

Abstract
8-Oxoguanine (8-oxoG), induced by reactive oxygen species and arguably one of the most important mutagenic DNA lesions, is prone to further oxidation. Its one-electron oxidation products include potentially mutagenic guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp) because of their mispairing with A or G. All three oxidized base-specific DNA glycosylases of Escherichia coli, namely endonuclease III (Nth), 8-oxoG-DNA glycosylase (MutM) and endonuclease VIII (Nei), excise Gh and Sp, when paired with C or G in DNA, although Nth is less active than the other two. MutM prefers Sp and Gh paired with C (kcat/Km of 0.24–0.26 min–1 nM–1), while Nei prefers G over C as the complementary base (kcat/Km – 0.15–0.17 min–1 nM–1). However, only Nei efficiently excises these paired with A. MutY, a 8-oxoG·A(G)-specific A(G)-DNA glycosylase, is inactive with Gh(Sp)·A/G-containing duplex oligonucleotide, in spite of specific affinity. It inhibits excision of lesions by MutM from the Gh·G or Sp·G pair, but not from Gh·C and Sp·C pairs. In contrast, MutY does not significantly inhibit Nei for any Gh(Sp) base pair. These results suggest a protective function for MutY in preventing mutation as a result of A (G) incorporation opposite Gh(Sp) during DNA replication.
INTRODUCTION
8-Oxoguanine (8-oxoG or G*) is one of the most common genomic lesions in normal tissues. It is readily induced in DNA by a variety of endogenous reactive oxygen species (ROS) which are generated during oxidative phosphorylation and inflammatory responses, and also by a variety of external agents (1,2). In fact, 8-oxoG is often used as the marker of oxidative stress in cells (3). Oxidative DNA damage has been implicated in the etiology of many degenerative diseases including cancer, and also in aging (4,5).
8-oxoG is mutagenic because of its propensity to mispair with A during replication by DNA polymerases (6,7). Incorporation of A opposite 8-oxoG leads to a G·C→T·A transversion mutation, which is the second most common somatic mutation in human cancers, and is especially frequent in the mutational spectrum of the p53 tumor suppressor gene (8). Thus, 8-oxoG is considered to be a major endogenous mutagenic lesion that may broadly contribute to spontaneous cellular transformation.
It has long been recognized that 8-oxoG is readily subjected to further oxidation, and is a likely target of endogenous oxidizing agents. There are several important models for oxidation of 8-oxoG: (i) alkaline KMnO4 or iodine-mediated oxidation of uric acid; (ii) electrochemical oxidation; and (iii) photo-oxidation (9). Electrochemical oxidation of 8-oxoG generates guanidinohydantoin (Gh) and related compounds (10). We have recently characterized the oxidation products of 8-oxoG using the one-electron oxidant IrCl62– (11). In previous mass spectrometric analysis of oligonucleotides, two IrCl62–-mediated oxidation products of 8-oxoG were obtained: the major product with mass M–10, and a minor product of mass M+16 in duplex DNA. M–10 was then identified as Gh, while spiroiminodihydantoin (Sp) could account for the M+16 product as shown in Figure 1 (12,13). In a nucleoside model study (W.Luo, J.G.Muller, E.M.Rachlin and C.J.Burrows, unpublished observation), the Gh lesion was found to be in pH-dependent equilibrium with its isomer iminoallantoin (Ia). The relative abundance of Gh and Ia in duplex DNA is currently unknown, but the reversibility of the Gh–Ia equilibrium ensures that synthetic oligomers will adjust to their most relevant duplex structures after annealing. Therefore, for convenience, the mixture of Gh + Ia isomers will be referred to as simply Gh throughout the text. The redox potential of IrCl62– is 0.90 V versus normal hydrogen electrode (14), and could be optimum for oxidation of only oxopurines and not of normal DNA bases, thus making it a convenient model for one-electron oxidation events in the cell. A number of transition metal complexes also trigger one-electron oxidation of purines, including those of carcinogenic nickel (15,16). Organonickel (II) complexes, particularly of peptide ligands, are redox active in the presence of certain oxidants, leading to oxidation of guanine and 8-oxoG in DNA (17). These oxidants have been studied both as models for nickel carcinogenesis and as probes for DNA (RNA) structural studies, because of their ability to oxidatively modify G residues (18).
Figure 1.

Structures of lesions investigated in this study. The (M–10) species is the sole product of 8-oxoG oxidation by Ir(IV) in duplex or single-stranded DNA at low temperature, while Sp predominates when 8-oxoG is present as the nucleoside or under condition of high temperature oxidation. The Gh lesion was found to be in pH-dependent equilibrium with its isomer Ia when present as the nucleoside.
We have recently shown that Gh as well as Sp in the template DNA directs incorporation of dAMP and dGMP by Escherichia coli Kf exo-DNA polymerase which suggests that both Gh and Sp are potential mutagenic lesions that could induce G→T and G→C transversion mutations (O.Kornyushyna and C.Burrows, unpublished observations; 11).
Repair of 8-oxoG in DNA occurs primarily via the base excision repair (BER) process, which is initiated by excision of the base lesion by ubiquitous 8-oxoG-DNA glycosylase (OGG). The major OGG in E.coli, named MutM or Fpg, removes 8-oxoG from DNA when paired with C, T or G but not with A (19). When A is incorporated opposite unrepaired 8-oxoG, MutY can remove this A and thus provides a second chance for correct pairing with C during subsequent repair replication. In the following step, 8-oxoG could be removed by OGG, thus restoring the original DNA. However, 8-oxoG is not only generated in DNA in situ, but may also be incorporated into DNA from ROS-induced 8-oxodGTP in the nucleotide pool (6,20,21). MutT, a ubiquitous 8-oxodGTPase, prevents incorporation of 8-oxodGMP by hydrolyzing the triphosphate (6).
Apart from MutM, two other oxidatively damaged base-specific DNA glycosylases/AP lyases, namely Nth and Nei, are present in E.coli. These enzymes excise oxidative base lesions, and subsequently cleave the phosphodiester backbone, in the first step of the repair process (22,23). Nth, discovered on the basis of its endonucleolytic activity on X-ray- and heavily UV-irradiated DNA (24,25), removes primarily oxidized pyrimidines. Nei, structurally homologous to MutM but not to Nth, was identified as a second pyrimidine-specific glycosylase/AP lyase (26). We have recently discovered that Nei also possesses significant OGG activity (27).
In an earlier study, excision of Gh and Sp in synthetic oligodeoxynucleotides by MutM was demonstrated (13). The present work confirms those results and presents a comparison of base excision of Gh and Sp by all three oxidative base-specific E.coli DNA glycosylases. This study further shows that these enzymes are differentially inhibited by MutY which binds to the lesion-containing duplex DNAs without utilizing them as substrates.
MATERIALS AND METHODS
Preparation of substrate DNA
The following oligodeoxynucleotides were synthesized with an Applied Biosystems 392B synthesizer following the manufacturer’s protocols and incorporating β-mercaptoethanol in the final deprotection step for DNA sequences containing 8-oxoG: 5′-TCATGGGTCXTCGGTATA-3′ (Seq. A, Fig. 2) and 5′-TATACCGANGACCCATGA-3′ (Seq. B, Fig. 2), where X = 8-oxoG or G, and N = C, G or A. The oligos were purified by PAGE using 20% polyacrylamide/7 M urea.
Figure 2.

Sequences of Gh, Sp or 8-oxoG-containing oligo substrates used in this study. Seq. A, 18mer oligo with Gh, Sp or 8-oxoG (indicated by X) at position 10; Seq. B, complementary strand of Seq. A, where N represents A, G or C; Seq. C, 9mer marker corresponding to the 5′ segment of Seq. A.
Oligodeoxynucleotides containing Gh or Sp were prepared by oxidation of a 50 µl solution containing 12 µM Seq. A (X = 8-oxoG) with Na2IrCl6 (100 µM final concentration) in 10 mM NaPO4 pH 7.0 and 100 mM NaCl at 4°C for 1 h which resulted in conversion of 8-oxoG to Gh. The reaction product was then dialyzed against water for 24 h in a dialysis bag with 2 kDa cut-off. The Sp-oligo was also produced from Seq. A in the same way except that the reaction was performed at 50°C (Fig. 1). The samples were analyzed by negative ion electrospray MS (Micromass Quattro II) as previously described (11,13), and their purity was estimated to be ∼95% based on the intensities of related molecular ions.
Purification of enzymes
Purification of recombinant Nei, Nth and MutY polypeptides to near homogeneity has been reported previously (27–30). For purification of MutM, E.coli HB101 containing a MutM expression plasmid cloned into the NdeI and XhoI site of pET22b vector, a gift of Dr Y.W.Kow, was used. The wild-type coding sequence of the mutM gene in the plasmid was first confirmed by sequencing. Expression of recombinant MutM in E.coli and its purification was carried out in the same way as for Nei (27). Briefly, the enzyme was purified from sonicated bacterial extract via a series of steps starting with the removal of nucleic acids by polymin P precipitation and followed by fractional precipitation of the enzyme in 30–60% saturated ammonium sulfate. The ammonium sulfate pellet was dialyzed in buffer A (20 mM Tris–HCl pH 7.5, 1 mM dithiotheritol,10% glycerol) containing 50 mM NaCl. The dialyzate was then sequentially subjected to chromatography through Hi Trap SP-Sepharose (Amersham Pharmacia) and Superdex 75. Active fractions were quickly frozen in liquid nitrogen and then stored at –80°C.
Assay of lesion-specific strand incision
The DNA glycosylases Nei, Nth and MutM used in this study have intrinsic AP lyase activity which causes DNA strand cleavage at the AP site generated after base excision catalyzed by these enzymes. Because the AP sites are better substrates for these glycosylases than the base lesions, no free AP site persists in the oligo substrates during enzymatic reaction (22). Thus, incision of the 5′ 32P-labeled lesion-containing strand in the duplex oligo was used to measure the combined DNA glycosylase/AP lyase activity of these enzymes. The incision assay was carried out at 37°C for 20 min in a 20 µl reaction mixture containing 14 nM duplex oligo (20–25 fmol 32P-labeled oligo), 2 nM enzyme in buffer B (20 mM Tris–HCl pH 7.5, 1 mM EDTA, 50 mM NaCl) containing 100 µg/ml BSA. After termination of the reaction, the substrate oligo and its 9mer cleaved product were separated by electrophoresis in a 20% denaturing polyacrylamide gel containing 7 M urea, 90 mM Tris–borate pH 8.3 and 2 mM EDTA. The radioactivity in gel bands was then quantified with a PhosphorImager (Molecular Dynamics). MutY activity was assayed as previously described (29,30).
Analysis of trapped enzyme complexes
DNA glycosylase/AP lyases excise base lesions by breaking the glycosylic bond via formation of a Schiff base intermediate, resulting in cleavage of the DNA phosphodiester backbone after β or β,δ elimination reaction (31,32). This transient covalent intermediate forms a stable product when reduced by NaBH4 or NaCNBH3, and is called the ‘trapped complex’. DNA trapping reactions were carried out at 37°C for 30 min in a 20 µl reaction mixture containing 15 nM substrate DNA (20 fmol 32P-labeled substrate oligo) and 5 nM enzyme in buffer B containing 50 mM NaCNBH3, as described previously (33). The reaction was terminated by heating at 100°C for 5 min, after addition of equal volume of 2× gel loading buffer. The trapped complexes were then analyzed by 12% SDS–PAGE, and the gels were dried on DE-81 paper for analysis of radioactivity.
Kinetics of Nei, Nth and MutM reactions with Gh- and Sp-containing duplex oligonucleotide substrates
The DNA glycosylases (1 nM active enzyme) were incubated at 37°C with Gh- or Sp-containing duplex oligonucleotide (5–15 nM) for 5 min. We had confirmed earlier that these enzymes maintain linear kinetics for at least 20 min under our experimental conditions. The reaction buffer (20 µl) was essentially the same as described earlier for assay of lesions. The reaction was stopped by heating at 65°C for 10 min after addition of 10 µl formamide/dye mixture (90% formamide, 5 mM EDTA, 0.05% xylene cyanol and 0.05% bromophenol blue) and the products were then resolved on a 20% polyacrylamide–urea gel. The radioactivity in the gel bands was quantified with a PhosphorImager and the data were fitted to the Michaelis–Menten equation by using data analysis/graphics application program of Kaleidagraph.
Gel mobility-shift assay for DNA binding
Duplex oligonucleotides containing Gh or Sp paired with A, G or C were prepared by labeling the lesion-containing single strand oligo with 5′-[γ-32P]ATP, and then annealing to its complementary strand containing A, G or C opposite the base lesion. The duplex oligo (0.5 nM) was incubated with 15 nM of MutY at 22°C for 25 min in a buffer containing 25 mM HEPES pH 7.8, 0.5 mM EDTA, 0.5 mM DTT, 12% glycerol, 50 mM NaCl, 10 µg/ml poly(dI-dC) and 100 µg/ml BSA. Reactions were performed in duplicate, with one set of reaction containing 50-fold excess of competitor duplex oligo whose sequence was identical to that of the substrate except for the presence of a G·C pair at the site of Gh(Sp)-containing pair. Protein–DNA complexes were separated in 5% non-denaturing polyacrylamide gels.
Inhibition of lesion excision by MutY
Substrate oligos (5 nM) were preincubated with MutY (10 or 100 nM) for 5 min at room temperature and then incubated with 5 nM of Nei, Nth or MutM at 37°C for 5 min in a reaction buffer (20 µl) as described earlier. The reaction was stopped by heating at 65°C for 5 min after addition of 10 µl formamide dye mix. The reaction products were resolved by electrophoresis, and the radioactivity was quantified as before.
RESULTS
Purification of enzymes
All of the DNA glycosylases (Nei, Nth and MutM) except MutY were expressed in, and purified from mutM nei E.coli to minimize possible cross-contamination with other enzymes, and were purified to apparent homogeneity (Fig. 3). For kinetic studies, the active fractions of these proteins were calculated as described previously (34). MutM, Nei and Nth preparations were found to contain ∼65, 60 and 50% of enzymatically active molecules, respectively.
Figure 3.
SDS–PAGE (12% polyacrylamide) analysis of purified DNA glycosylases. M, prestained low-range molecular weight marker (Bio-Rad).
Repair of Gh and Sp by Nei, Nth and MutM proteins and the effect of lesion base pairing on excision activity
We tested the incision activity of Nei, Nth and MutM for Gh and Sp lesions paired to C, G and A in duplex oligos. It is evident from Figure 4 that both Gh- and Sp-containing oligo strands were incised efficiently by all three enzymes when C and G were present in the duplex oligos opposite those lesions. However, when A was present opposite either Gh or Sp, MutM did not excise these lesions to a detectable extent under our experimental conditions, and Nth did so only poorly. In contrast, Nei had strong activity towards both Gh·A and Sp·A pairs. It is also evident from these results that MutM prefers C over G opposite the lesion, whereas Nei prefers G as the complementary base.
Figure 4.

Incision of Gh- and Sp-containing duplex oligos containing the lesions opposite G, C or A with Nth, Nei and MutM as indicated in the figure. Nth, Nei or MutM (2 nM) was incubated with substrate oligo (14 nM) at 37°C for 20 min and the reaction products were separated in 20% polyacrylamide gel containing 7 M urea. Lanes 1–4, Sp·G; lanes 5–8, Gh·G; lanes 9–11, Sp·C; lanes 12–14, Gh·C; lanes 15–17, Sp·A; lanes 18–20, Gh·A. Lower panel, quantitative comparison of activity. The amount of product (fmol) is represented in the bar diagram.
We examined the nature of the DNA cleavage products generated by these enzymes (Fig. 5). As expected, based on its reaction with other substrates, Nth carried out β elimination (Fig. 5, lane 1), generating a 3′-phospho α,β unsaturated aldehyde. In contrast, Nei and MutM, which are known to carry out β,δ elimination with standard substrate lesions, generated 3′-phosphates at the abasic site, characteristic of such a reaction (Fig. 5, lanes 3 and 5). These different termini could be distinguished by characteristic electrophoretic mobilities of the oligo fragments containing these ends (33). Figure 5 shows that the upstream fragment contained 3′-phosphate for both MutM and Nei, as the mobility of their reaction products was identical to that of the product of a 8-oxoG·C duplex treated with MutM (Fig. 5, lane 7). The Nth-treated product, on the other hand, migrated more slowly, suggesting β elimination for Nth. This was further confirmed by treating the reaction products of these enzymes with E.coli endonuclease IV (Nfo). Nfo-treated products in all cases contain 3′-OH termini (Fig. 5, lanes 2, 4 and 6), on the basis of their identical mobility to the 9mer (3′-OH) marker (Fig. 5, lane 9). Such oligos migrate in between β,δ and β elimination reaction products as we and others have shown previously (33).
Figure 5.
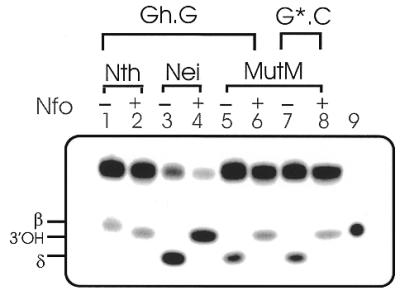
Characterization of 3′-termini at the cleavage sites of Gh-containing oligo duplexes generated by DNA glycosylases. Reaction products of Gh·G (100–150 fmol) with 50 fmol of Nth (lanes 1 and 2), Nei (lanes 3 and 4) and MutM (lanes 5 and 6) in the absence (–) or presence (+) of Nfo (5 ng, Trevigen); lane 7, MutM reaction product of 8-oxoG·C oligo was used as a marker for the β,δ elimination product; lane 8, 3′-OH-containing marker generated by further Nfo digestion of incubation of MutM product in lane 7; lane 9, 5′-32P-labeled 9mer marker with 3′-OH terminus (Seq. C). The positions of β, 3′-OH and β,δ elimination products of the Gh oligo are indicated.
We confirmed that no free AP sites were present in the reaction products of Nth, Nei or MutM. These would be generated as a result of base excision, but without further cleavage of the resulting AP site. After phenol/chloroform extraction, the enzyme reaction products were treated with Nfo; the near equivalent amounts of Nfo-treated and untreated reaction products indicated that the glycosylase and lyase acted in concert so that no free AP sites were generated (data not shown).
Schiff base intermediates
Nth, Nei and MutM formed trapped complexes with Sp- and Gh-containing substrates (Fig. 6). Although we tested only Gh·G and Sp·G pair-containing duplex oligos, it is reasonable to assume that similar trapped complexes would be obtained if Gh and Sp were paired with either C, T or A. Such trapped complexes are expected to be produced by these enzymes, all of which have AP lyase activity. These results indicate that all three enzymes function as DNA glycosylase/AP lyases for Gh and Sp residues in DNA.
Figure 6.

Analysis of Nth, Nei and MutM trapped complexes with cleaved products of Sp- and Gh-containing oligos at 37°C for 30 min as described in Materials and Methods; the trapped complexes were analyzed by 12% SDS–PAGE. The enzymes (5 nM) were incubated with different Sp- and Gh-containing duplex oligos (15 nM). Oligos without enzyme (lanes 1 and 2), with Nth (lanes 3 and 4), with Nei (lanes 5 and 6) and with MutM (lanes 7 and 8). The positions of trapped complexes and free DNA are indicated.
Kinetic properties of Nei, Nth and MutM
We have determined the kinetic parameters of these DNA glycosylases for Gh·N and Sp·N (N denotes C, G or A)-containing duplex oligo substrates (Table 1). These enzymes incised Gh and Sp in a dose- and time-dependent manner. The Km and kcat values were determined in the linear range of enzyme activity, and active enzyme concentrations were used to calculate the kinetic parameters. Such analysis indicates that Nei preferred G as the complementary base, followed by A and C, while MutM prefered C compared to G. Taken together, the activity of MutM follows the order of Gh(Sp)·C > G >> A, and of Nei Gh(Sp)·G > A > C. This dichotomy mirrors the pattern of 8-oxoG repair by MutM and Nei when this lesion is paired with different bases (19,27).
Table 1. Kinetic parameters of Nei, Nth and MutM for Gh- and Sp-containing duplex oligo substratesa.
| Substrate | Nei | Nth | MutM |
| |
Km (nM) |
kcat (min–1) |
kcat/Km (min–1 nM–1) |
Km (nM) |
kcat (min–1) |
kcat/Km (min–1 nM–1) |
Km (nM) |
kcat (min–1) |
kcat/Km (min–1 nM–1) |
| Sp·C |
30 ± 4.0 |
1.6 |
0.05 |
32 ± 4.5 |
0.50 |
0.015 |
25 ± 3.0 |
6.0 |
0.24 |
| Sp·G |
38 ± 5.2 |
5.8 |
0.15 |
34 ± 5.0 |
1.14 |
0.030 |
75 ± 8.0 |
1.5 |
0.02 |
| Sp·A |
40 ± 6.0 |
2.5 |
0.06 |
|
ND |
|
|
ND |
|
| Gh·C |
40 ± 5.8 |
1.0 |
0.03 |
39 ± 3.8. |
0.39 |
0.010 |
22 ± 2.5 |
5.9 |
0.26 |
| Gh·G |
35 ± 3.8 |
6.1 |
0.17 |
42 ± 4.8 |
0.50 |
0.012 |
65 ± 7.0 |
1.7 |
0.03 |
| Gh·A | 45 ± 5.0 | 3.0 | 0.07 | ND | ND |
aAll substrate oligo duplexes have identical sequence except for Gh or Sp residue at position 10.
ND, not determined.
These data are in qualitative agreement with our earlier study on the kinetics of excision of Sp and Gh lesions with MutM (13). However, a quantitative comparison with those results is not possible because we have used conditions of steady state reaction of the enzymes in the present study. Nth did not show significant base discrimination, and had an overall poor activity toward these substrates compared to MutM and Nei. Kinetic parameters of MutM and Nth for the Sp(Gh)·A base pairs were not determined because of their negligible activity with these base pairs.
MutY binds to duplex oligo containing Sp or Gh
Since MutY primarily removes A or G when these are paired with 8-oxoG (21,30,35), we tested the activity of MutY in excising the base paired with Gh or Sp in the duplex oligo. MutY did not excise the base paired with Gh or Sp (data not shown), but it formed specific, stable complexes with A, C or G opposite Sp or Gh in the duplex oligos (Fig. 7). The affinity of MutY for Sp(Gh)·C-containing oligo was weaker than for oligos with G or A opposite the lesion. However, the binding was specific for the lesion because the complex was not competed with a 50-fold excess control oligo (Fig. 7). As expected, the radioactivity in the complex was reduced when excess unlabeled lesion-containing oligo was included in the binding reaction (data not shown). The control oligo was identical in sequence to the lesion-containing oligos except for substitution of the Gh or Sp bases with G, which is paired with C in the complementary strand.
Figure 7.

Gel mobility shift analysis of MutY (15 nM) complex with duplex oligos (0.5 nM) containing Gh and Sp, in the presence (lanes 3, 6, 9, 12, 15 and 18) or absence of competitor DNA (lanes 2, 5, 8, 11, 14 and 17). Competitor oligo DNA (50-fold molar excess) has the same sequence as the labeled substrate except for the presence of G in place of Sp or Gh. No enzyme (lanes 1, 4, 7, 10, 13 and 16). The positions of free DNA and protein–DNA complexes are indicated.
Inhibitory effects of MutY on the repair of Gh or Sp by E.coli DNA glycosylases
Since MutY binds Sp- or Gh-containing duplex oligos efficiently, and could potentially interfere with excision of Sp and Gh by Nei, Nth or MutM, we tested the possible consequences of competition between MutY and the latter enzymes during repair of these lesions. It is evident from Figures 8 and 9 that MutM was significantly inhibited by 10 nM MutY during repair of Sp or Gh lesion when G, but not C, was present as the complementary base. On the contrary, Nei was not inhibited significantly by the same concentration of MutY, regardless of whether the opposite base was C, A or G. However, MutY did inhibit Nei significantly but a much higher concentration (100 nM). On the other hand, Nth was inhibited by MutY irrespective of the complementary base.
Figure 8.

Inhibition of Nth, Nei and MutM activity by MutY for Gh-containing oligo substrates. Upper panel, analysis of cleaved products of Gh·C (lanes 1–9), Gh·G (lanes 10–18) and Gh·A (lanes 19–21) generated by Nei (lanes 1–3, 10–12 and 19–21), Nth (lanes 4–6 and 13–15) and MutM (lanes 7–9 and 16–18) in the absence (–) or presence of MutY (+, 10 nM; ++, 100 nM). Lower panel, quantitative comparison of MutY-dependent inhibition of strand cleavage for different substrates. Identical lane numbers are used in two panels. Other details are as described in Figure 4.
Figure 9.

MutY inhibition on the catalytic activity of Nei, Nth and MutM with an Sp-containing duplex oligo. The bar graph (lower panel) provides a quantitative comparison of inhibition as described in Figure 8.
DISCUSSION
8-oxoG is one of the most common oxidized DNA base lesions because of the ease of its formation by all types of ROS. However, the inherent instability of 8-oxoG and its further oxidation have been recognized previously. Several groups reported oxidation of 8-oxoG and an array of products have already been identified under various oxidative conditions (9,36–38). Furthermore, the ability of an electron hole to migrate over long distances in duplex DNA (39) and finally equilibriating at an 8-oxoG site, suggests that 8-oxoG oxidation products from one-electron pathways could be easily induced. Therefore, we deemed it important to investigate the repair processes of one-electron oxidation products of 8-oxoG in DNA.
One-electron oxidation processes can be triggered in DNA by a variety of redox-active transition metals, photochemistry, ionizing radiation or addition of an oxygen radical followed by loss of an oxyanion. We have characterized Gh and Sp as two major one-electron oxidation products of 8-oxoG (11–13). Although these have not yet been identified in cellular genomes, they are likely to be formed in vivo, and their identification in DNA from oxidatively stressed cells is currently under way. We observed earlier that Gh and Sp have high mispairing potential because only G and A are inserted opposite these lesions during in vitro synthesis by E.coli Kf exo-DNA polymerase (O.Kornyushyna and C.Burrows, unpublished observations; 11). Recent studies have indicated that G·C→C·G and G·C→T·A transversions are predominant among mutations induced by ROS, which are more likely to arise from mispairing of G oxidation products rather than derivatives of C (40–43). Duarte et al. (44) have identified oxazolone as one of the one-electron and OH-mediated oxidation products of G, and further showed that it directed dAMP incorporation during DNA synthesis by E.coli Kf exo-DNA polymerase. G·C→C·G transversions were the major mutations induced by singlet oxygen-mediated DNA damage both in vivo and in vitro (42,43). Because both singlet oxygen and one-electron oxidation processes induce other oxidative modifications of G in addition to 8-oxoG (43), it is possible that many of the transversion mutations at G·C base pair sites in DNA result from such oxidation products, including Sp and Gh. G possesses the lowest ionization potential among the standard Watson–Crick bases and the ionization potential of G depends on its neighboring sequence in duplex DNA (45,46). It is thus likely that some of these Gs are converted to Gh or Sp. Schulz et al. (43) have recently reported that preferential induction of G·C→C·G transversions is predominant at those G residues which are flanked by another purine on their 3′ side. Such G residues also have the lowest ionization potential. Taken together, these results suggest that in ROS-mediated DNA damage, Gh or Sp could be a significant contributor to G to C transversion mutations.
Because oxidative base lesions and other small DNA adducts are repaired primarily via the BER process, which is initiated by excision of such lesions by specific DNA glycosylases, we investigated the ability of oxidized base-specific DNA glycosylases of E.coli, namely Nth, Nei and MutM, to repair Gh and Sp residues in DNA. Although these DNA glycosylases have broad and somewhat overlapping substrate specificity, they have distinct preferences for specific classes of lesions. The relative activities of MutM and Nei for 8-oxoG- and Sp(Gh)-containing oligos determined in the previous and present studies, respectively, could not be accurately compared because of the difference in size and sequence of 8-oxoG oligos used in the earlier studies. Nevertheless, the kcat/Km value (140 min–1 pM–1) of MutM for the 8-oxoG·C pair (47) is comparable to 240 min–1 pM–1 for the Gh·C pair determined in this study. The kcat values for these substrates are also comparable. In contrast, Nei appears to have a higher catalytic specificity (150 min–1 pM–1) for the Gh·G pair than for the 8-oxoG·G pair (6 min–1 pM–1) (27). These results suggest that MutM will be efficient in repairing Gh and Sp lesions in unreplicated DNA, and Nei will be more active in repairing these lesions compared to 8-oxoG when all of them mispair with G (or A) during DNA replication.
All three DNA glycosylases were active in excising both Sp and Gh from DNA and in carrying out the AP lyase reaction, although Nth had generally lower substrate specificity and catalytic activity than Nei and MutM. MutM is inactive with 8-oxoG when it is paired with A. This lack of activity was rationalized in the ‘GO model’, which postulated that repair of 8-oxoG opposite A would be mutagenic when A is incorporated in the nascent strand (21). We extended the GO model and predicted the need for repairing 8-oxoG from an 8-oxoG·A pair, specifically when A is in the template strand and directs incorporation of 8-oxoG in the nascent strand from the deoxynucleotide pool (27,33).
MutY, a DNA glycosylase, removes A (or G) when paired with 8-oxoG; its in vivo function is to prevent mutation due to misincorporation of A, as proposed in the GO model (21,35). It was somewhat surprising that MutY did not excise A or G when paired with either Gh or Sp while MutM is active on both 8-oxoG and these lesions. However, this result can be rationalized by postulating that removal of A or G by MutY from a Gh (Sp) base pair would be futile because in this situation, where the enzyme removes A or G paired with the lesion, unlike in the case of 8-oxoG, a C residue will never be incorporated opposite Sp or Gh to restore the wild-type sequence. Thus, MutY would not provide a second chance to prevent mutation as hypothesized in the GO model (21). Nevertheless, MutY was found to bind efficiently to the duplex oligo containing Gh or Sp when these were paired with G or A and not as strongly with C. This could explain why MutY did not inhibit MutM or Nei activity for excision of Sp and Gh when these lesions were paired with C. However, with Sp(Gh)·G pair-containing oligos, MutY strongly inhibited MutM, but did not inhibit Nei activity at least under the same reaction condition (Figs 8 and 9). Remarkably, a nearly identical pattern of differential inhibition of MutM and Nei by MutY was observed with the 8-oxoG substrate when it was paired with C, G or A (48). Similar results on inhibition of MutM by MutY for excision of 8-oxoG paired with various complementary bases have recently been reported by Li et al. (49).
We would like to propose that the above results reflect the mutation avoidance processes of E.coli, as schematically outlined in Figure 10. Sp and Gh (as well as 8-oxoG) are paired with C in the cellular genome when these lesions are generated from G in situ. Their repair by MutM, Nth or Nei will prevent mutation and thus should not be inhibited by MutY. When A or G is incorporated opposite 8-oxoG, repair of these normal bases by MutY will also prevent mutation as explained in the GO model. However, in the case of unrepaired Sp or Gh in the parental DNA, which may pair only with A or G but not with C during replication, excision of the incorporated base will not prevent mutation. Thus, in this situation, MutY binds to the base pair and provides a protective function by preventing excision of the lesion by DNA glycosylases. It is tempting to speculate that such protection of Sp or Gh by MutY then triggers another repair process, possibly homologous recombination, which would be the only mutation-avoidance process available in this situation. Recently, Samrakandi and Pasta (50) have shown that hyperrecombination in Streptococcus pneumoniae depends on MutY, suggesting that MutY is somehow involved in recombination. Furthermore, Valentine et al. (51) have shown that G·C→C·G and G·C→T·A mutations are significantly higher in the recA mutant than in wild-type E.coli, again suggesting a role of recombination in mutation avoidance.
Figure 10.

A model for antimutagenic repair of Sp and Gh (shown as X) in E.coli. (Left) The DNA lesions formed by ROS in situ are repaired by MutM, Nei or Nth prior to replication. Only G (or A) can be incorporated opposite Gh (Sp) and mutagenic repair of the lesion by the DNA glycosylases is prevented by binding of MutY. Homologous recombination restores the original DNA sequence. (Right) Sp (Gh) could be generated by ROS in the deoxynucleotide pool from which it is incorporated opposite G (A). Only Nei but not MutM excises the lesion to prevent subsequent mutation. Dashed line indicates nascent strand.
The fact that only Nei is able to excise Sp (or Gh) when these are paired with A suggests that the primary role of Nei is to remove such lesions which may be incorporated from the deoxynucleotide pool in the nascent strand opposite G or A, as we had proposed in the case of 8-oxoG (27,33). Deoxynucleoside triphosphate of Sp is expected to be produced by oxidation of 8-oxodGTP in the pool. 8-oxodG was indeed shown to be a preferred precursor of the Sp deoxynucleoside (12).
In a recent report we showed that MutM, at a higher concentration, was able to excise Sp or Gh when these were paired with A (or G) in a duplex oligo (13). Such repair will cause mutation in vivo if A or G is incorporated opposite the lesion during DNA replication. While this could explain the observed G·C→T·A (or C·G) mutation, we expect that the major function of the repair process is to prevent mutation which may involve complex interaction among proteins including MutY.
We have investigated the effect of the opposite base on repair of Gh and Sp lesions by these DNA glycosylases. It is quite interesting that the orders of their preference for the complementary base for these lesions are identical. Thus, MutM does not excise either the hydantoins or 8-oxoG when the opposite base is A, while Nei prefers G or A as the opposite base regardless of whether the substrate is 8-oxoG, Sp or Gh. These results suggest a critical role of the opposite base in substrate recognition for both enzymes which could be elucidated by solving the structure of the enzyme–substrate complex by X–ray crystallography.
Finally, in view of the likely presence of Sp and Gh in genomes of all organisms, it is important to test whether the mammalian orthologs of E.coli DNA glycosylases are able to repair these hydantoins.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr David Konkel for critically reading the manuscript and Ms Wanda Smith for secretarial assistance. We also thank Dr Wah Kow for a gift of plasmids. This research was supported by the US Public Health Service grants R01 CA81063 (to S.M.), Welch Foundation H1402 (to R.S.L.), NSF CHE9818484 (to C.J.B.) and by the NIEHS Center grant ES06676.
References
- 1.Grisham M.B. and McCord,J.M. (1986) Physiology of Oxygen Radicals. Waverly Press, Baltimore, MD, pp. 1–18.
- 2.Ward J.F. (1994) The complexity of DNA damage: relevance to biological consequences. Int. J. Radiat. Biol., 66, 427–432. [DOI] [PubMed] [Google Scholar]
- 3.Helbock H.J., Beckman,K.B. and Ames,B.N. (1999) 8-Hydroxydeoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzymol., 300, 156–166. [DOI] [PubMed] [Google Scholar]
- 4.Ames B.N., Shigenaga,M.K. and Hagen,T.M. (1993) Oxidants, antioxidants and the degenerative diseases of aging. Proc. Natl Acad. Sci. USA, 90, 7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasai H. and Nishimura,S. (1991) In Sies,H. (ed), Oxidative Stress: Oxidants and Antioxidants. Academic Press, Ltd, London, pp. 99–116.
- 6.Maki H. and Sekiguchi,M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature, 355, 273–275. [DOI] [PubMed] [Google Scholar]
- 7.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 8.Hollstein M., Shomer,B., Greenblatt,M., Soussi,T., Hovig,E., Montesano,R. and Harris,C.C. (1996) Somatic point mutations in the p53 gene of human tumors and cell lines: updated compilation. Nucleic Acids Res., 24, 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niles J.C., Burney,S., Singh,S.P., Wishnok,J.S. and Tannenbaum,S.R. (1999) Peroxynitrite reaction products of 3′,5′-di-O-acetyl-8-oxo-7,8-dihydro-2′-deoxyguanosine. Proc. Natl Acad. Sci. USA, 96, 11729–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goyal R.N. and Dryhurst,G. (1982) Redox chemistry of guanine and 8-oxoguanine and a comparison of the perioxidase catalyzed and electrochemical oxidation of 8-oxoguanine. J. Electroanal. Chem., 135, 75–91. [Google Scholar]
- 11.Duarte V., Muller,J.G. and Burrows,C.J. (1999) Insertion of dGMP and dAMP during in vitro DNA synthesis opposite an oxidized form of 7,8-dihydro-8-oxoguanine. Nucleic Acids Res., 27, 2247–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo W., Muller,J.G., Rachlin,E.M. and Burrows,C.J. (2000) Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org. Lett., 2, 613–616. [DOI] [PubMed] [Google Scholar]
- 13.Leipold M.D., Muller,J.G., Burrows,C.J. and David,S.S. (2000) Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry, 39, 14984–14992. [DOI] [PubMed] [Google Scholar]
- 14.Muller J.G., Duarte,V., Hickerson,R.P. and Burrows,C.J. (1998) Gel electrophoretic detection of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine via oxidation by Ir (IV). Nucleic Acids Res., 26, 2247–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oller A.R., Costa,M. and Oberdorster,G. (1997) Carcinogenicity assessment of selected nickel compounds. Toxicol. Appl. Pharmacol., 143, 152–166. [DOI] [PubMed] [Google Scholar]
- 16.Thorp H.H. (1995) Electron-, energy- and atom-transfer reactions between metal complexes and DNA. Adv. Inorg. Chem., 43, 127–175. [Google Scholar]
- 17.Muller J.G., Hickerson,R.P., Perez,R.J. and Burrows,C.J. (1997) DNA damage from sulfite autooxidation catalyzed by a nickel (II) peptide. J. Am. Chem. Soc., 119, 1501–1506. [Google Scholar]
- 18.Burrows C.J. and Rokita,S.E. (1996) Nickel complexes as probes of guanine sites in nucleic acid folding. In Sigel,A. and Sigel,H. (eds), Metal Ions in Biological Systems. Dekker, New York, NY, Vol. 33, pp. 537–559. [PubMed]
- 19.Tchou J., Kasai,H., Shibutani,S., Chung,M.-H., Laval,J., Grollman,A.P. and Nishimura,S. (1991) 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl Acad. Sci. USA, 88, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 21.Michaels M.L. and Miller,J.H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol., 174, 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kow Y. and Wallace,S.S. (1987) Mechanism of action of Escherichia coli endonuclease III. Biochemistry, 26, 8200–8206. [DOI] [PubMed] [Google Scholar]
- 23.Melamede R.J., Hatahet,Z., Kow,Y.W., Ide,H. and Wallace,S.S. (1994) Isolation and characterization of endonuclease VIII from Escherichia coli. Biochemistry, 33, 1255–1269. [DOI] [PubMed] [Google Scholar]
- 24.Gates F.T. and Linn,S. (1977) Endonuclease from Escherichia coli that acts specifically upon duplex DNA damaged by ultraviolet light, osmium tetroxide, acid, or x-rays. J. Biol. Chem., 252, 2802–2807. [PubMed] [Google Scholar]
- 25.Radman M. (1976) An endonuclease from Escherichia coli that introduces single polynucleotide chain scissions in ultraviolet-irradiated DNA. J. Biol. Chem., 251, 1438–1445. [PubMed] [Google Scholar]
- 26.Jiang D., Hatahet,Z., Melamede,R.J., Kow,Y.W. and Wallace,S. (1997) Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem., 272, 32230–32239. [DOI] [PubMed] [Google Scholar]
- 27.Hazra, T.K., Izumi,T., Venkataraman,R., Kow,Y.W., Dizdaroglu,M. and Mitra,S. (2000) Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J. Biol. Chem., 275, 27762–27767. [DOI] [PubMed] [Google Scholar]
- 28.Asahara H., Wistort,P.M., Bank,J.F., Bakerian,R.H. and Cunningham,R.P. (1989) Purification and characterization of Escherichia coli endonuclease III from the cloned nth gene. Biochemistry, 28, 4444–4449. [DOI] [PubMed] [Google Scholar]
- 29.Manuel, R.C., Czerwinski,E.W. and Lloyd,R.S. (1996) Identification of the structural and functional domains of MutY, an Escherichia coli DNA mismatch repair enzyme. J. Biol. Chem., 271, 16218–16226. [DOI] [PubMed] [Google Scholar]
- 30.Manuel, R.C. and Lloyd,R.S. (1997) Cloning, overexpression and biochemical characterization of the catalytic domain of MutY. Biochemistry, 36, 11140–11152. [DOI] [PubMed] [Google Scholar]
- 31.Dodson M.L., Michaels,M.L. and Lloyd,R.S. (1994) Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem., 269, 32709–32712. [PubMed] [Google Scholar]
- 32.Nash H.M., Bruner,S.D., Scharer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 33.Hazra T.K., Izumi,T., Maidt,L., Floyd,R.A. and Mitra,S. (1998) The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Res., 26, 5116–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda S., Biswas,T., Roy,R., Izumi,T., Boldogh,I., Kurosky,A., Sarker,A.H., Seki,S. and Mitra,S. (1998) Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem., 273, 21585–21593. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Q.M., Ishikawa,N., Nakahara,T. and Yonei,S. (1998) Escherichia coli MutY protein has a guanine-DNA glycosylase that acts on 7,8-dihydro-8-oxoguanine:guanine mispairs to prevent spontaneous G:C→C:G transversions. Nucleic Acids Res., 26, 4669–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adam W., Saha-Moller,C.R., Schonberger,A., Berger,M. and Cadet,J. (1995) Formation of 7,8-dihydro-8-oxoguanine in the 1,2-dioxetane-induced oxidation of calf thymus DNA: evidence for photosensitized DNA damage by thermally generated triplet ketones in the dark. Photochem. Photobiol., 62, 231–238. [DOI] [PubMed] [Google Scholar]
- 37.Goyal R.N., Jain,N. and Garg,D.K. (1997) Electrochemical and enzymic oxidation of guanosine and 8-hydroxyguanosine and the effects of oxidation products in mice. Bioelectrochem. Bioenerg., 43, 104–114. [Google Scholar]
- 38.Sheu C. and Foote,C.S. (1995) Photosensitized oxygenation of a 7,8-dihydro-8-oxoguanosine derivative. Formation of dioxetane and hydroperoxide intermediates. J. Am. Chem. Soc., 117, 474–477. [Google Scholar]
- 39.Hall D.B., Holmlin,R.E. and Barton,J.K. (1996) Oxidative DNA damage through long-range electron transfer. Nature, 382, 731–735. [DOI] [PubMed] [Google Scholar]
- 40.McBride T.J., Preston,B.D. and Loeb,L.A. (1991) Mutagenic spectrum resulting from DNA damage by oxygen radicals. Biochemistry, 30, 207–213. [DOI] [PubMed] [Google Scholar]
- 41.McBride T.J., Schneider,J.E., Floyd,R.A. and Loeb,L.A. (1992) Mutations induced by methylene blue plus light in single-stranded M13mp2. Proc. Natl Acad. Sci. USA, 89, 6866–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Oliveira R.C., Ribeiro,D.T., Nigro,R.G., Di Mascio,P. and Menck,C.F.M. (1992) Singlet oxygen induced mutation spectrum in mammalian cells. Nucleic Acids Res., 20, 4319–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulz I., Mahler,H.-C., Boiteux,S. and Epe,B. (2000) Oxidative DNA base damage induced by singlet oxygen and photosensitization: recognition by repair endonucleases and mutagenicity. Mutat. Res., 401, 145–150. [DOI] [PubMed] [Google Scholar]
- 44.Duarte V., Gasparutto,D., Jaquinad,M. and Cadet,J. (2000) In vitro DNA synthesis opposite oxazolone and repair of this DNA damage using modified oligonucleotides. Nucleic Acids Res., 28, 1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito I., Takayama,M., Sugiyama,H., Nakatani,K., Tsuchida,A. and Yamamoto,M. (1995) Photoinduced DNA cleavage via electron transfer: demonstration that guanine residues located 5′ to guanine are the most electron-donating sites. J. Am. Chem. Soc., 117, 6406–6407. [Google Scholar]
- 46.Sugiyama H. and Saito,I. (1996) Theoretical studies of GG-specific photocleavage of DNA via electron transfer: significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA. J. Am. Chem. Soc., 118, 7063–7068. [Google Scholar]
- 47.Asagoshi K., Yamada,T., Terato,H., Ohyama,Y., Monden,Y., Arai,T., Nishimura,S., Aburatani,H., Lindahl,T. and Ide,H. (2000) Distinct repair activities of human 7,8-dihydro-8-oxoguanine DNA glycosylase and formamidopyrimidine DNA glycosylase for formamidopyrimidine and 7,8-dihydro-8-oxoguanine. J. Biol. Chem., 275, 4956–5964. [DOI] [PubMed] [Google Scholar]
- 48.Hazra T.K., Hill,J.W., Izumi,T. and Mitra,S. (2001) Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. In Proceedings DNA Base Excision Repair Workshop 2000. Special issue of Prog. Nucleic Acid Res. Mol. Biol., in press. [DOI] [PubMed] [Google Scholar]
- 49.Li X., Wright,P.M. and Lu,A.-L. (2000) The C-terminal domain of MutY glycosylase determines the 7,8-dihydro-8-oxo-guanine specificity and is crucial for mutation avoidance. J. Biol. Chem., 275, 8448–8455. [DOI] [PubMed] [Google Scholar]
- 50.Samrakandi M.M. and Pasta,F. (2000) Hyperrecombination in Streptococcus pneumoniae depends on an atypical mutY homologue. J. Bacteriol., 182, 3353–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valentine M.R., Rodriquez,H. and Termini,J. (1998) Mutagenesis by peroxy radical is dominated by transversions at deoxyguanosine: evidence for the lack of involvement of 8-oxo-dG1 and/or abasic site formation. Biochemistry, 37, 7030–7038. [DOI] [PubMed] [Google Scholar]