left singular vectors across the three layers (i.e., support is an integer that cannot exceed the data dimension). Smaller support values suggest a sparser model. Table 1 shows that HSSVD, FIT-SSVD and LSHM yield similar levels of sparsity in the gene signal, whereas SVD is not sparse, as expected. Fig. 4 shows checkerboard plots of rank-three approximations by the four methods. Patients are placed on the vertical axis, and the patient order is the same for all images. Patients within the same subtype are stacked together and different subtypes are separated by white lines. Within each image, genes are laid on the horizontal axis and are ordered by the value of 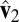 (10). We can see a clear block structure in both the FIT-SSVD and HSSVD methods, indicating biclustering. The block structure suggests we can discriminate the four cancer subtypes using either the FIT-SSVD or HSSVD methods, whereas LSHM and SVD are unable to achieve such separation among subtypes.
(10). We can see a clear block structure in both the FIT-SSVD and HSSVD methods, indicating biclustering. The block structure suggests we can discriminate the four cancer subtypes using either the FIT-SSVD or HSSVD methods, whereas LSHM and SVD are unable to achieve such separation among subtypes.
Table 1.
Cardinality of union support of the first three singular vectors for different methods applied on lung cancer data
| Support | HSSVD | FIT-SSVD | LSHM | SVD |
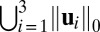 |
4,689 | 4,686 | 4,655 | 12,625 |
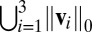 |
56 | 56 | 56 | 56 |
Fig. 4.

Checkerboard plots for four methods. We plot the rank-three approximation for each method. Within each image, samples are laid in rows, and genes are in columns. We order the samples by subtype for all images (top to bottom: carcinoid, colon, normal, and small cell), and different subtypes are separated by white lines. Genes are sorted by the estimated second right singular vector 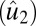 , and we only included genes that are in the support (defined in Table 1). Across all methods, the HSSVD and FIT-SSVD methods provide the clearest block structure reflecting biclusters.
, and we only included genes that are in the support (defined in Table 1). Across all methods, the HSSVD and FIT-SSVD methods provide the clearest block structure reflecting biclusters.
Simulation Study
To evaluate the performance of HSSVD quantitatively, we conducted a simulation study. We compared HSSVD with the most relevant existing biclustering methods, FIT-SSVD and LSHM (9, 10). HSSVD includes a rank estimation component, whereas the other methods do not automatically include this. For this reason, we will use a fixed oracle rank (at the true value) for the non-HSSVD methods. For comparison, we also evaluate HSSVD with fixed oracle rank (HSSVD-O).
The performance of these methods on simulated data was evaluated on four criteria. The first criterion is “sparsity of estimation,” defined as the ratio between the size of the correctly identified background cluster and the size of the true background cluster. The second criterion is “biclustering detection rate,” defined as the ratio of the intersection of the estimated bicluster and the true bicluster over their union (also known as the Jaccard index). For the first two criteria, larger values indicate better performance. The third and fourth criteria are “overall matrix approximation errors” for mean and variance biclusters, consisting of the scaled recovery error for the low-rank mean signal matrix 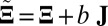 , computed via
, computed via
and the scaled recovery error for the low-rank variance signal matrix 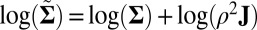 , computed via
, computed via
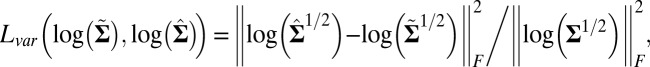 |
with 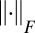 being the Frobenius norm.
being the Frobenius norm.
The simulated data comprise a 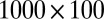 matrix with independent entries. The background entries follow a normal distribution with mean 1 and SD 2. We denote the distribution as
matrix with independent entries. The background entries follow a normal distribution with mean 1 and SD 2. We denote the distribution as 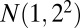 , where
, where 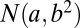 represents a normal random variable with mean a and SD b. There are five nonoverlapping rectangular- shaped biclusters: bicluster 1, bicluster 2, and bicluster 5 are mean clusters, bicluster 3 is a mean and small variance cluster, and bicluster 4 is a large variance cluster. More precisely, bicluster 1 (size
represents a normal random variable with mean a and SD b. There are five nonoverlapping rectangular- shaped biclusters: bicluster 1, bicluster 2, and bicluster 5 are mean clusters, bicluster 3 is a mean and small variance cluster, and bicluster 4 is a large variance cluster. More precisely, bicluster 1 (size 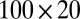 ) is generated from
) is generated from 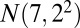 , bicluster 2 (size
, bicluster 2 (size 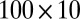 ) is generated from
) is generated from 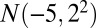 , bicluster 3 (size
, bicluster 3 (size 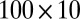 ) is generated from
) is generated from 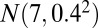 , bicluster 4 (size
, bicluster 4 (size 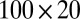 ) is generated from
) is generated from 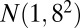 , and bicluster 5 (size
, and bicluster 5 (size 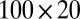 ) is generated from
) is generated from 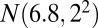 . The biclustering results are shown in Table 2: HSSVD and HSSVD-O can detect both mean and variance biclusters, whereas FIT-SSVD-O and LSHM-O can only detect mean biclusters (where “O” stands for oracle input bicluster number). For mean bicluster detection, all methods performed well because the biclustering detection rates are all greater than 0.7. For variance bicluster detection, HSSVD and HSSVD-O deliver a similar biclustering detection rate. On average, the computation time of LSHM-O is about 30 times that of HSSVD and 60 times that of FIT-SSVD-O.
. The biclustering results are shown in Table 2: HSSVD and HSSVD-O can detect both mean and variance biclusters, whereas FIT-SSVD-O and LSHM-O can only detect mean biclusters (where “O” stands for oracle input bicluster number). For mean bicluster detection, all methods performed well because the biclustering detection rates are all greater than 0.7. For variance bicluster detection, HSSVD and HSSVD-O deliver a similar biclustering detection rate. On average, the computation time of LSHM-O is about 30 times that of HSSVD and 60 times that of FIT-SSVD-O.
Table 2.
Comparison of four methods in the simulation study
| Criteria | HSSVD | HSSVD-O | FITSSVD-O | LSHM-O | ||||
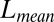 |
0.013 | (0.01) | 0.013 | (0.01) | 0.081 | (0.01) | 0.019 | (0.01) |
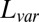 |
0.157 | (0.03) | 0.156 | (0.03) | NA | NA | ||
| Sparsity | 0.950 | (0.04) | 0.950 | (0.03) | 0.988 | (0.02) | 0.997 | (0.01) |
| BLK1 (mean) | 0.861 | (0.10) | 0.862 | (0.10) | 0.818 | (0.08) | 0.872 | (0.08) |
| BLK2 (mean) | 0.934 | (0.18) | 0.936 | (0.17) | 0.939 | (0.18) | 0.976 | (0.01) |
| BLK3 (mean) | 0.972 | (0.10) | 0.974 | (0.10) | 0.971 | (0.11) | 0.987 | (0.01) |
| BLK5 (mean) | 0.977 | (0.11) | 0.948 | (0.11) | 0.977 | (0.11) | 0.996 | (0.01) |
| BLK3 (var) | 0.977 | (0.02) | 0.977 | (0.02) | NA | NA | ||
| BLK4 (var) | 0.628 | (0.25) | 0.633 | (0.24) | NA | NA | ||
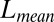 and
and 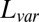 measure the difference between the approximated signal and the true signal, and so smaller is better. For the other measures of accuracy of bicluster detection, the larger the better. The rows BLK1 to BLK5 represent the biclustering detection rate for each bicluster.“-O” indicates that the oracle rank is provided.
measure the difference between the approximated signal and the true signal, and so smaller is better. For the other measures of accuracy of bicluster detection, the larger the better. The rows BLK1 to BLK5 represent the biclustering detection rate for each bicluster.“-O” indicates that the oracle rank is provided.
Both FIT-SSVD and LSHM are provided with the oracle rank as input. We also evaluated an automated rank version for these methods, but determined the performance was worse than the corresponding oracle rank version (results not shown). Note that the input data are standardized to mean 0 and SD 1 element-wisely for FIT-SSVD-O and LSHM-O. Although this step is not mentioned in the original papers (9, 10), this simple procedure is critical for accurate mean bicluster detection. From Table 2, we can see that HSSVD-O provides the best overall performance, while HSSVD is close to the best; however, in practice, the oracle rank is unknown. For this reason, HSSVD is the only fully automated approach which delivers robust mean and variance detection in the present of unknown heterogeneous residual variance among those considered.
Conclusion and Discussion
In this paper, we introduced HSSVD, a statistical framework and its implementation, to detect biclusters with potentially heterogeneous variances. Compared with existing methods, HSSVD is both scale invariant and rotation invariant (as the quantity for scaling is the same for all matrix entries and does not vary by row or column). HSSVD also has the advantage of working on the log scale (Materials and Methods) in estimating the variance components: the log scale makes detection of low-variance (less than 1) biclusters possible, and any traditional SSVD method can be naturally used in our variance detection steps. This method confirms the existence of methylation hypervariability in the methylation data example. Although we use the FIT-SSVD method in our implementation, other low-rank matrix approximation methods are applicable. Moreover, the software implementing our proposed approach was computationally comparable to the other approaches we evaluated.
A potential shortcoming of SVD-based methods is their inability to detect overlapping biclusters. We investigate this problem in the first paragraph of SI Materials and Methods. We show that our method can serve as a denoising process for overlapping bicluster detection. In particular, we can first apply the HSSVD method on the raw data to obtain the mean approximation. Then we can apply a suitable approach, such as the widely used plaid model (16, 23), on the mean approximation to detect overlapping biclusters. This combined procedure improves on the performance of the plaid model when the overlapping biclusters have heterogeneous variance. Hence, our method remains useful in the present of overlapping biclusters.
Another potential issue for HSSVD is the question of whether a low-rank mean approximation plus a low-rank variance approximation could be alternatively represented by a higher-rank mean approximation. In other words, is it possible to detect variance biclusters through mean biclusters only, even though the mean clusters that form the variance clusters would be pseudomean clusters? A detailed discussion of this issue can be found in the second paragraph of SI Materials and Methods. Our conclusion is that the variance detection step in HSSVD is necessary for the following two reasons: First, pseudomean biclusters are completely unable to capture small variance biclusters. Second, although pseudomean biclusters are able to capture some structure from large variance biclusters, such structure is much less accurate than that provided by HSSVD, and can be confounded with one or more true mean biclusters.
Although HSSVD works well in practice, there are a number of open questions that are important to address in future studies. For example, it would be worthwhile to modify the method to allow nonnegative matrix approximations to better handle count data such as next-generation sequencing data (RNA-seq). Additionally, the ability to incorporate data from multiple “omic” platforms is becoming increasingly important in current biomedical research, and it would be useful to extend this work to simultaneous analysis of methylation, gene expression, and microRNA data.
Materials and Methods
Model Assumptions for HSSVD.
We define biclusters as subsets of the data matrix which have the same mean and variance. We assume that there exists a dominate null cluster in which all elements have a common mean and variance and that all other biclusters are restricted to rectangular structures which have either a distinct mean or variance compared with the null cluster. We can also express our model in the framework of a random effect model wherein
where X and  are the same structures given in the traditional model 1, and where we require
are the same structures given in the traditional model 1, and where we require 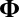 , an
, an 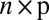 matrix, to have i.i.d. random components with mean 0 and variance 1. Moreover, the “×” in [2] is defined element-wisely: see the next section for details. Added components in the model include
matrix, to have i.i.d. random components with mean 0 and variance 1. Moreover, the “×” in [2] is defined element-wisely: see the next section for details. Added components in the model include 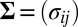 , an
, an 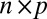 matrix representing the heterogeneous variance signal;
matrix representing the heterogeneous variance signal; 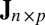 , an
, an 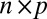 matrix with all values equal to 1; ρ, a finite positive number serving as a common scale factor; and b, a finite number serving as a common location factor. We also make the sparsity assumption that the majority of
matrix with all values equal to 1; ρ, a finite positive number serving as a common scale factor; and b, a finite number serving as a common location factor. We also make the sparsity assumption that the majority of 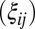 values are 0 and the majority of
values are 0 and the majority of 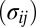 values are 1. Further, just as we assumed for the mean structure
values are 1. Further, just as we assumed for the mean structure  , we also assume that the variance structure
, we also assume that the variance structure 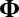 is low rank.
is low rank.
From the definitions, the traditional model 1 is a special case of our model 2, with 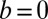 ,
, 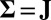 , and
, and 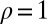 . The presence of b and ρ in the model allows the corresponding method to be scale invariant, while the presence of
. The presence of b and ρ in the model allows the corresponding method to be scale invariant, while the presence of 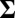 enables us to incorporate heterogeneous variance signals.
enables us to incorporate heterogeneous variance signals.
HSSVD Method.
We propose HSSVD based on the model 2 with a hierarchical structure for signal recovery. First, we properly scale the matrix elements to minimize false detection of pseudomean biclusters which can arise as artifacts of high-variance clusters. This motivates us to add the quadratic rescaling step in the procedure. Then we can detect mean biclusters based on the scaled data and later detect variance biclusters based on the logarithm of the squared residual data after subtracting out the mean biclusters. The quadratic rescaling step works well in practice, as shown in the simulation studies and data analysis. The pseudocode for the algorithm is provided as follows:
-
1.
Input step: Input the raw data matrix
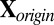 . Standardize
. Standardize 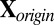 (treat each cell as i.i.d.) to have mean 0 and variance 1. Denote the overall mean of
(treat each cell as i.i.d.) to have mean 0 and variance 1. Denote the overall mean of 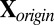 as
as 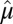 and the overall SD as
and the overall SD as 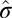 , and let the standardized matrix be defined as
, and let the standardized matrix be defined as 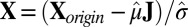 .
. -
2.
Quadratic rescaling: Apply SSVD on
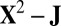 to obtain the approximation matrix U.
to obtain the approximation matrix U. -
3.
Mean search: Let
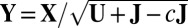 , where c is a small nonpositive constant to ensure that
, where c is a small nonpositive constant to ensure that 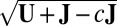 exists. Then, apply SSVD on Y to obtain the approximation matrix
exists. Then, apply SSVD on Y to obtain the approximation matrix 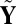 .
. -
4.
Variance search: Let
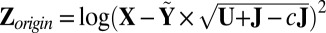 , center
, center 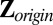 to have mean 0, and denote the centered version as Z. Perform SSVD on Z to obtain the approximation matrix
to have mean 0, and denote the centered version as Z. Perform SSVD on Z to obtain the approximation matrix 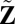 .
. -
5.
Background estimation: Let
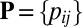 denote the
denote the 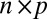 matrix of indicators of whether the corresponding cells belong to the background cluster, with
matrix of indicators of whether the corresponding cells belong to the background cluster, with 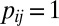 if both
if both 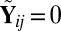 and
and 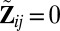 , and
, and 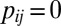 otherwise. Based on the assumption that most elements in the matrix should be in the null cluster, we can estimate
otherwise. Based on the assumption that most elements in the matrix should be in the null cluster, we can estimate 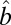 with
with 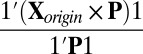 and
and 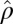 with
with 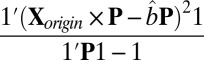 , where 1 is a vector with all elements equal to 1.
, where 1 is a vector with all elements equal to 1. -
6.
Scale back: Define
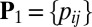 , with
, with 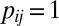 if
if 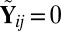 ,
, 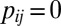 otherwise. Similarly, define
otherwise. Similarly, define 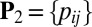 , with
, with 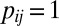 if
if 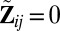 ,
, 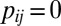 otherwise. The mean
otherwise. The mean 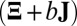 approximation is computed with
approximation is computed with 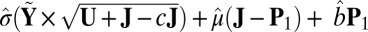 , and the variance
, and the variance 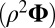 approximation is computed with
approximation is computed with 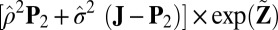 .
.
The operators ×, /, 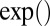 ,
, ,
, 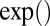 ,
, 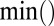 , and
, and 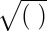 used above are defined element-wisely when they are applied to the matrix, e.g.,
used above are defined element-wisely when they are applied to the matrix, e.g., 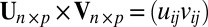 . In all steps involving SSVD, we implement the FIT-SSVD method (10). We use FIT-SSVD because it is computationally fast and has similar or superior performance compared with other competing methods under the homogeneous variance assumption (10). The matrix
. In all steps involving SSVD, we implement the FIT-SSVD method (10). We use FIT-SSVD because it is computationally fast and has similar or superior performance compared with other competing methods under the homogeneous variance assumption (10). The matrix 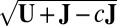 provides a working variance level estimate of the data and makes our method more robust. Note that the reason for working on the log scale for the variance detection is twofold. First, working on the log scale makes the detection of the deflated variance (less than 1) bicluster possible. Intuitively, as variance measures deviance from the mean, we can work on the squared residuals to find the variance structure. For the deflated variance bicluster setting, if the mean structure is estimated correctly, the residuals within the bicluster are close to zero. The SSVD-based methods shrink the small nonzero elements to zero to achieve sparsity. As a result, if we work on the squared residuals directly, the SSVD based methods will fail to detect the low variance structure. Second, to use the well-established SSVD method in the variance detection steps we need to work on the log scale. To see this, we can rewrite the equation in [2] as
provides a working variance level estimate of the data and makes our method more robust. Note that the reason for working on the log scale for the variance detection is twofold. First, working on the log scale makes the detection of the deflated variance (less than 1) bicluster possible. Intuitively, as variance measures deviance from the mean, we can work on the squared residuals to find the variance structure. For the deflated variance bicluster setting, if the mean structure is estimated correctly, the residuals within the bicluster are close to zero. The SSVD-based methods shrink the small nonzero elements to zero to achieve sparsity. As a result, if we work on the squared residuals directly, the SSVD based methods will fail to detect the low variance structure. Second, to use the well-established SSVD method in the variance detection steps we need to work on the log scale. To see this, we can rewrite the equation in [2] as 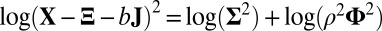 , which is similar to the model in [1]. Consequently, we can apply any methods which are applicable to [1] in our variance detection step if we work on the log scale and
, which is similar to the model in [1]. Consequently, we can apply any methods which are applicable to [1] in our variance detection step if we work on the log scale and 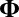 is low rank. We also want to point out that results obtained directly from FIT-SSVD are relative to the location and scale of the background cluster. In addition, we have scaled the data in the “input step.” To provide a correct mean and variance approximation of the original data, we need the “scale back” step. Assuming that the detection of null clusters is close to the truth, then the pooled mean and variance estimates based on elements exclusively from the identified null cluster (
is low rank. We also want to point out that results obtained directly from FIT-SSVD are relative to the location and scale of the background cluster. In addition, we have scaled the data in the “input step.” To provide a correct mean and variance approximation of the original data, we need the “scale back” step. Assuming that the detection of null clusters is close to the truth, then the pooled mean and variance estimates based on elements exclusively from the identified null cluster (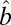 and
and 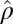 ) are more accurate than estimates based on all elements of the matrix (
) are more accurate than estimates based on all elements of the matrix (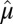 and
and 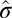 ). As a result, we need to use the comprehensive formula proposed in the scale back step.
). As a result, we need to use the comprehensive formula proposed in the scale back step.
The FIT-SSVD method, as well as any other SVD-based method, requires an approximation of the rank of the matrix (which is essentially the number of true biclusters) as input. We adapt the bicross validation method (BCV) by ref. 24 for rank estimation, and we notice that in some cases the rank is underestimated. For this reason, we introduce additional steps following a BCV rank estimation of rank k: First, we approximate the data with a sparse matrix 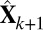 (rank = k + 1), where
(rank = k + 1), where 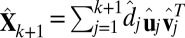 . Define the proportion of variance explained by the top i rank sparse matrix as
. Define the proportion of variance explained by the top i rank sparse matrix as 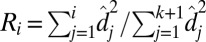 (25).
(25). 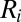 is between 0 and 1 and is increasing with i, and we believe that the redundant components of the sparse matrix should not contribute much to the total variance. The final rank estimation for HSSVD is the smallest integer r which satisfies
is between 0 and 1 and is increasing with i, and we believe that the redundant components of the sparse matrix should not contribute much to the total variance. The final rank estimation for HSSVD is the smallest integer r which satisfies 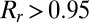 , and
, and 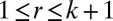 . Note that FIT-SSVD (10) used the modified BCV method for rank estimation; however, the authors require that most rows (the whole row) and most columns (the whole column) are sparse, which appears to be too restrictive. In practice, this assumption is violated if the data are block diagonal or have certain other commonly assumed data structures. For this reason, we use the original BCV method as our starting point.
. Note that FIT-SSVD (10) used the modified BCV method for rank estimation; however, the authors require that most rows (the whole row) and most columns (the whole column) are sparse, which appears to be too restrictive. In practice, this assumption is violated if the data are block diagonal or have certain other commonly assumed data structures. For this reason, we use the original BCV method as our starting point.
Supplementary Material
Acknowledgments
The authors thank the editor and two referees for helpful comments. The authors also thank Dr. Dan Yang for sharing part of her code. This work was supported in part by Grant P01 CA142538 from the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. X.S. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304376110/-/DCSupplemental.
References
- 1. Hastie T, Tibshirani R, Friedman J (2009) The Elements of Statistical Learning. (Springer, New York), Vol 1, pp 460–462.
- 2.Witten DM, Tibshirani R. A framework for feature selection in clustering. J Am Stat Assoc. 2010;105(490):713–726. doi: 10.1198/jasa.2010.tm09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma Z (2013) Sparse principal component analysis and iterative thresholding. Ann Statist 41(2):772–801.
- 4.Shen H, Huang J. Sparse principal component analysis via regularized low rank matrix approximation. J Multivariate Anal. 2008;99(6):1015–1034. [Google Scholar]
- 5.Zou H, Hastie T, Tibshirani R. Sparse principal component analysis. J Comput Graph Statist. 2006;15(2):265–286. [Google Scholar]
- 6.Kriegel HP, Kröger P, Zimek A. Clustering high-dimensional data: A survey on subspace clustering, pattern-based clustering, and correlation clustering. ACM Trans Knowl Discov Data. 2009;3(1):1–58. [Google Scholar]
- 7.Cheng Y, Church GM. Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol. 2000;8:93–103. [PubMed] [Google Scholar]
- 8.Busygin S. Biclustering in data mining. Comput Oper Res. 2008;35(9):2964–2987. [Google Scholar]
- 9.Lee M, Shen H, Huang JZ, Marron JS. Biclustering via sparse singular value decomposition. Biometrics. 2010;66(4):1087–1095. doi: 10.1111/j.1541-0420.2010.01392.x. [DOI] [PubMed] [Google Scholar]
- 10. Yang D, Ma Z, Buja A (2011) A sparse SVD method for high-dimensional data. arXiv: 1112.2433.
- 11.Witten DM, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009;10(3):515–534. doi: 10.1093/biostatistics/kxp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoff PD. Model averaging and dimension selection for the singular value decomposition. J Am Stat Assoc. 2006;102(478):674–685. [Google Scholar]
- 13.Johnstone IM, Lu AY. On consistency and sparsity for principal components analysis in high dimensions. J Am Stat Assoc. 2009;104(486):682–693. doi: 10.1198/jasa.2009.0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc, B. 1996;58(1):267–288. [Google Scholar]
- 15.Fan J, Li R. Variable selection via nonconcave penalized likelihood and its oracle properties. J Am Stat Assoc. 2001;96(456):1348–1360. [Google Scholar]
- 16.Lazzeroni L, Owen A. Plaid models for gene expression data. Statist Sinica. 2002;12:61–86. [Google Scholar]
- 17.Shabalin AA, Weigman VJ, Perou CM, Nobel AB. Finding large average submatrices in high dimensional data. Ann Appl Stat. 2009;3(3):985–1012. [Google Scholar]
- 18. Allen GI, Grosenick L, Taylor J (2011) A generalized least squares matrix decomposition. arXiv: 1102.3074.
- 19.Hansen KD, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Hayes DN, Nobel A, Marron JS. Statistical significance of clustering for high-dimension, low-sample size data. J Am Stat Assoc. 2008;103(483):1281–1293. [Google Scholar]
- 22.Bhattacharjee A, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA. 2001;98(24):13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner H, Bailey T, Krzanowski W. Improved biclustering of microarray data demonstrated through systematic performance tests. Comput Stat Data Anal. 2005;48(2):235–254. [Google Scholar]
- 24.Owen AB, Perry PO. Bi-cross-validation of the SVD and the nonnegative matrix factorization. Ann Appl Stat. 2009;3(2):564–594. [Google Scholar]
- 25.Allen GI, Maletić-Savatić M. Sparse non-negative generalized PCA with applications to metabolomics. Bioinformatics. 2011;27(21):3029–3035. doi: 10.1093/bioinformatics/btr522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.